

Abstract
Over the past 70 years, diffuse axonal injury (DAI) has emerged as one of the most common and important pathological features of traumatic brain injury (TBI). Axons in the white matter appear to be especially vulnerable to injury due to the mechanical loading of the brain during TBI. As such, DAI has been found in all severities of TBI and may represent a key pathologic substrate of mild TBI (concussion). Pathologically, DAI encompasses a spectrum of effects from primary mechanical breaking of the axonal cytoskeleton, to transport interruption, swelling and proteolysis, through secondary physiological changes. Depending on the severity and extent of injury, these changes can manifest acutely as immediate loss of consciousness or confusion and persist as coma and/or cognitive dysfunction. In addition, recent evidence suggests that TBI may induce long-term neurodegenerative processes, such as insidiously progressive axonal pathology. Indeed, axonal degeneration has been found to continue even years after injury in humans, and appears to play a role in the development of Alzheimer’s disease-like pathological changes. Here we review the current understanding of DAI as a uniquely mechanical injury, its histopathological identification, and its acute and chronic pathogenesis following TBI.
Keywords: Axon, Diffuse axonal Injury, DAI, axonal pathology, traumatic brain injury, TBI, neurodegeneration, head injury, rotational acceleration, microtubule
Introduction
Although historically ignored as a major health issue, traumatic brain injury (TBI) is a leading cause of morbidity and mortality internationally, with significant socio-economic implications. In the US alone, over 1.7M individuals suffer a TBI each year (Faul, et al., 2010) at an estimated cost of over $60 billion (Finkelstein E, et al., 2006). Moreover, there is considerable evidence indicating just a single TBI may be associated with the later onset of neurodegenerative disorders, including Alzheimer’s disease (AD) (Fleminger, et al., 2003, Graves, et al., 1990, Guo, et al., 2000, Johnson, et al., Molgaard, et al., 1990, Mortimer, et al., 1985, Mortimer, et al., 1991, O’Meara, et al., 1997, Plassman, et al., 2000, Salib and Hillier, 1997, Schofield, et al., 1997).
While the neuropathological consequences of TBI are heterogeneous, one of the most common across all severities of closed head injury is diffuse axonal injury (DAI) (Adams, et al., 1989, Adams, et al., 1982, Graham, et al., 1988, Povlishock, 1992, Povlishock and Becker, 1985, Povlishock, et al., 1983, Povlishock and Katz, 2005, Smith and Meaney, 2000), which may reflect the selective vulnerability of white matter axons to damage from mechanical loading of the brain during rapid head accelerations. After TBI, axonal degeneration arising from DAI is conventionally recognized as a progression from disruption in axonal transport leading to axonal swelling followed by secondary disconnection and, finally, Wallerian degeneration. Traditionally this process was thought to be limited to the acute and sub-acute periods following trauma. However, recent evidence has identified axonal degeneration in human brain material years following injury, suggesting TBI may precipitate a progressive, long-term neurodegenerative process, in part reflected in axonal degeneration (Chen, et al., 2009). Of particular note, axonal pathology may have a role in the development of Alzheimer-like pathologies both in the acute phase following injury as well as with longer term survival (Chen, et al., 2009, Chen, et al., 2004, Johnson, et al., 2010, Marklund, et al., 2009, Smith, et al., 2003, Smith, et al., 1999, Smith, et al., 2003, Stone, et al., 2002, Tran, et al.). Here we explore the current understanding of the short- and long-term pathological sequelae of axonal degeneration following TBI.
Historical Perspective of DAI
Classical descriptions of diffuse axonal injury are of a clinicopathological syndrome manifest as a patient unconscious from the time of injury where, on subsequent autopsy examination of the brain, there is widespread axonal injury in the cerebral hemispheres, cerebellum and brainstem. Approaching the middle of last century, the pathological appearances of trauma related axonal injury were first described in human tissue where, in addition to the previously well-known macroscopic focal lesions, microscopic examination revealed TBI could also induce widespread, subtle and yet seemingly important pathological changes throughout the brain parenchyma (Rand and Courville, 1946). Apparently unique to TBI, it was proposed that these pathological white matter changes occur as a direct consequence of the mechanical forces experienced at the time of injury.
This hypothesis evolved from experimental observations by Holbourn who induced rapid rotational forces on gelatin molds of brain sections and observed that rotational accelerations, at levels akin to those calculated for head rotations in human TBI, could induce widely distributed shear and tensile strains throughout the surrogate brain material (Holbourn, 1943, Holbourn, 1945). Following detailed histopathological examination of tissue from severely injured TBI patients, it was proposed that these shear strains at the moment of injury were likely to be responsible for the observed morphological alterations in white matter (Strich, 1956, Strich, 1961).
Further neuropathological assessments emerged in support of this concept, with axonal pathology identified at very early time points post-injury (Nevin, 1967). In addition, it became clear that the presence of swollen axonal profiles could be identified across a range of injury severities (Oppenheimer, 1968, Peerless and Rewcastle, 1967), though in these early studies, the injuries would now be regarded as in the moderate to severe range (i.e. Glasgow coma scales at presentation less than 13).
Subsequently, Adams and colleagues performed extensive characterization of the extent and distribution of axonal pathology in a large series of TBI cases and introduced the now universally recognized term “diffuse axonal injury” (Adams, et al., 1982, Gennarelli, et al., 1982). In addition to confirming the presence of axonal pathology in patients dying shortly after a moderate to severe injury, DAI was identified following various causes of injury including rapid acceleration/deceleration, such as can occur in motor vehicle collisions, and direct impacts, such as from falls or assaults (Gennarelli, et al., 1982). It was these data that led Adams and colleagues to develop and refine a grading system for DAI based on the extent and distribution of pathology (Adams JH, 1989, Adams, et al., 1982).
Diagnosis of DAI, other than through histopathological examination, has remained a major challenge. As a consequence of its microscopic and disseminated nature, the axonal pathology of DAI is not readily discernable with standard non-invasive techniques, such as conventional CT or MRI. Therefore, this ‘stealth’ pathology is often missed or regarded as a diagnosis of exclusion based on symptoms in the absence of overt changes with conventional neuroimaging following TBI. To further complicate matters, the term “diffuse axonal injury” is itself somewhat of a misnomer, since technically the distribution is not diffuse, but is instead stereotypically multifocal; preferentially involving midline white matter tracts such as the corpus callosum, internal capsules, brainstem and cerebellar peduncles (Adams JH, et al., 1989, Adams, et al., 1991). Moreover, the term diffuse axonal injury has occasionally been adopted in other fields where widespread white matter pathology may be encountered, such as demyelinating diseases and in reference to hypoxic/ischemic injury. However, while there have been efforts to underline the etiology of the axonal injury in trauma, using terms such as “diffuse traumatic axonal injury (dTAI)” or “traumatic axonal injury (TAI)”, the clinical designation in relation to trauma induced diffuse axonal damage remains “DAI”, which will be the terminology used in this review.
DAI: Pathological Features and Identification
A primary outcome of dynamic deformation of white matter tracts during trauma is the interruption of axonal transport, resulting in accumulation of transported materials as axonal swellings within just hours of trauma (Christman, et al., 1994, Povlishock and Becker, 1985, Smith, et al., 1999). Commonly, these swellings appear in a periodic arrangement along the length of an axon at the site of injury, classically referred to as “axonal varicosities” (Figure 1a,c-d). A more widely recognized, but not necessarily more common, axonal pathology found shortly after TBI is a large single swelling described as an “axonal bulb” (previously referred to as a “retraction ball”), which likely represents complete axonal disconnection (Figure 1a-b) (Adams, et al., 1989, Adams, et al., 1984, Adams, et al., 1982, Cajal, 1928, Chen, et al., 1999, Povlishock, 1992, Povlishock and Becker, 1985, Povlishock, et al., 1983, Povlishock, et al., 1999, Povlishock and Katz, 2005, Rand and Courville, 1946, Smith and Meaney, 2000, Smith and Meaney, 2000, Smith, et al., 2003, Strich, 1956).
Figure 1. Representative Images of Axonal Pathology Following TBI in Humans Identified Using APP Immunohistochemistry.
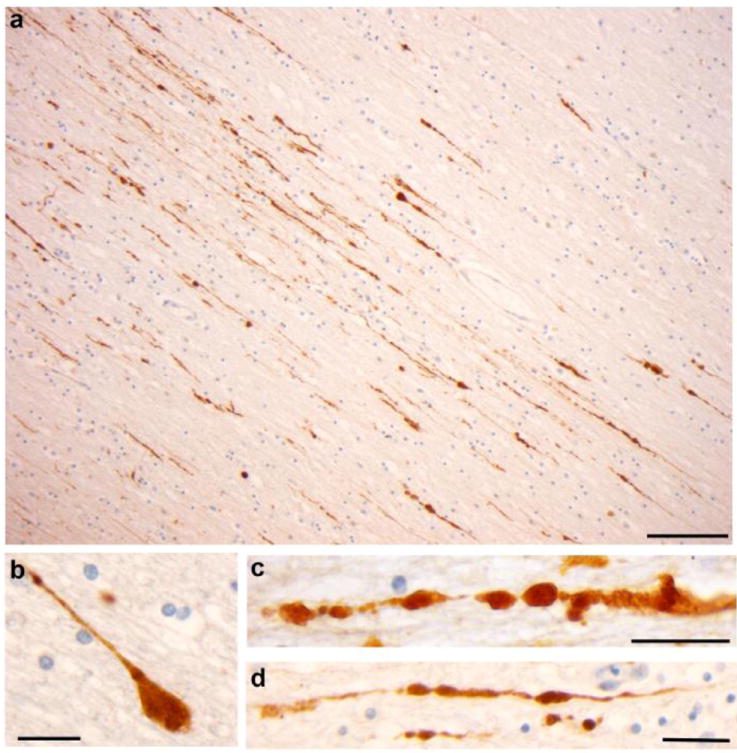
(a) Extensive axonal pathology with classic varicosities and axonal bulb formation in a region of the corpus callosum of an young male who died 10 hours following blunt force trauma to the head. Scale bar: 100μm (b) High magnification of a single axon immunoreactive for APP displaying the classic morphology of an axonal bulb. Scale Bar 15 μm. (c-d) High magnification of a single axon accumulating APP. Axons are morphologically varicose, exhibiting multiple points of transport interruption to give the appearance of beads on a string. Scale bars: 30μm.
