

Abstract
While a history of a single traumatic brain injury (TBI) is associated with the later development of syndromes of cognitive impairment such as Alzheimer's disease, the long‐term pathology evolving after single TBI is poorly understood. However, a progressive tauopathy, chronic traumatic encephalopathy, is described in selected cohorts with a history of repetitive concussive/mild head injury. Here, post‐mortem brains from long‐term survivors of just a single TBI (1–47 years survival; n = 39) vs. uninjured, age‐matched controls (n = 47) were examined for neurofibrillary tangles (NFTs) and amyloid‐β (Aβ) plaques using immunohistochemistry and thioflavine‐S staining. Detailed maps of findings permitted classification of pathology using semiquantitative scoring systems. NFTs were exceptionally rare in young, uninjured controls, yet were abundant and widely distributed in approximately one‐third of TBI cases. In addition, Aβ‐plaques were found in a greater density following TBI vs. controls. Moreover, thioflavine‐S staining revealed that while all plaque‐positive control cases displayed predominantly diffuse plaques, 64% of plaque‐positive TBI cases displayed predominantly thioflavine‐S‐positive plaques or a mixed thioflavine‐S‐positive/diffuse pattern. These data demonstrate that widespread NFT and Aβ plaque pathologies are present in up to a third of patients following survival of a year or more from a single TBI. This suggests that a single TBI induces long‐term neuropathological changes akin to those found in neurodegenerative disease.
Keywords: amyloid beta plaques, head injury, long‐term survival, neurodegeneration, neurofibrillary tangles, traumatic brain injury
INTRODUCTION
Representing a major health issue, 1.7 million people in the United States suffer a traumatic brain injury (TBI) each year (9). In addition to the acute effects, there is growing concern that a single TBI may initiate long‐term processes that further damage the brain (20). Indeed, in epidemiological studies, TBI is recognized as a major risk factor for the later development of syndromes of cognitive impairment, such as Alzheimer's disease (AD) 14, 16, 26, 27, 28, 30, 33, 38, 39.
While a long‐term pathological link between a single TBI and neurodegenerative disease has not been identified, a tau pathology, currently referred to as chronic traumatic encephalopathy (CTE; formerly dementia pugilistica), has been described in the brains of individuals exposed to repetitive, often mild or concussive, head injury such as boxers 7, 8, 10, 35, 43 and, more recently, professional American football players 23, 24, 31, 32. In contrast, thus far, neurofibrillary tangles (NFTs) have not been identified following a single TBI in humans; although the only study examining this was limited to patients dying within 4 weeks of injury (40). Thus, it remains unclear whether tau pathology may appear as a late phenomenon following a single TBI or if it is a manifestation unique to repetitive injury.
In contrast to NFTs, amyloid‐beta (Aβ) plaques, a hallmark pathology of AD, have been identified following a single TBI in approximately 30% of patients 6, 18, 19, 36, 37, 41, 45. Notably, plaques found acutely following TBI are typically diffuse in nature and in contrast to the thioflavine‐S‐positive neuritic plaques characteristic of advanced AD. While similarly diffuse plaques can be observed as part of so called “normal aging”12, 34, 37, 44, TBI appears capable of inducing diffuse plaque formation within hours of injury, even in children 13, 19, 37. However, few plaques are observed following survival of several months from injury (6), perhaps indicating that the plaques observed initially following TBI may be a limited acute phase response. Alternatively, it is possible that increased severity of injury associated with early mortality is an important determinant of plaque pathology. Nonetheless, as with tau pathologies, Aβ plaques have not been evaluated years after a single TBI.