The histopathological identification of DAI is dependent upon the visualization of abnormal axonal profiles as described above. Historically, standard tinctorial stains, such as Hematoxlylin and eosin (H&E), and various silver impregnation techniques, such as Palmgren’s, were successfully applied to identify damaged axons. However, the introduction of immunohistochemical methods revealed that such techniques often under-represent the extent of axonal pathology and are limited in their capacity to identify damaged axons in individuals with very short survival times. Following immunohistochemical examination of multiple candidate proteins accumulating in injured axons (Grady, et al., 1993, Gultekin and Smith, 1994, Ng, et al., 1994, Sherriff, et al., 1994), immunoreactivity to amyloid precursor protein (APP) has emerged as a highly sensitive and robust technique for the detection of DAI (Gentleman, et al., 1993, Sherriff, et al., 1994) (Figure 1). Transported by fast axonal transport, APP may be identified accumulating in damaged axons within 2 hours following injury. Moreover, when directly compared to silver impregnation techniques, APP staining reveals far more extensive axonal pathology (Gentleman, et al., 1995). As such, APP immunohistochemistry remains the gold-standard for the clinico-pathological identification of axonal pathology. However, accumulation of APP in axons is not exclusive to trauma and has been described following other mechanisms of brain injury including hypoxic/ischemic injury (Graham et al 2004; Reichard et al 2005). In this regard, careful assessment of the extent and distribution of pathology, as well as its evolution in relation to survival time, is critical in the assessment of DAI.
While diagnostic confirmation of DAI is currently only possible with histopathological examination of post-mortem brain tissue, increasing evidence suggests novel advanced neuroimaging techniques may be useful in the assessment of white matter tracts in vivo. In particular, diffusion tensor imaging (DTI) has emerged as a promising technique for assessing white matter integrity via the measurement of the anisotropic diffusion of water molecules (for review see (Hunter, et al., 2011). Indeed, early evidence indicates DTI may be useful in assessing patients with even mild TBI (Bazarian, et al., 2007, Bazarian, et al., Mayer, et al., Miles, et al., 2008, Wilde, et al., 2008). However, considerable work remains before DTI can be reasonably included as part of the routine clinical diagnosis of TBI. Notably, it remains unclear what the neuropathological substrates are for changes observed using DTI, and thus whether findings are representative of the axonal pathology of DAI.
DAI Genesis and the Link with Coma and Transient Loss of Consciousness
Animal models have been instrumental in confirming that the principal mechanical force responsible for DAI is rotational acceleration of the brain, resulting from unrestricted head movement inducing dynamic shear, tensile, and compressive strains within the tissue (Gennarelli, et al., 1982, Meaney, et al., 1996, Ommaya and Hirsch, 1971, Smith, et al., 1997, Thibault, et al., 1990). As such, the size of the human brain plays an important role in the development of DAI, as the substantial mass effects occurring during injury can result in high shear strains between regions of tissue (Smith and Meaney, 2000, Smith, et al., 1996). Few clinically relevant models of DAI in gyrencephalic animals have been characterized. This reflects the difficulty of developing a model system that replicates the dynamics of diffuse injury, such as the inertial loading conditions produced in automotive crashes or at the moment of head impact (Smith, et al., 1996, Smith, 2003).
The first model of DAI was developed at the University of Pennsylvania in the 1980s by Gennarelli and colleagues (Gennarelli, et al., 1982). In landmark studies, they demonstrated that head rotational acceleration in non-human primates could induce extensive axonal pathology throughout the white matter with identical characteristics to human DAI. Moreover, Gennarelli and colleagues found that DAI was responsible for immediate and prolonged post-traumatic coma, independent of a mass lesion. Non-human primates were originally chosen due to their large brain mass with extensive white matter domains, similar to the human brain. Nonetheless, despite their relatively sizable brains (approximately 95g), far greater rotational accelerations were necessary to generate the same tissue deformations calculated for the larger human brain in TBI (Holbourn, 1943, Margulies, et al., 1990). These data were the first to conclusively link dynamic mechanical deformation of the brain during trauma with the selective pathology of DAI. In addition, the concurrence of DAI with immediate and prolonged coma in this animal model was directly extrapolated to transform the clinical diagnosis of DAI in TBI patients.
Subsequently, using the same injury device, a similar model was developed using miniature swine which have gyrencephalic brains of similar size to non-human primates (Meaney, et al., 1995, Smith, et al., 1997). Studies using this model demonstrated that the plane of head rotational acceleration in reference to the brainstem is critical in determining the induction and duration of loss of consciousness following injury. Specifically, rotation, transverse to the brainstem, was associated with coma, whereas equivalent rotation circumferential to the brainstem was not (Smith, et al., 2000). Notably, even at mild parameters of injury, loss of consciousness was induced following rotations transverse to the brainstem (Browne, et al., 2011). The duration of loss of consciousness or coma following injury was directly related to the extent of axonal pathology in the brainstem, indicating brainstem damage is a key anatomic substrate for immediate loss of consciousness in TBI (Smith, et al., 2000). However, when the brainstem was relatively spared, even extensive axonal pathology throughout the hemispheric white matter produced little or no loss of consciousness. Thus, while there is a link between DAI and immediate post-traumatic loss of consciousness, it appears that the distribution, rather than the overall extent, of axonal pathology is important in determining consciousness immediately following TBI (Browne, et al., 2011).
Potential Primary Mechanical Damage due to Axonal Trauma
With evidence that mechanical damage to axons can directly account for clinical symptoms, the biomechanical nature of TBI was increasingly recognized as an important and unique feature. Specifically, the viscoelastic properties of the brain emerged as a potential liability during the rapid mechanical loading conditions of TBI. White matter axons appear especially vulnerable to injury under such circumstances, potentially as a result of their highly anisotropic arrangement and/or their inherent structural design. In normal circumstances, axons are compliant and ductile under stretch, readily relaxing back to their original length when the stretch is removed. However, with rapid application of tissue strain, such as at the moment of head impact, axons behave differently, essentially becoming brittle (Smith and Meaney, 2000, Smith, et al., 1999). Nonetheless, disconnection of axons at the moment of injury, known as “primary axotomy”, is considered a relatively rare occurrence. Instead, in the majority of cases, the swelling that follows cytoskeletal disruption can induce “secondary axotomy” (Christman, et al., 1994). Due to the critical role of mechanical damage in the development of DAI, evaluation of the effects of dynamic tissue deformation on the axonal cytoskeleton during trauma has been the subject of various avenues of investigation both from a structural and functional perspective (Chung, et al., 2005, Jafari, et al., 1997, Jafari, et al., 1998, Maxwell, et al., 2003, Maxwell and Graham, 1997, Maxwell, et al., 1999, Maxwell, et al., 1995, Pettus and Povlishock, 1996, Povlishock and Pettus, 1996, Staal, et al., 2009, Staal, et al., 2010).
Direct evidence of primary mechanical damage of axons has been shown using an in vitro model of dynamic stretch injury of micropatterned axons spanning two populations of cortical neurons (Iwata, et al., 2004, Smith, et al., 1999, Stys and Jiang, 2002, Tang-Schomer, et al., 2010, Wolf, et al., 2001). Within seconds of dynamic axonal stretch, axons temporarily become undulated and misaligned due to loss of elasticity and underlying cytoskeletal damage (Smith, et al., 1999). Notably, axonal undulations are also a common feature of acute TBI in humans, suggesting primary cytoskeletal failure due to mechanical trauma (Tang-Schomer, et al., 2011).
Recently, in vitro studies have shown that primary breaking of axonal microtubules underlies the observed posttraumatic axonal undulations. Specifically, twisting and misalignment of broken microtubules at multiple sites along injured axons appears to impede relaxation of axons back to their original straight orientation. Although subsequent depolymerization of the microtubules from the break points can allow gradual relaxation of the axons, it comes at a cost, by interrupting axonal transport and inducing progressive swellings and degeneration (Tang-Schomer, et al., 2010). These observations may explain the apparent loss of axonal microtubules previously found in a feline model of TBI and a guinea pig model of dynamic optic nerve stretch injury (Maxwell and Graham, 1997, Pettus and Povlishock, 1996). Notably, by manipulating microtubule stability in the in vitro stretch-injury model using the microtubule-stabilizing drug Taxol, subsequent axonal degeneration post-injury could be mitigated (Tang-Schomer, et al., 2010).
In addition to the inherent vulnerability of white matter axons to damage in TBI, there is a differential sensitivity of axon subtypes. Indeed, several studies indicate that myelinated fibers are more tolerant to mechanical strains compared to unmyelinated fibers, with both in vivo and in vitro TBI models (Reeves, et al., 2007, Reeves, et al., 2005, Staal and Vickers, 2011). Specifically, in vivo, smaller unmyelinated axons were found more likely to suffer irreversible dysfunction of conduction pathways, while an in vitro model of axon stretch-injury showed that non-myelinated axons were more prone to secondary disconnection when compared to myelinated axons. However, it remains unknown how these differences are related to structural variation between these populations of neurons.
These observations provide a glimpse of the mechanical genesis of selective axonal pathology following trauma leading to cytoskeletal failure and disconnection. Ultimately, it is thought that disconnected axons undergo Wallerian degeneration. However, the possibility that many swollen or otherwise damaged axons may undergo repair remains an intriguing concept. Conceivably, axonal repair could range anywhere from restoration of simple ionic homeostasis through direct replacement of the damaged cytoskeleton such as turnover of microtubules or neurofilaments (Chen, et al., 1999, Tang-Schomer, et al., 2011). For example, it was recently demonstrated that the periodic swellings that comprise axonal varicosities represent a form of “partial interruption of axonal transport” resulting from a staggering of break points between microtubules within axons. This damage at the level of individual microtubules induces only limited derailment and accumulation of transported cargoes at periodic regions of the axon, thereby creating the varicose appearance (Tang-Schomer, et al., 2011). Thus, if none of the swellings grow to the point of inducing disconnection, repair of the microtubule lattice may provide an opportunity for injured axons to contend with the residual protein accumulation. Examination of the mechanisms of axonal repair after trauma will be important for future considerations of therapeutic interventions.
Secondary Chemical Cascades Following TBI
During TBI, all axons within a white matter tract are thought to suffer relatively similar dynamic deformations. Yet, even in severe TBI, only a small percentage of axons within a given tract undergo transport interruption as classically identified by accumulation of transported cargoes in swellings. For the remaining axons that do not display appreciable interruption to transport following dynamic deformation, they may nonetheless suffer important pathophysiological changes capable of contributing to axonal dysfunction, such as the altered conduction velocities observed in animal models, even in mTBI (Baker, et al., 2002, Reeves, et al., 2007, Reeves, et al., 2005). Such secondary cascades may result in degeneration of axons or, conversely, render axons viable yet functionally impaired.
A multitude of diverse secondary cascades detrimental to axons have been investigated following TBI. Specifically, alterations to mitochondria, including mictochondrial permeability transition are potentially detrimental to normal energy metabolism and thus axonal integrity (Buki, et al., 1999, Maxwell, et al., 2003, Okonkwo and Povlishock, 1999). In addition, oxidative stress and lipid peroxidation have long been implicated in the pathophysiology of TBI and have been associated with both mitochondrial dysfunction and cytoskeletal degradation in vivo, a process that can be mitigated via treatment using free radical scavengers (Deng, et al., 2007, Deng-Bryant, et al., 2008, Fujita, et al., Mustafa, et al., Mustafa, et al.). Increasing evidence suggests that neuroinflammation and microglial activation in the white matter may also contribute to cellular damage (Loane and Byrnes, 2010) and remarkably, can persist for even years after injury in humans (Chen, et al., 2009, Gentleman, et al., 2004).
Of particular note, ionic imbalance after axonal trauma has been thought to play a central role post-injury in both axonal degeneration and the persistent dysfunction of otherwise intact axons. Specifically, it was long-suspected that elevated intra-axonal Ca2+ levels ([Ca2+]i) play a pivotal role in the secondary damage to axons following mechanical deformation (Banik, et al., 1987, Benz, et al., 1997, Buki, et al., 2003, Buki, et al., 1999, Gitler and Spira, 1998, Ichiya, et al., 1991, Maxwell, et al., 1999, Maxwell, et al., 1995, Maxwell, et al., 1991, Povlishock, 1992, Povlishock, et al., 1999, Wolf, et al., 2001). Maxwell and colleagues found indirect evidence of post-traumatic calcium influx into axons via changes in calcium-ATPase activity after optic nerve stretch injury (Maxwell, et al., 1999, Maxwell, et al., 1995, Maxwell, et al., 1991). Subsequently, using an in vitro axon stretch model, direct visual evidence of calcium entry into axons was seen immediately following trauma (Wolf, et al., 2001). Surprisingly, this post-traumatic rise in [Ca2+]i was found to be dependent on entry of sodium via voltage-gated channels and reversal of the Na+/Ca2+ exchanger (Iwata, et al., 2004). Recent work using another in vitro axon stretch injury model indicates that acute increases in [Ca2+] may also, in part, originate from release of intracellular stores of [Ca2+](Staal, et al., 2010). In addition, both in vitro and in vivo models have shown that increases in the [Ca2+] - activated protease, calpain, was demonstrated to cause a range of subtle to catastrophic damage to the axonal cytoskeleton and ion channels (Buki, et al., 1999, Huh, et al., 2006, Iwata, et al., 2004, Kampfl, et al., 1996, Kupina, et al., 2001, McGinn, et al., 2009, Saatman, et al., 2003, Saatman, et al., 1996, Saatman, et al., 1996, von Reyn, et al., 2009). For example, activated calpain appears responsible for degradation of the inactivation gate of sodium channels, which can cause a deleterious feed-forward process of unmitigated sodium influx, in turn, inducing progressively increasing intraaxonal calcium levels and related pathogenesis (Iwata, et al., 2004, von Reyn, et al., 2009). Inhibition of either calpain and another calcium-activated enzyme, calcineurin was found to mitigate axonal degeneration following injury in vivo (Marmarou and Povlishock, 2006, Reeves, et al., 2007, Saatman, et al., 2000, Singleton, et al., 2001), the latter of which may additionally promote recovery via the promotion of axonal sprouting observed following stretch injury in vitro (Staal, et al., 2010).
Limited clinical and experimental evidence suggests that demyelination may also play an important role in the pathophysiology of TBI. Experimental studies demonstrate disruptions of the myelin sheath acutely following stretch-injury of the guinea pig optic nerve (Maxwell, et al., 2003, Maxwell, et al., 1999). In addition, histochemical analysis of the rat brain following fluid percussion injury showed a loss of myelin staining as indicated by luxol fast blue in association with progressive white matter atrophy up to 1 year post injury (Bramlett and Dietrich, 2002). Similar histochemical analysis of DAI in humans revealed acute myelin globoids in association with axonal pathology (Ng, et al., 1994). However, it is unclear whether damage to the myelin sheath occurs only as a direct consequence of axonal degeneration. Interestingly, apoptotic oligodendrocytes have been observed acutely and chronically following TBI clinically (Shaw, et al., 2001). Such a loss of oligodendrocytes may result in insufficient myelination and potentially compromise the integrity or function of axons. Examination of the time course and mechanistic basis of demyelination following TBI will be an important future consideration.
Evidence for persistent Axonal Degeneration following TBI
Although both overt and subtle pathological changes to axons may play a role in the immediate loss of consciousness and/ or cognitive dysfunction that characterizes TBI, the relative contributions of differing forms of axonal pathologies over time have yet to be determined. Using APP as a marker of DAI, axonal pathology is observed to increase to a peak in the initial 24 hours following injury, thereafter leveling off (Gultekin and Smith, 1994). Although this represents the peak of pathology, damaged axons have been observed weeks to months and, in a small number of cases, even years following TBI (Blumbergs, et al., 1989, Chen, et al., 2009), perhaps heralding a persistent white matter degeneration instigated by TBI in a proportion of cases. In support of these observations, neuroradiological evidence indicates selective white matter loss post-injury (Gale, et al., 1995). Notably, in the limited descriptions of axonal pathology in long-term survivors thus far, damaged axons displayed the phenotype of axonal bulbs, potentially representing complete disconnection, rather than that of varicose, connected axons (Chen, et al., 2009), indicating a distinct mechanism. However, appropriately extensive characterisation of long-term axonal pathologies has yet to be performed.
It is true that there may be many other subtle pathological changes that affect functional outcome, such as dendritic alterations, imbalances in neurotransmitters, or changes in brain metabolism, as previously suggested (Monnerie, et al., 2010, Yuen, et al., 2009). These considerations highlight potential difficulties in designating all subtle pathological changes following traumatic injury as “DAI”. Further investigation of the role that chronic axonal pathologies play in neurodegeneration is of particular interest in light of emerging observations between TBI and syndromes of cognitive impairment including Alzheimer’s disease (AD).
TBI and the Link with Neurodegenerative Diseases
The association between TBI and chronic progressive neurodegeneration emerged from observation of boxers at the turn of last century when the term “punch drunk syndrome” was first used to describe a progressive dementing disorder in participants of the sport (Martland, 1928). Now called dementia pugilistica, the disorder is believed to occur as a consequence of repetitive mild TBI and is neuropathologically characterized by abnormal intracellular accumulations of hyperphosphorylated forms of the microtubule associated protein tau, known as neurofibrillary tangles (NFTs) and neuropil threads (NTs). (Braak and Braak, 1991, Corsellis, et al., 1973, Dale, et al., 1991, Forman, et al., 2004, Geddes, et al., 1999, Roberts, et al., 1990, Selkoe, 2001, Tokuda, et al., 1991). Extracellular AD-like beta-amyloid (Aβ) plaques and vascular Aβ deposits later emerged as a potentially important finding (Roberts, et al., 1990, Tokuda, et al., 1991). While much of this work has focused on boxers, recent evidence suggests that repetitive TBI experienced from playing professional American Football can result in increased rates of late-life cognitive impairment (Guskiewicz, et al., 2005). Furthermore, small selected studies examining the pathological studies of the brains of former players in the National Football League in the USA revealed NFTs and, in some cases, Aβ plaques (McKee, et al., 2009, Omalu, et al., 2006, Omalu, et al., 2005).
In addition, several studies have identified a history of just a single TBI as an epigenetic risk factor for the later development of clinical syndromes of cognitive impairment such as AD (Fleminger, et al., 2003, Graves, et al., 1990, Guo, et al., 2000, Molgaard, et al., 1990, Mortimer, et al., 1985, Mortimer, et al., 1991, O’Meara, et al., 1997, Plassman, et al., 2000, Salib and Hillier, 1997, Schofield, et al., 1997). Moreover, recent data indicate TBI may accelerate the onset of dementia (Gedye, et al., 1989, Nemetz, et al., 1999, Schofield, et al., 1997, Sullivan, et al., 1987). The first pathological link between the pathologies of a single TBI and AD was the observation that Aβ plaques are present in up to 30% of patients dying acutely following injury (Chen, et al., 2008, DeKosky, et al., 2007, Gentleman, et al., 1997, Huber A, 1993, Ikonomovic, et al., 2004, Roberts, et al., 1991, Roberts, et al., 1994, Smith, et al., 2003, Uryu, et al., 2007). Notably, plaques following TBI were observed rapidly, within hours of injury, and across the age spectrum, even in children35. It was also found that genetic polymorphisms of both the apolipoprotein E and neprilysin genes are associated with acute Aβ plaques following TBI, suggesting certain individuals may be at increased risk for the development of acute post-traumatic AD-like pathologies (Johnson, et al., 2009, Nicoll, et al., 1995). Moreover, while plaques were observed to diminish in the months following injury (Chen, et al., 2009), with survival of more than a year, plaques re-emerged being present to a greater extent than in uninjured controls (Johnson, et al., 2011). While NFTs, were not observed acutely following TBI, recent work indicates NFTs can be found at an increased extent and frequency, even in young individuals, long-term (>1 year) following just a single moderate – severe TBI in humans (Johnson, et al., 2011).
Injured Axons as a Source of Aβ
Although it is possible that multiple sources contribute to Aβ-plaque formation after TBI, the rapid accumulation of the precursor of Aβ, APP, in damaged axons, represents an intriguing potential source of Aβ for further investigation. Indeed, upon close examination of axonal bulbs in DAI, APP is seen to co-accumulate with the enzymes necessary for its cleavage to Aβ peptides, including presenelin-1 and beta-site APP-cleaving enzyme. Observed in both the pig model of DAI (Chen, et al., 2004) and, subsequently, in humans (Chen, et al., 2008, Uryu, et al., 2007), these data suggest trauma may create a situation whereby the substrates for Aβ formation are forced to co-exist in the same place and at the same time (Figure 2.). Indeed, abundant Aβ was confirmed within these axon bulbs, again both in humans and in animal models (Chen, et al., 2008, Chen, et al., 2004, Ikonomovic, et al., 2004, Smith, et al., 2003, Smith, et al., 1999, Uryu, et al., 2007).
Figure 2. Accumulation of Aβ and Associated Proteins in Axonal Bulbs Following TBI in Humans.