METHODS
We examined the brains of long‐term survivors (1–47 years) following a single TBI to evaluate the presence of NFTs and Aβ‐plaques. Brains were selected from the comprehensive TBI brain archive of the Department of Neuropathology, Southern General Hospital, Glasgow, UK. Approval for the use of tissue was granted by the South Glasgow and Clyde Research Ethics Committee. Cases were selected with a history of a single TBI (n = 39) and were compared to age‐matched controls (n = 47). Detailed reports from the diagnostic post‐mortem and/or forensic reports were available for all cases and confirmed a history of a single moderate to severe TBI, which was confirmed at diagnostic post‐mortem. Following a single TBI, all cases were discharged from hospitalization following recovery and, ultimately, died from causes of death unrelated to TBI or trauma. None were in a persistent vegetative state prior to death. Cases with a history of amateur or professional boxing were excluded. From the same archive, age‐matched controls were selected with no documented history of neurological disease, neurodegenerative disease or TBI, and no autopsy evidence to support previous TBI (Table 1). All material was obtained following routine diagnostic autopsy examination at the same institution serving a distinct regional location.
Table 1.
Demographic and clinical data for cases. Abbreviations: PM = post‐mortem; TBI = traumatic brain injury; SUDEP = sudden unexpected death in epilepsy; GI = gastrointestinal; ARDS = acute respiratory distress syndrome; GSW = gun shot wound; MVC = motor vehicle collision; * = 3 unknown.
| TBI cases (n = 39) | Controls (n = 47) | |||
|---|---|---|---|---|
| Mean age (Range) (Years) | 53 (19–89) | 47 (14–92) | ||
| Males | 35 (89.7%) | 30 (64.8%) | ||
| Mean PM delay (Range) (Hours) | 64 (12–184)* | 57 (5–264) | ||
| Mean survival interval (Range) (Years) | 8.2 (1–47) | Not applicable | ||
| Cause of TBI | Assault | 8 (20.5%) | Not applicable: controls had no known history of TBI | |
| Fall | 16 (41.0%) | |||
| MVC | 8 (20.5%) | |||
| Unknown | 7 (17.9%) | |||
| Cause of death | Pulmonary (Including pneumonia) | 9 (23.1%) | SUDEP | 14 (29.8%) |
| Chronic heart failure | 6 (15.4%) | Pulmonary (Including pneumonia) | 9 (19.1%) | |
| SUDEP | 6 (15.4%) | Acute cardiovascular | 7 (14.9%) | |
| Acute cardiovascular | 4 (10.3%) | Drug overdose | 4 (8.5%) | |
| GI disease (Including liver failure) | 3 (7.7%) | Chronic heart failure | 4 (8.5%) | |
| Renal disease (Including pyelonephritis) | 3 (7.7%) | Sepsis | 2 (4.3%) | |
| Unknown | 3 (7.7%) | Malignancy | 2 (4.3%) | |
| Malignancy | 2 (5.1%) | Hypothermia | 1 (2.1%) | |
| Acute intracerebral hemorrhage | 1 (2.6%) | Vasculitis | 1 (2.1%) | |
| ARDS | 1 (2.6%) | GSW—chest | 1 (2.1%) | |
| Hypothermia | 1 (2.6%) | GI disease (Including liver failure) | 1 (2.1%) | |
| Myasthenia gravis | 1 (2.1%) | |||
Brain tissue preparation
Whole brains were immersion fixed in 10% formol saline for a minimum of 3 weeks then dissected, sampled following a standardized block selection protocol and processed to paraffin using standard techniques. Three blocks from the coronal plane at midthalamic level were selected to include: (i) hippocampus at the level of the lateral geniculate nucleus extending through the entorhinal cortex including the inferior temporal gyrus; (ii) corpus callosum and cingulate gyrus; and (iii) insula. Regions of analysis were standardized based on the hypothesis that TBI is capable of inducing widespread neuropathological changes long‐term following injury, regardless of the location of any focal lesion. Specific regions were selected based on previous published observations of acute plaque formation following injury and brain regions with known vulnerability in both TBI and neurodegenerative disorders.