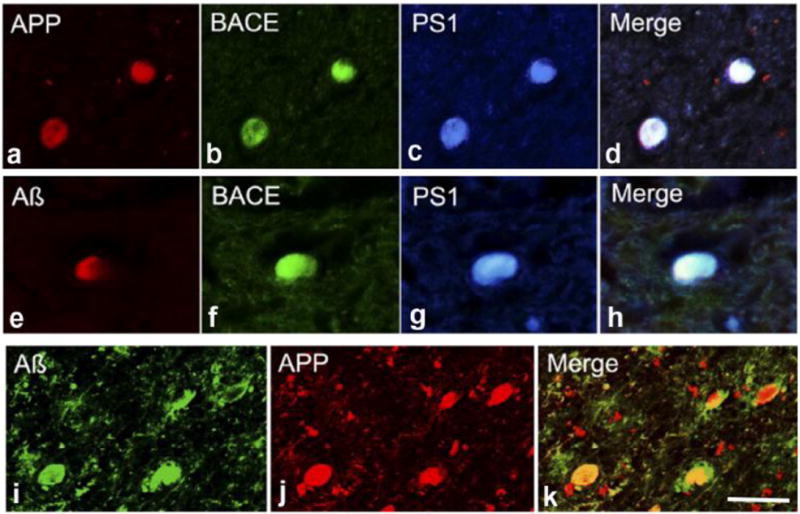
(a-c) Double immunofluorescent labeling showing co-accumulation of APP with Aβ in multiple axonal bulbs following TBI. Further immunohistochemical analyses showing co-accumulation of BACE and PS-1 with APP (d–g) and Aβ (h–k) in axonal bulbs. Scale bar: 50 μm.
Interestingly, interruption of axonal transport has also been implicated in APP processing and Aβ formation in AD. Specifically, studies have described Aβ genesis via BACE and PS-1 within the axonal membrane of murine peripheral nerves (Kamal, et al., 2001, Kamal, et al., 2000). The authors suggested that APP, β-secretase and PS-1 are transported within the axonal compartment via interaction with kinesin-1(Kamal, et al., 2001, Kamal, et al., 2000). The observation of Aβ within the axonal compartment led to the suggestion that amyloidogenic cleavage of APP can occur during transportation (Kamal, et al., 2001). However, these findings have been refuted by another study which failed to detect Aβ peptides within the same murine peripheral nerve (Lazarov, et al., 2005). However, more recent data using a well-established AD mouse model, indicates disruption of axonal transport via decreased kinesin-1 can result in increased axonal pathology with associated elevations in intraneuronal Aβ accumulation as well as extracellular Aβ deposition (Stokin, et al., 2005).
The eventual lysis and breakdown of damaged axons following trauma may permit the expulsion of accumulating Aβ into the parenchyma where it can aggregate to form plaques (Chen, et al., 2004, Smith, et al., 1999). In addition to providing a mechanism for the acute formation of Aβ following trauma, the possibility of persistent axonal pathology observed even years after injury may offer a potential mechanism of chronic Aβ genesis (Johnson, et al., 2011). Moreover, given that even mild TBI appears capable of inducing axonal injury (Blumbergs, et al., 1995, Browne, et al., 2011), the mechanistic role of axonal pathologies following single and repetitive mild TBI and associated disorders such as dementia pugilistica and chronic traumatic encephalopathy would be of interest to examine, both with regard to Aβ plaque formation and the accumulation of the microtubule associated-protein tau.
Elucidating the magnitude of persistent axonal degeneration after TBI and uncovering the mechanisms governing the protracted disconnection and degeneration of axons may provide a direct means of slowing the production of AD-associated proteins following injury, perhaps also providing insight into possible therapies more widely applicable to neurodegenerative disease.
Conclusions
Recently, there has been mounting evidence of the substantial pathological consequences of DAI due to TBI. Occurring as a direct consequence of mechanical injury, DAI has been identified as responsible for immediate and persistent coma following injury and is independently a significant cause of morbidity and mortality. However, more recently it has emerged that DAI may induce a multitude of functionally detrimental effects over a far greater range of severity than previously considered. Specifically, the classical understanding of DAI as morphologically altered axons due to impaired transport may represent just one pathological subset of damaged axons. In contrast, morphologically intact axons with disrupted physiology may contribute to the pathological milieu leading to clinical dysfunction across a wide range of injury severities, including mild TBI. Investigation of these pathways may represent important therapeutic targets in the treatment of TBI and potentially the mitigation of chronic neurodegeneration.
Acknowledgments
This work is supported by the National Institute of Health grants: NS056202, NS038104 (all DHS).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Adams JH, Doyle D, Ford I, Gennarelli TA, Graham DI, DR M. Diffuse axonal injury in head injury: definition, diagnosis and grading. Histopathology. 1989;15:49–59. doi: 10.1111/j.1365-2559.1989.tb03040.x. [DOI] [PubMed] [Google Scholar]
- 2.Adams JH, D D, Ford I, Gennarelli TA, Graham DI, McLellan DR. Diffuse axonal injury in head injury: definition, diagnosis and grading. Histopathology. 1989;15:49–59. doi: 10.1111/j.1365-2559.1989.tb03040.x. [DOI] [PubMed] [Google Scholar]
- 3.Adams JH, Doyle D, Ford I, Gennarelli TA, Graham DI, McClellan DR. Diffuse axonal injury in head injury: definition, diagnosis, and grading. Histopathol. 1989;15:49–59. doi: 10.1111/j.1365-2559.1989.tb03040.x. [DOI] [PubMed] [Google Scholar]
- 4.Adams JH, Doyle D, Ford I, Gennarelli TA, Graham DI, McLellan DR. Diffuse axonal injury in head injury: definition, diagnosis and grading. Histopathology. 1989;15:49–59. doi: 10.1111/j.1365-2559.1989.tb03040.x. [DOI] [PubMed] [Google Scholar]
- 5.Adams JH, Doyle D, Graham DI, et al. Diffuse axonal injury in head injuries caused by a fall. Lancet. 1984;2:1420–1422. doi: 10.1016/s0140-6736(84)91620-9. [DOI] [PubMed] [Google Scholar]
- 6.Adams JH, Graham DI, Gennarelli TA, Maxwell WL. Diffuse axonal injury in non-missile head injury. J Neurol Neurosurg Psychiatry. 1991;54:481–483. doi: 10.1136/jnnp.54.6.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Adams JH, Graham DI, Murray LS, Scott G. Diffuse axonal injury due to nonmissile head injury in humans: an analysis of 45 cases. Ann Neurol. 1982;12:557–563. doi: 10.1002/ana.410120610. [DOI] [PubMed] [Google Scholar]
- 8.Adams JH, Graham DI, Murray LS, Scott G. Diffuse axonal injury due to nonmissile head injury in humans: An analysis of 45 cases. Ann Neurol. 1982;12:557–563. doi: 10.1002/ana.410120610. [DOI] [PubMed] [Google Scholar]
- 9.Baker AJ, Phan N, Moulton RJ, Fehlings MG, Yucel Y, Zhao M, Liu E, Tian GF. Attenuation of the electrophysiological function of the corpus callosum after fluid percussion injury in the rat. J Neurotrauma. 2002;19:587–599. doi: 10.1089/089771502753754064. [DOI] [PubMed] [Google Scholar]
- 10.Banik NL, Hogan EL, Hsu CY. The multimolecular cascade of spinal cord injury. Studies on prostanoids, calcium, and proteinases. Neurochem Pathol. 1987;7:57–77. doi: 10.1007/BF02834292. [DOI] [PubMed] [Google Scholar]
- 11.Bazarian JJ, Zhong J, Blyth B, Zhu T, Kavcic V, Peterson D. Diffusion tensor imaging detects clinically important axonal damage after mild traumatic brain injury: a pilot study. J Neurotrauma. 2007;24:1447–1459. doi: 10.1089/neu.2007.0241. [DOI] [PubMed] [Google Scholar]
- 12.Bazarian JJ, Zhu T, Blyth B, Borrino A, Zhong J. Subject-specific changes in brain white matter on diffusion tensor imaging after sports-related concussion. Magn Reson Imaging. doi: 10.1016/j.mri.2011.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Benz I, Beck W, Kraas W, Stoll D, Jung G, Kohlhardt M. Two types of modified cardiac Na+ channels after cytosolic interventions at the alpha-subunit capable of removing Na+ inactivation. Eur Biophys J. 1997;25:189–200. doi: 10.1007/s002490050031. [DOI] [PubMed] [Google Scholar]
- 14.Blumbergs PC, Jones NR, North JB. Diffuse axonal injury in head trauma. J Neurol Neurosurg Psychiatry. 1989;52:838–841. doi: 10.1136/jnnp.52.7.838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Blumbergs PC, Scott G, Manavis J, Wainwright H, Simpson DA, McLean AJ. Topography of axonal injury as defined by amyloid precursor protein and the sector scoring method in mild and severe closed head injury. J Neurotrauma. 1995;12:565–572. doi: 10.1089/neu.1995.12.565. [DOI] [PubMed] [Google Scholar]
- 16.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol (Berl) 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 17.Bramlett HM, Dietrich WD. Quantitative structural changes in white and gray matter 1 year following traumatic brain injury in rats. Acta Neuropathol. 2002;103:607–614. doi: 10.1007/s00401-001-0510-8. [DOI] [PubMed] [Google Scholar]
- 18.Browne KD, Chen XH, Meaney DF, Smith DH. Mild Traumatic Brain Injury and Diffuse Axonal Injury in Swine. J Neurotrauma. 2011 doi: 10.1089/neu.2011.1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Buki A, Farkas O, Doczi T, Povlishock JT. Preinjury administration of the calpain inhibitor MDL-28170 attenuates traumatically induced axonal injury. J Neurotrauma. 2003;20:261–268. doi: 10.1089/089771503321532842. [DOI] [PubMed] [Google Scholar]
- 20.Buki A, Koizumi H, Povlishock JT. Moderate posttraumatic hypothermia decreases early calpain-mediated proteolysis and concomitant cytoskeletal compromise in traumatic axonal injury. Exp Neurol. 1999;159:319–328. doi: 10.1006/exnr.1999.7139. [DOI] [PubMed] [Google Scholar]
- 21.Buki A, Okonkwo DO, Povlishock JT. Postinjury cyclosporin A administration limits axonal damage and disconnection in traumatic brain injury. J Neurotrauma. 1999;16:511–521. doi: 10.1089/neu.1999.16.511. [DOI] [PubMed] [Google Scholar]
- 22.Buki A, Siman R, Trojanowski JQ, Povlishock JT. The role of calpain-mediated spectrin proteolysis in traumatically induced axonal injury. J Neuropathol Exp Neurol. 1999;58:365–375. doi: 10.1097/00005072-199904000-00007. [DOI] [PubMed] [Google Scholar]
- 23.Cajal SM. Degeneration and regeneration in the nervous system. Oxford university press; Oxforf, UK: 1928. [Google Scholar]
- 24.Chen X-H, Meaney DF, Xu B-N, Nonaka M, McIntosh TK, Wolf JA, Saatman KE, Smith DH. Evolution of neurofilament subtype accumulation in axons following diffuse brain injury in the pig. J Neuropathol Exp Neurol. 1999;58:588–596. doi: 10.1097/00005072-199906000-00003. [DOI] [PubMed] [Google Scholar]
- 25.Chen XH, Johnson VE, Uryu K, Trojanowski JQ, Smith DH. A Lack of Amyloid beta Plaques Despite Persistent Accumulation of Amyloid beta in Axons of Long-Term Survivors of Traumatic Brain Injury. Brain Pathol. 2008 doi: 10.1111/j.1750-3639.2008.00176.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen XH, Johnson VE, Uryu K, Trojanowski JQ, Smith DH. A lack of amyloid beta plaques despite persistent accumulation of amyloid beta in axons of long-term survivors of traumatic brain injury. Brain Pathol. 2009;19:214–223. doi: 10.1111/j.1750-3639.2008.00176.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen XH, Meaney DF, Xu BN, Nonaka M, McIntosh TK, Wolf JA, Saatman KE, Smith DH. Evolution of neurofilament subtype accumulation in axons following diffuse brain injury in the pig. J Neuropathol Exp Neurol. 1999;58:588–596. doi: 10.1097/00005072-199906000-00003. [DOI] [PubMed] [Google Scholar]
- 28.Chen XH, Siman R, Iwata A, Meaney DF, Trojanowski JQ, Smith DH. Long-term accumulation of amyloid-beta, beta-secretase, presenilin-1, and caspase-3 in damaged axons following brain trauma. Am J Pathol. 2004;165:357–371. doi: 10.1016/s0002-9440(10)63303-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Christman CW, Grady MS, Walker SA, Holloway KL, Povlishock JT. Ultrastructural studies of diffuse axonal injury in humans. J Neurotrauma. 1994;11:173–186. doi: 10.1089/neu.1994.11.173. [DOI] [PubMed] [Google Scholar]
- 30.Chung RS, Staal JA, McCormack GH, Dickson TC, Cozens MA, Chuckowree JA, Quilty MC, Vickers JC. Mild axonal stretch injury in vitro induces a progressive series of neurofilament alterations ultimately leading to delayed axotomy. J Neurotrauma. 2005;22:1081–1091. doi: 10.1089/neu.2005.22.1081. [DOI] [PubMed] [Google Scholar]
- 31.Corsellis JA, Bruton CJ, Freeman-Browne D. The aftermath of boxing. Psychol Med. 1973;3:270–303. doi: 10.1017/s0033291700049588. [DOI] [PubMed] [Google Scholar]
- 32.Dale GE, Leigh PN, Luthert P, Anderton BH, Roberts GW. Neurofibrillary tangles in dementia pugilistica are ubiquitinated. J Neurol Neurosurg Psychiatry. 1991;54:116–118. doi: 10.1136/jnnp.54.2.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.DeKosky ST, Abrahamson EE, Ciallella JR, Paljug WR, Wisniewski SR, Clark RS, Ikonomovic MD. Association of increased cortical soluble abeta42 levels with diffuse plaques after severe brain injury in humans. Arch Neurol. 2007;64:541–544. doi: 10.1001/archneur.64.4.541. [DOI] [PubMed] [Google Scholar]
- 34.Deng Y, Thompson BM, Gao X, Hall ED. Temporal relationship of peroxynitrite-induced oxidative damage, calpain-mediated cytoskeletal degradation and neurodegeneration after traumatic brain injury. Exp Neurol. 2007;205:154–165. doi: 10.1016/j.expneurol.2007.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Deng-Bryant Y, Singh IN, Carrico KM, Hall ED. Neuroprotective effects of tempol, a catalytic scavenger of peroxynitrite-derived free radicals, in a mouse traumatic brain injury model. J Cereb Blood Flow Metab. 2008;28:1114–1126. doi: 10.1038/jcbfm.2008.10. [DOI] [PubMed] [Google Scholar]
- 36.Faul M, Xu L, Wald MM, Coronado VG. Traumatic brain injury in the United States: emergency department visits, hospitalizations, and deaths. Centers for Disease Control and Prevention, National Center for Injury Prevention and Control; Atlanta (GA): 2010. [Google Scholar]
- 37.Finkelstein E, Corso P, M T. The Incidence and Economic Burden of Injuries in the United States. Oxford University Press; New York (NY): 2006. [Google Scholar]
- 38.Fleminger S, Oliver DL, Lovestone S, Rabe-Hesketh S, Giora A. Head injury as a risk factor for Alzheimer’s disease: the evidence 10 years on; a partial replication. J Neurol Neurosurg Psychiatry. 2003;74:857–862. doi: 10.1136/jnnp.74.7.857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Forman MS, Trojanowski JQ, Lee VM. Neurodegenerative diseases: a decade of discoveries paves the way for therapeutic breakthroughs. Nat Med. 2004;10:1055–1063. doi: 10.1038/nm1113. [DOI] [PubMed] [Google Scholar]
- 40.Fujita M, Oda Y, Wei EP, Povlishock JT. The combination of either tempol or FK506 with delayed hypothermia: implications for traumatically induced microvascular and axonal protection. J Neurotrauma. 28:1209–1218. doi: 10.1089/neu.2011.1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gale SD, Johnson SC, Bigler ED, Blatter DD. Nonspecific white matter degeneration following traumatic brain injury. J Int Neuropsychol Soc. 1995;1:17–28. doi: 10.1017/s1355617700000060. [DOI] [PubMed] [Google Scholar]
- 42.Geddes JF, Vowles GH, Nicoll JA, Revesz T. Neuronal cytoskeletal changes are an early consequence of repetitive head injury. Acta Neuropathol. 1999;98:171–178. doi: 10.1007/s004010051066. [DOI] [PubMed] [Google Scholar]
- 43.Gedye A, Beattie BL, Tuokko H, Horton A, Korsarek E. Severe head injury hastens age of onset of Alzheimer’s disease. J Am Geriatr Soc. 1989;37:970–973. doi: 10.1111/j.1532-5415.1989.tb07283.x. [DOI] [PubMed] [Google Scholar]
- 44.Gennarelli TA, Thibault LE, Adams JH, Graham DI, Thompson CJ, Marcincin RP. Diffuse axonal injury and traumatic coma in the primate. Ann Neurol. 1982;12:564–574. doi: 10.1002/ana.410120611. [DOI] [PubMed] [Google Scholar]
- 45.Gentleman SM, Greenberg BD, Savage MJ, Noori M, Newman SJ, Roberts GW, Griffin WS, Graham DI. A beta 42 is the predominant form of amyloid beta-protein in the brains of short-term survivors of head injury. Neuroreport. 1997;8:1519–1522. doi: 10.1097/00001756-199704140-00039. [DOI] [PubMed] [Google Scholar]
- 46.Gentleman SM, Leclercq PD, Moyes L, Graham DI, Smith C, Griffin WS, Nicoll JA. Long-term intracerebral inflammatory response after traumatic brain injury. Forensic Sci Int. 2004;146:97–104. doi: 10.1016/j.forsciint.2004.06.027. [DOI] [PubMed] [Google Scholar]
- 47.Gentleman SM, Nash MJ, Sweeting CJ, Graham DI, Roberts GW. Beta-amyloid precursor protein (beta APP) as a marker for axonal injury after head injury. Neurosci Lett. 1993;160:139–144. doi: 10.1016/0304-3940(93)90398-5. [DOI] [PubMed] [Google Scholar]
- 48.Gentleman SM, Roberts GW, Gennarelli TA, Maxwell WL, Adams JH, Kerr S, Graham DI. Axonal injury: a universal consequence of fatal closed head injury? Acta Neuropathol. 1995;89:537–543. doi: 10.1007/BF00571509. [DOI] [PubMed] [Google Scholar]
- 49.Gitler D, Spira ME. Real time imaging of calcium-induced localized proteolytic activity after axotomy and its relation to growth cone formation. Neuron. 1998;20:1123–1135. doi: 10.1016/s0896-6273(00)80494-8. [DOI] [PubMed] [Google Scholar]
- 50.Grady MS, McLaughlin MR, Christman CW, Valadka AB, Fligner CL, Povlishock JT. The use of antibodies targeted against the neurofilament subunits for the detection of diffuse axonal injury in humans. J Neuropathol Exp Neurol. 1993;52:143–152. doi: 10.1097/00005072-199303000-00007. [DOI] [PubMed] [Google Scholar]
- 51.Graham DI, Adams JH, Gennarelli TA. Mechanisms of non-penetrating head injury. Prog Clin Biol Res. 1988;234:159–168. [PubMed] [Google Scholar]
- 52.Graves AB, White E, Koepsell TD, Reifler BV, van Belle G, Larson EB, Raskind M. The association between head trauma and Alzheimer’s disease. Am J Epidemiol. 1990;131:491–501. doi: 10.1093/oxfordjournals.aje.a115523. [DOI] [PubMed] [Google Scholar]
- 53.Gultekin SH, Smith TW. Diffuse axonal injury in craniocerebral trauma. A comparative histologic and immunohistochemical study. Arch Pathol Lab Med. 1994;118:168–171. [PubMed] [Google Scholar]
- 54.Guo Z, Cupples LA, Kurz A, Auerbach SH, Volicer L, Chui H, Green RC, Sadovnick AD, Duara R, DeCarli C, Johnson K, Go RC, Growdon JH, Haines JL, Kukull WA, Farrer LA. Head injury and the risk of AD in the MIRAGE study. Neurology. 2000;54:1316–1323. doi: 10.1212/wnl.54.6.1316. [DOI] [PubMed] [Google Scholar]
- 55.Guskiewicz KM, Marshall SW, Bailes J, McCrea M, Cantu RC, Randolph C, Jordan BD. Association between recurrent concussion and late-life cognitive impairment in retired professional football players. Neurosurgery. 2005;57:719–726. doi: 10.1093/neurosurgery/57.4.719. discussion 719-726. [DOI] [PubMed] [Google Scholar]
- 56.Holbourn AHS. Mechanics of Head Injury. The Lancet. 1943;242:438–441. [Google Scholar]
- 57.Holbourn AHS. Mechanics of Brain Injuries. Brit Med Bull. 1945;3:147–149. [Google Scholar]
- 58.Huber A, G K, Kelemen J, Cervod-Navarro J. Desity of amyloid plaques in brains after head trauma. J Neurotrauma. 1993;10(Suppl):S180. [Google Scholar]
- 59.Huh JW, Franklin MA, Widing AG, Raghupathi R. Regionally distinct patterns of calpain activation and traumatic axonal injury following contusive brain injury in immature rats. Dev Neurosci. 2006;28:466–476. doi: 10.1159/000094172. [DOI] [PubMed] [Google Scholar]
- 60.Hunter JV, Wilde EA, Tong KA, Holshouser BA. Emerging Imaging Tools for Use with Traumatic Brain Injury Research. J Neurotrauma. 2011 doi: 10.1089/neu.2011.1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ichiya Y, Kuwabara Y, Otsuka M, Tahara T, Yoshikai T, Fukumura T, Jingu K, Masuda K. Assessment of response to cancer therapy using fluorine-18-fluorodeoxyglucose and positron emission tomography. J Nucl Med. 1991;32:1655–1660. [PubMed] [Google Scholar]
- 62.Ikonomovic MD, Uryu K, Abrahamson EE, Ciallella JR, Trojanowski JQ, Lee VM, Clark RS, Marion DW, Wisniewski SR, DeKosky ST. Alzheimer’s pathology in human temporal cortex surgically excised after severe brain injury. Exp Neurol. 2004;190:192–203. doi: 10.1016/j.expneurol.2004.06.011. [DOI] [PubMed] [Google Scholar]
- 63.Iwata A, Stys PK, Wolf JA, Chen XH, Taylor AG, Meaney DF, Smith DH. Traumatic axonal injury induces proteolytic cleavage of the voltage-gated sodium channels modulated by tetrodotoxin and protease inhibitors. J Neurosci. 2004;24:4605–4613. doi: 10.1523/JNEUROSCI.0515-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jafari SS, Maxwell WL, Neilson M, Graham DI. Axonal cytoskeletal changes after non-disruptive axonal injury. J Neurocytol. 1997;26:207–221. doi: 10.1023/a:1018588114648. [DOI] [PubMed] [Google Scholar]
- 65.Jafari SS, Nielson M, Graham DI, Maxwell WL. Axonal cytoskeletal changes after nondisruptive axonal injury. II. Intermediate sized axons. J Neurotrauma. 1998;15:955–966. doi: 10.1089/neu.1998.15.955. [DOI] [PubMed] [Google Scholar]
- 66.Johnson VE, Stewart W, Smith DH. Widespread Tau and Amyloid-Beta Pathology Many Years After a Single Traumatic Brain Injury in Humans. Brain Pathol. doi: 10.1111/j.1750-3639.2011.00513.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Johnson VE, Stewart W, Smith DH. Traumatic brain injury and amyloid-beta pathology: a link to Alzheimer’s disease? Nat Rev Neurosci. 2010;11:361–370. doi: 10.1038/nrn2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Johnson VE, Stewart W, Smith DH. Amyloid and Tau Pathologies Many Years Following Single Traumatic Brain Injury in Humans. Brain Pathology. 2011 doi: 10.1111/j.1750-3639.2011.00513.x. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Johnson VE, Stewart W, Smith DH. Widespread Amyloid and Tau Pathologies Following a Single Traumatic Brain Injury in Humans. Brain Pathol (Accepted) 2011 doi: 10.1111/j.1750-3639.2011.00513.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Johnson VE, Stewart W, Stewart JE, Graham DI, Praestgaard AH, Smith DH. A Neprilysin Polymorphism and Amyloid-beta Plaques Following Traumatic Brain Injury. J Neurotrauma. 2009 doi: 10.1089/neu.2008.0843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kamal A, Almenar-Queralt A, LeBlanc JF, Roberts EA, Goldstein LS. Kinesin-mediated axonal transport of a membrane compartment containing beta-secretase and presenilin-1 requires APP. Nature. 2001;414:643–648. doi: 10.1038/414643a. [DOI] [PubMed] [Google Scholar]
- 72.Kamal A, Stokin GB, Yang Z, Xia CH, Goldstein LS. Axonal transport of amyloid precursor protein is mediated by direct binding to the kinesin light chain subunit of kinesin-I. Neuron. 2000;28:449–459. doi: 10.1016/s0896-6273(00)00124-0. [DOI] [PubMed] [Google Scholar]
- 73.Kampfl A, Posmantur R, Nixon R, Grynspan F, Zhao X, Liu SJ, Newcomb JK, Clifton GL, Hayes RL. mu-calpain activation and calpain-mediated cytoskeletal proteolysis following traumatic brain injury. J Neurochem. 1996;67:1575–1583. doi: 10.1046/j.1471-4159.1996.67041575.x. [DOI] [PubMed] [Google Scholar]
- 74.Kupina NC, Nath R, Bernath EE, Inoue J, Mitsuyoshi A, Yuen PW, Wang KK, Hall ED. The novel calpain inhibitor SJA6017 improves functional outcome after delayed administration in a mouse model of diffuse brain injury. J Neurotrauma. 2001;18:1229–1240. doi: 10.1089/089771501317095269. [DOI] [PubMed] [Google Scholar]
- 75.Lazarov O, Morfini GA, Lee EB, Farah MH, Szodorai A, DeBoer SR, Koliatsos VE, Kins S, Lee VM, Wong PC, Price DL, Brady ST, Sisodia SS. Axonal transport, amyloid precursor protein, kinesin-1, and the processing apparatus: revisited. J Neurosci. 2005;25:2386–2395. doi: 10.1523/JNEUROSCI.3089-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Loane DJ, Byrnes KR. Role of microglia in neurotrauma. Neurotherapeutics. 2010;7:366–377. doi: 10.1016/j.nurt.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Margulies SS, Thibault LE, Gennarelli TA. Physical model simulations of brain injury in the primate. J Biomech. 1990;23:823–836. doi: 10.1016/0021-9290(90)90029-3. [DOI] [PubMed] [Google Scholar]
- 78.Marklund N, Blennow K, Zetterberg H, Ronne-Engstrom E, Enblad P, Hillered L. Monitoring of brain interstitial total tau and beta amyloid proteins by microdialysis in patients with traumatic brain injury. J Neurosurg. 2009;110:1227–1237. doi: 10.3171/2008.9.JNS08584. [DOI] [PubMed] [Google Scholar]
- 79.Marmarou CR, Povlishock JT. Administration of the immunophilin ligand FK506 differentially attenuates neurofilament compaction and impaired axonal transport in injured axons following diffuse traumatic brain injury. Exp Neurol. 2006;197:353–362. doi: 10.1016/j.expneurol.2005.10.003. [DOI] [PubMed] [Google Scholar]
- 80.Martland H. Punch drunk. J Am Med Assoc. 1928;91:1103–1107. [Google Scholar]
- 81.Maxwell WL, Domleo A, McColl G, Jafari SS, Graham DI. Post-acute alterations in the axonal cytoskeleton after traumatic axonal injury. J Neurotrauma. 2003;20:151–168. doi: 10.1089/08977150360547071. [DOI] [PubMed] [Google Scholar]
- 82.Maxwell WL, Graham DI. Loss of axonal microtubules and neurofilaments after stretch-injury to guinea pig optic nerve fibers. J Neurotrauma. 1997;14:603–614. doi: 10.1089/neu.1997.14.603. [DOI] [PubMed] [Google Scholar]
- 83.Maxwell WL, Kosanlavit R, McCreath BJ, Reid O, Graham DI. Freeze-fracture and cytochemical evidence for structural and functional alteration in the axolemma and myelin sheath of adult guinea pig optic nerve fibers after stretch injury. J Neurotrauma. 1999;16:273–284. doi: 10.1089/neu.1999.16.273. [DOI] [PubMed] [Google Scholar]
- 84.Maxwell WL, McCreath BJ, Graham DI, Gennarelli TA. Cytochemical evidence for redistribution of membrane pump calcium-ATPase and ecto-Ca-ATPase activity, and calcium influx in myelinated nerve fibres of the optic nerve after stretch injury. J Neurocytol. 1995;24:925–942. doi: 10.1007/BF01215643. [DOI] [PubMed] [Google Scholar]
- 85.Maxwell WL, Watt C, Pediani JD, Graham DI, Adams JH, Gennarelli TA. Localisation of calcium ions and calcium-ATPase activity within myelinated nerve fibres of the adult guinea-pig optic nerve. J Anat. 1991;176:71–79. [PMC free article] [PubMed] [Google Scholar]
- 86.Mayer AR, Ling J, Mannell MV, Gasparovic C, Phillips JP, Doezema D, Reichard R, Yeo RA. A prospective diffusion tensor imaging study in mild traumatic brain injury. Neurology. 74:643–650. doi: 10.1212/WNL.0b013e3181d0ccdd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.McGinn MJ, Kelley BJ, Akinyi L, Oli MW, Liu MC, Hayes RL, Wang KK, Povlishock JT. Biochemical, structural, and biomarker evidence for calpain-mediated cytoskeletal change after diffuse brain injury uncomplicated by contusion. J Neuropathol Exp Neurol. 2009;68:241–249. doi: 10.1097/NEN.0b013e3181996bfe. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.McKee AC, Cantu RC, Nowinski CJ, Hedley-Whyte ET, Gavett BE, Budson AE, Santini VE, Lee HS, Kubilus CA, Stern RA. Chronic traumatic encephalopathy in athletes: progressive tauopathy after repetitive head injury. J Neuropathol Exp Neurol. 2009;68:709–735. doi: 10.1097/NEN.0b013e3181a9d503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Meaney DF, Smith DH, Shreiber DI, Bain AC, Miller RT, Ross DT, Gennarelli TA. Biomechanical analysis of experimental diffuse axonal injury. J Neurotrauma. 1995;12:689–694. doi: 10.1089/neu.1995.12.689. [DOI] [PubMed] [Google Scholar]
- 90.Meaney DF, Smith DH, Shreiber DI, Bain AC, Miller RT, Ross DT, Gennarelli TA. Biomechanical analysis of experimental diffuse axonal injury. In: Bandak FA, Eppinger RH, Ommaya AK, editors. Traumatic Brain Injury Bioscience and Mechanics. Mary Ann Liebert, Inc.; 1996. pp. 167–180. [Google Scholar]
- 91.Miles L, Grossman RI, Johnson G, Babb JS, Diller L, Inglese M. Short-term DTI predictors of cognitive dysfunction in mild traumatic brain injury. Brain Inj. 2008;22:115–122. doi: 10.1080/02699050801888816. [DOI] [PubMed] [Google Scholar]
- 92.Molgaard CA, Stanford EP, Morton DJ, Ryden LA, Schubert KR, Golbeck AL. Epidemiology of head trauma and neurocognitive impairment in a multi-ethnic population. Neuroepidemiology. 1990;9:233–242. doi: 10.1159/000110778. [DOI] [PubMed] [Google Scholar]
- 93.Monnerie H, Tang-Schomer MD, Iwata A, Smith DH, Kim HA, Le Roux PD. Dendritic alterations after dynamic axonal stretch injury in vitro. Exp Neurol. 2010;224:415–423. doi: 10.1016/j.expneurol.2010.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Mortimer JA, French LR, Hutton JT, Schuman LM. Head injury as a risk factor for Alzheimer’s disease. Neurology. 1985;35:264–267. doi: 10.1212/wnl.35.2.264. [DOI] [PubMed] [Google Scholar]
- 95.Mortimer JA, van Duijn CM, Chandra V, Fratiglioni L, Graves AB, Heyman A, Jorm AF, Kokmen E, Kondo K, Rocca WA, et al. Head trauma as a risk factor for Alzheimer’s disease: a collaborative re-analysis of case-control studies EURODEM Risk Factors Research Group. Int J Epidemiol. 1991;20(Suppl 2):S28–35. doi: 10.1093/ije/20.supplement_2.s28. [DOI] [PubMed] [Google Scholar]
- 96.Mustafa AG, Singh IN, Wang J, Carrico KM, Hall ED. Mitochondrial protection after traumatic brain injury by scavenging lipid peroxyl radicals. J Neurochem. 114:271–280. doi: 10.1111/j.1471-4159.2010.06749.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Mustafa AG, Wang JA, Carrico KM, Hall ED. Pharmacological inhibition of lipid peroxidation attenuates calpain-mediated cytoskeletal degradation after traumatic brain injury. J Neurochem. 117:579–588. doi: 10.1111/j.1471-4159.2011.07228.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemetz PN, Leibson C, Naessens JM, Beard M, Kokmen E, Annegers JF, Kurland LT. Traumatic brain injury and time to onset of Alzheimer’s disease: a population-based study. Am J Epidemiol. 1999;149:32–40. doi: 10.1093/oxfordjournals.aje.a009724. [DOI] [PubMed] [Google Scholar]
- 99.Nevin NC. Neuropathological changes in the white matter following head injury. J Neuropathol Exp Neurol. 1967;26:77–84. doi: 10.1097/00005072-196701000-00006. [DOI] [PubMed] [Google Scholar]
- 100.Ng HK, Mahaliyana RD, Poon WS. The pathological spectrum of diffuse axonal injury in blunt head trauma: assessment with axon and myelin strains. Clin Neurol Neurosurg. 1994;96:24–31. doi: 10.1016/0303-8467(94)90025-6. [DOI] [PubMed] [Google Scholar]
- 101.Nicoll JA, Roberts GW, Graham DI. Apolipoprotein E epsilon 4 allele is associated with deposition of amyloid beta-protein following head injury. Nat Med. 1995;1:135–137. doi: 10.1038/nm0295-135. [DOI] [PubMed] [Google Scholar]
- 102.O’Meara ES, Kukull WA, Sheppard L, Bowen JD, McCormick WC, Teri L, Pfanschmidt M, Thompson JD, Schellenberg GD, Larson EB. Head injury and risk of Alzheimer’s disease by apolipoprotein E genotype. Am J Epidemiol. 1997;146:373–384. doi: 10.1093/oxfordjournals.aje.a009290. [DOI] [PubMed] [Google Scholar]
- 103.Okonkwo DO, Povlishock JT. An intrathecal bolus of cyclosporin A before injury preserves mitochondrial integrity and attenuates axonal disruption in traumatic brain injury. J Cereb Blood Flow Metab. 1999;19:443–451. doi: 10.1097/00004647-199904000-00010. [DOI] [PubMed] [Google Scholar]
- 104.Omalu BI, DeKosky ST, Hamilton RL, Minster RL, Kamboh MI, Shakir AM, Wecht CH. Chronic traumatic encephalopathy in a national football league player: part II. Neurosurgery. 2006;59:1086–1092. doi: 10.1227/01.NEU.0000245601.69451.27. discussion 1092-1083. [DOI] [PubMed] [Google Scholar]
- 105.Omalu BI, DeKosky ST, Minster RL, Kamboh MI, Hamilton RL, Wecht CH. Chronic traumatic encephalopathy in a National Football League player. Neurosurgery. 2005;57:128–134. doi: 10.1227/01.neu.0000163407.92769.ed. discussion 128-134. [DOI] [PubMed] [Google Scholar]
- 106.Ommaya AK, Hirsch A. Tolerances for cerebral concussion from head impact and whiplash in primates. J Biomechanics. 1971;4:13–21. doi: 10.1016/0021-9290(71)90011-x. [DOI] [PubMed] [Google Scholar]
- 107.Oppenheimer DR. Microscopic lesions in the brain following head injury. J Neurol Neurosurg Psychiatry. 1968;31:299–306. doi: 10.1136/jnnp.31.4.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Peerless SJ, Rewcastle NB. Shear injuries of the brain. Can Med Assoc J. 1967;96:577–582. [PMC free article] [PubMed] [Google Scholar]
- 109.Pettus EH, Povlishock JT. Characterization of a distinct set of intra-axonal ultrastructural changes associated with traumatically induced alteration in axolemmal permeability. Brain Res. 1996;722:1–11. doi: 10.1016/0006-8993(96)00113-8. [DOI] [PubMed] [Google Scholar]
- 110.Plassman BL, Havlik RJ, Steffens DC, Helms MJ, Newman TN, Drosdick D, Phillips C, Gau BA, Welsh-Bohmer KA, Burke JR, Guralnik JM, Breitner JC. Documented head injury in early adulthood and risk of Alzheimer’s disease and other dementias. Neurology. 2000;55:1158–1166. doi: 10.1212/wnl.55.8.1158. [DOI] [PubMed] [Google Scholar]
- 111.Povlishock JT. Traumatically induced axonal injury: pathogenesis and pathobiological implications. Brain Pathol. 1992;2:1–12. [PubMed] [Google Scholar]
- 112.Povlishock JT. Traumatically induced axonal injury: pathogenesis and pathobiological implications. Brain Pathol. 1992;2:1–12. [PubMed] [Google Scholar]
- 113.Povlishock JT, Becker DP. Fate of reactive axonal swellings induced by head injury. Lab Invest. 1985;52:540–552. [PubMed] [Google Scholar]
- 114.Povlishock JT, Becker DP. Fate of reactive axonal swellings induced by head injury. Lab Invest. 1985;52:540–552. [PubMed] [Google Scholar]
- 115.Povlishock JT, Becker DP, Cheng CLY, Vaughan GW. Axonal change in minor head injury. J Neuropathol Exp Neurol. 1983;42:225–242. doi: 10.1097/00005072-198305000-00002. [DOI] [PubMed] [Google Scholar]
- 116.Povlishock JT, Buki A, Koiziumi H, Stone J, Okonkwo DO. Initiating mechanisms involved in the pathobiology of traumatically induced axonal injury and interventions targeted at blunting their progression. Acta Neurochir (Suppl)(Wien) 1999;73:15–20. doi: 10.1007/978-3-7091-6391-7_3. [DOI] [PubMed] [Google Scholar]
- 117.Povlishock JT, Buki A, Koiziumi H, Stone J, Okonkwo DO. Initiating mechanisms involved in the pathobiology of traumatically induced axonal injury and interventions targeted at blunting their progression. Acta Neurochir Suppl. 1999;73:15–20. doi: 10.1007/978-3-7091-6391-7_3. [DOI] [PubMed] [Google Scholar]
- 118.Povlishock JT, Katz DI. Update of neuropathology and neurological recovery after traumatic brain injury. J Head Trauma Rehabil. 2005;20:76–94. doi: 10.1097/00001199-200501000-00008. [DOI] [PubMed] [Google Scholar]
- 119.Povlishock JT, Pettus EH. Traumatically induced axonal damage: evidence for enduring changes in axolemmal permeability with associated cytoskeletal change. Acta Neurochir Suppl. 1996;66:81–86. doi: 10.1007/978-3-7091-9465-2_15. [DOI] [PubMed] [Google Scholar]
- 120.Rand CW, Courville CB. Histologic changes in the brain in cases of fatal injury to the head; alterations in nerve cells. Arch Neurol Psychiatry. 1946;55:79–110. doi: 10.1001/archneurpsyc.1946.02300130003001. [DOI] [PubMed] [Google Scholar]
- 121.Reeves TM, Phillips LL, Lee NN, Povlishock JT. Preferential neuroprotective effect of tacrolimus (FK506) on unmyelinated axons following traumatic brain injury. Brain Res. 2007;1154:225–236. doi: 10.1016/j.brainres.2007.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Reeves TM, Phillips LL, Povlishock JT. Myelinated and unmyelinated axons of the corpus callosum differ in vulnerability and functional recovery following traumatic brain injury. Exp Neurol. 2005;196:126–137. doi: 10.1016/j.expneurol.2005.07.014. [DOI] [PubMed] [Google Scholar]
- 123.Roberts GW, Allsop D, Bruton C. The occult aftermath of boxing. J Neurol Neurosurg Psychiatry. 1990;53:373–378. doi: 10.1136/jnnp.53.5.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Roberts GW, Gentleman SM, Lynch A, Graham DI. beta A4 amyloid protein deposition in brain after head trauma. Lancet. 1991;338:1422–1423. doi: 10.1016/0140-6736(91)92724-g. [DOI] [PubMed] [Google Scholar]
- 125.Roberts GW, Gentleman SM, Lynch A, Murray L, Landon M, Graham DI. Beta amyloid protein deposition in the brain after severe head injury: implications for the pathogenesis of Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 1994;57:419–425. doi: 10.1136/jnnp.57.4.419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Saatman KE, Abai B, Grosvenor A, Vorwerk CK, Smith DH, Meaney DF. Traumatic axonal injury results in biphasic calpain activation and retrograde transport impairment in mice. J Cereb Blood Flow Metab. 2003;23:34–42. doi: 10.1097/01.WCB.0000035040.10031.B0. [DOI] [PubMed] [Google Scholar]
- 127.Saatman KE, Bozyczko-Coyne D, Marcy V, Siman R, McIntosh TK. Prolonged calpain-mediated spectrin breakdown occurs regionally following experimental brain injury in the rat. J Neuropathol Exp Neurol. 1996;55:850–860. doi: 10.1097/00005072-199607000-00010. [DOI] [PubMed] [Google Scholar]
- 128.Saatman KE, Murai H, Bartus RT, Smith DH, Hayward NJ, Perri BR, McIntosh TK. Calpain inhibitor AK295 attenuates motor and cognitive deficits following experimental brain injury in the rat. Proc Natl Acad Sci U S A. 1996;93:3428–3433. doi: 10.1073/pnas.93.8.3428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Saatman KE, Zhang C, Bartus RT, McIntosh TK. Behavioral efficacy of posttraumatic calpain inhibition is not accompanied by reduced spectrin proteolysis, cortical lesion, or apoptosis. J Cereb Blood Flow Metab. 2000;20:66–73. doi: 10.1097/00004647-200001000-00010. [DOI] [PubMed] [Google Scholar]
- 130.Salib E, Hillier V. Head injury and the risk of Alzheimer’s disease: a case control study. Int J Geriatr Psychiatry. 1997;12:363–368. doi: 10.1002/(sici)1099-1166(199703)12:3<363::aid-gps515>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 131.Schofield PW, Tang M, Marder K, Bell K, Dooneief G, Chun M, Sano M, Stern Y, Mayeux R. Alzheimer’s disease after remote head injury: an incidence study. J Neurol Neurosurg Psychiatry. 1997;62:119–124. doi: 10.1136/jnnp.62.2.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Selkoe DJ. Alzheimer’s disease: genes, proteins, and therapy. Physiol Rev. 2001;81:741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- 133.Shaw K, MacKinnon MA, Raghupathi R, Saatman KE, McLntosh TK, Graham DI. TUNEL-positive staining in white and grey matter after fatal head injury in man. Clin Neuropathol. 2001;20:106–112. [PubMed] [Google Scholar]
- 134.Sherriff FE, Bridges LR, Sivaloganathan S. Early detection of axonal injury after human head trauma using immunocytochemistry for beta-amyloid precursor protein. Acta Neuropathol (Berl) 1994;87:55–62. doi: 10.1007/BF00386254. [DOI] [PubMed] [Google Scholar]
- 135.Singleton RH, Stone JR, Okonkwo DO, Pellicane AJ, Povlishock JT. The immunophilin ligand FK506 attenuates axonal injury in an impact-acceleration model of traumatic brain injury. J Neurotrauma. 2001;18:607–614. doi: 10.1089/089771501750291846. [DOI] [PubMed] [Google Scholar]
- 136.Smith DH, Chen X-H, Xu B-N, McIntosh TK, Gennarelli TA, Meaney DF. Characterization of diffuse axonal pathology and selective hippocampal damage following inertial brain trauma in the pig. J Neuropathol Exp Neurol. 1997;56:822–834. [PubMed] [Google Scholar]
- 137.Smith DH, Chen XH, Iwata A, Graham DI. Amyloid beta accumulation in axons after traumatic brain injury in humans. J Neurosurg. 2003;98:1072–1077. doi: 10.3171/jns.2003.98.5.1072. [DOI] [PubMed] [Google Scholar]
- 138.Smith DH, Chen XH, Nonaka M, Trojanowski JQ, Lee VM, Saatman KE, Leoni MJ, Xu BN, Wolf JA, Meaney DF. Accumulation of amyloid beta and tau and the formation of neurofilament inclusions following diffuse brain injury in the pig. J Neuropathol Exp Neurol. 1999;58:982–992. doi: 10.1097/00005072-199909000-00008. [DOI] [PubMed] [Google Scholar]
- 139.Smith DH, Chen XH, Xu BN, McIntosh TK, Gennarelli TA, Meaney DF. Characterization of diffuse axonal pathology and selective hippocampal damage following inertial brain trauma in the pig. Journal of Neuropathology & Experimental Neurology. 1997;56:822–834. [PubMed] [Google Scholar]
- 140.Smith DH, Meaney DF. Axonal damage in traumatic brain injury. The Neuroscientist. 2000;6:483–495. [Google Scholar]
- 141.Smith DH, Meaney DF. Axonal damage in traumatic brain injury. The neuroscientist. 2000;6:483–495. [Google Scholar]
- 142.Smith DH, Meaney DF, McIntosh TK. Experimental models of traumatic brain injury, Discoveries in Head Trauma. IBC. 1996:5–25. [Google Scholar]
- 143.Smith DH, Meaney DF, Shull WH. Diffuse axonal injury in head trauma. J Head Trauma Rehabil. 2003;18:307–316. doi: 10.1097/00001199-200307000-00003. [DOI] [PubMed] [Google Scholar]
- 144.Smith DH, Meaney DF, Shull WS. Diffuse axonal injury in head trauma. J Head Trauma Rehab. 2003;18:307–316. doi: 10.1097/00001199-200307000-00003. [DOI] [PubMed] [Google Scholar]
- 145.Smith DH, Nonaka M, Miller R, Leoni M, Chen XH, Alsop D, Meaney DF. Immediate coma following inertial brain injury dependent on axonal damage in the brainstem. J Neurosurg. 2000;93:315–322. doi: 10.3171/jns.2000.93.2.0315. [DOI] [PubMed] [Google Scholar]
- 146.Smith DH, Uryu K, Saatman KE, Trojanowski JQ, McIntosh TK. Protein accumulation in traumatic brain injury. Neuromolecular Med. 2003;4:59–72. doi: 10.1385/NMM:4:1-2:59. [DOI] [PubMed] [Google Scholar]
- 147.Smith DH, Wolf JA, Lusardi TA, Lee VM, Meaney DF. High tolerance and delayed elastic response of cultured axons to dynamic stretch injury. J Neurosci. 1999;19:4263–4269. doi: 10.1523/JNEUROSCI.19-11-04263.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Smith DH, Wolf JA, Lusardi TA, Lee VM-Y, Meaney DF. High tolerance and delayed elastic response of cultured axons to dynamic stretch injury. J Neurosci. 1999;19:4263–4269. doi: 10.1523/JNEUROSCI.19-11-04263.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Staal JA, Dickson TC, Chung RS, Vickers JC. Disruption of the ubiquitin proteasome system following axonal stretch injury accelerates progression to secondary axotomy. J Neurotrauma. 2009;26:781–788. doi: 10.1089/neu.2008.0669. [DOI] [PubMed] [Google Scholar]
- 150.Staal JA, Dickson TC, Gasperini R, Liu Y, Foa L, Vickers JC. Initial calcium release from intracellular stores followed by calcium dysregulation is linked to secondary axotomy following transient axonal stretch injury. J Neurochem. 2010;112:1147–1155. doi: 10.1111/j.1471-4159.2009.06531.x. [DOI] [PubMed] [Google Scholar]
- 151.Staal JA, Vickers JC. Selective vulnerability of non-myelinated axons to stretch injury in an in vitro co-culture system. J Neurotrauma. 2011;28:841–847. doi: 10.1089/neu.2010.1658. [DOI] [PubMed] [Google Scholar]
- 152.Stokin GB, Lillo C, Falzone TL, Brusch RG, Rockenstein E, Mount SL, Raman R, Davies P, Masliah E, Williams DS, Goldstein LS. Axonopathy and transport deficits early in the pathogenesis of Alzheimer’s disease. Science. 2005;307:1282–1288. doi: 10.1126/science.1105681. [DOI] [PubMed] [Google Scholar]
- 153.Stone JR, Okonkwo DO, Singleton RH, Mutlu LK, Helm GA, Povlishock JT. Caspase-3-mediated cleavage of amyloid precursor protein and formation of amyloid Beta peptide in traumatic axonal injury. J Neurotrauma. 2002;19:601–614. doi: 10.1089/089771502753754073. [DOI] [PubMed] [Google Scholar]
- 154.Strich SJ. Diffuse degeneration of the cerebral white matter in severe dementia following head injury. J Neurol Neurosurg Psychiatry. 1956;19:163–185. doi: 10.1136/jnnp.19.3.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Strich SJ. Sharing of the nerve fibers as a cause of brain damage due to head injury: A pathological study of 20 cases. The Lancet. 1961;278:443–448. [Google Scholar]
- 156.Stys PK, Jiang Q. Calpain-dependent neurofilament breakdown in anoxic and ischemic rat central axons. Neurosci Lett. 2002;328:150–154. doi: 10.1016/s0304-3940(02)00469-x. [DOI] [PubMed] [Google Scholar]
- 157.Sullivan P, Petitti D, Barbaccia J. Head trauma and age of onset of dementia of the Alzheimer type. Jama. 1987;257:2289–2290. doi: 10.1001/jama.1987.03390170045014. [DOI] [PubMed] [Google Scholar]
- 158.Tang-Schomer MD, Johnson VE, Baas PW, Stewart W, Smith DH. Partial interruption of axonal transport due to microtubule breakage accounts for the formation of periodic varicosities after traumatic axonal injury. Exp Neurol. 2011 doi: 10.1016/j.expneurol.2011.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Tang-Schomer MD, Patel AR, Baas PW, Smith DH. Mechanical breaking of microtubules in axons during dynamic stretch injury underlies delayed elasticity, microtubule disassembly, and axon degeneration. FASEB J. 2010;24:1401–1410. doi: 10.1096/fj.09-142844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Thibault LE, Gennarelli TA, Margulies SS, Marcus J, Eppinger R. The Strain Dependent Pathophysiological Consequences of Inertial Loading on Central Nervous System Tissue; Proceedings of the International Conference on the Biomechanics of Impact; Lyon, France. 1990. pp. 191–202. [Google Scholar]
- 161.Tokuda T, Ikeda S, Yanagisawa N, Ihara Y, Glenner GG. Re-examination of ex-boxers’ brains using immunohistochemistry with antibodies to amyloid beta-protein and tau protein. Acta Neuropathol (Berl) 1991;82:280–285. doi: 10.1007/BF00308813. [DOI] [PubMed] [Google Scholar]
- 162.Tokuda T, Ikeda S, Yanagisawa N, Ihara Y, Glenner GG. Re-examination of ex-boxers’ brains using immunohistochemistry with antibodies to amyloid beta-protein and tau protein. Acta Neuropathol. 1991;82:280–285. doi: 10.1007/BF00308813. [DOI] [PubMed] [Google Scholar]
- 163.Tran HT, Laferla FM, Holtzman DM, Brody DL. Controlled Cortical Impact Traumatic Brain Injury in 3xTg-AD Mice Causes Acute Intra-Axonal Amyloid-{beta} Accumulation and Independently Accelerates the Development of Tau Abnormalities. J Neurosci. 31:9513–9525. doi: 10.1523/JNEUROSCI.0858-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Uryu K, Chen XH, Martinez D, Browne KD, Johnson VE, Graham DI, Lee VM, Trojanowski JQ, Smith DH. Multiple proteins implicated in neurodegenerative diseases accumulate in axons after brain trauma in humans. Exp Neurol. 2007;208:185–192. doi: 10.1016/j.expneurol.2007.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.von Reyn CR, Spaethling JM, Mesfin MN, Ma M, Neumar RW, Smith DH, Siman R, Meaney DF. Calpain mediates proteolysis of the voltage-gated sodium channel alpha-subunit. J Neurosci. 2009;29:10350–10356. doi: 10.1523/JNEUROSCI.2339-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Wilde EA, McCauley SR, Hunter JV, Bigler ED, Chu Z, Wang ZJ, Hanten GR, Troyanskaya M, Yallampalli R, Li X, Chia J, Levin HS. Diffusion tensor imaging of acute mild traumatic brain injury in adolescents. Neurology. 2008;70:948–955. doi: 10.1212/01.wnl.0000305961.68029.54. [DOI] [PubMed] [Google Scholar]
- 167.Wolf JA, Stys PK, Lusardi T, Meaney D, Smith DH. Traumatic axonal injury induces calcium influx modulated by tetrodotoxin-sensitive sodium channels. J Neurosci. 2001;21:1923–1930. doi: 10.1523/JNEUROSCI.21-06-01923.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Yuen TJ, Browne KD, Iwata A, Smith DH. Sodium channelopathy induced by mild axonal trauma worsens outcome after a repeat injury. J Neurosci Res. 2009;87:3620–3625. doi: 10.1002/jnr.22161. [DOI] [PMC free article] [PubMed] [Google Scholar]