Immunohistochemistry
Following deparaffinization and rehydration to dH2O, sections were immersed in 3% aqueous H2O2 (10 minutes) to quench endogenous peroxidase activity. Antigen retrieval on all sections (8 µm) was performed via microwave. Additional antigen retrieval for Aβ staining included 5 minutes immersion in 77% formic acid. Blocking was then performed using one drop of normal horse serum (Vector Labs, Burlingame, CA, USA) per 5 mL of Optimax buffer (BioGenex, San Ramon, CA, USA) for 30 minutes. Incubation with the primary antibody was then performed for 20 h at 4°C. Immunostaining for tau was performed with a polyclonal antibody (Dako, Carpinteria, CA, USA; 1:7500) and for Aβ, the antibody 6F3D specific for the N‐terminal epitope of the peptide was used (Dako; 1:500). Visualization was achieved using a DAB kit (Vector Labs) and sections analyzed using a Leica DMRB light microscope (Leica Microsystems, Wetzlar, Germany).
Thioflavine‐S staining
To identify beta‐pleated sheet conformations of tau and Aβ, a modified thioflavine‐S staining technique was performed (15). Briefly, 8‐µm sections of tissue were deparaffinized, rehydrated to dH2O and immersed in phosphate‐buffered saline (PBS) for 5 minutes. Sections were then immersed in 0.05% KMnO4 in PBS for 20 minutes. Following rinsing, tissue was destained in 0.2% K2S2O5/0.2% Oxalic acid/PBS and subsequently immersed in 0.0125% thioflavine‐S (Sigma‐Aldrich, St Louis, MO, USA; in 40% EtOH/60% PBS). Tissue was then differentiated in 50% EtOH/50% PBS. Following rinsing, sections were cover‐slipped using a fluorescence mounting medium (Dako) and analyzed using a Leitz Laberlux‐K microscope (Leica Microsystems, Wetzlar, Germany) using a fluorescein isothiocyanate filter.
Pathological classification of findings
Observations were conducted blind to the demographic and clinical information for all cases. Detailed maps of NFTs and plaques were generated for each region to classify each case in accordance with standardized scoring systems.
NFTs
NFTs revealed by tau immunohistochemistry were semiquantitatively assessed and classified based on their extent and distribution. Group 1 pathology comprised cases with NFTs of sparse to moderate density in the transentorhinal cortex with or without small numbers in the CA1 sector of the hippocampus. Group 2 pathology included NFTs as described in Group 1 together with NFTs extending into the fusiform gyrus (lateral to entorhinal cortex) and sparse tangles in the isocortex (cingulate/insular blocks). Group 3 pathology contained NFTs in more widespread sectors of the hippocampus and subiculum and with extensive isocortical involvement.
Aβ plaques
Semiquantitative assessments of age‐adjusted plaque density were generated based on the mean plaque score across the three regions assessed. Thus, using standard protocols for assessment of plaque density with respect to age (25), sections with no (0), sparse (1), moderate (2) and frequent (3) plaques were identified.
Statistical analysis
Statistical analysis comparing cohorts for the presence of pathology was performed using the chi‐square test.
RESULTS
NFT pathology
NFT pathology was observed more widely across the age spectrum following TBI than in controls, being present in cases aged as young as 27 years (1, 2). In addition, NFTs were more common following TBI, with 18 of 39 (46%) cases containing tangles compared with 16 of 47 (34%) controls (Figure 2A); although in assessing the full cohort, including material from individuals at all ages, this difference did not reach statistical significance. However, in comparison with TBI cases, virtually all the controls displaying NFT pathology were aged over 60 years. As such, when material from patients aged greater than 60 is excluded from analysis, a clear difference is observed in the presence of NFTs following TBI vs. controls. Specifically, 11 of the 32 (34%) TBI cases aged 60 years or younger displayed NFT pathology compared with just three of the 32 (9%) controls (P = 0.015; chi‐square) (Figure 2A). Of these three control cases, two were aged 60 and one was aged 55.
Figure 1.
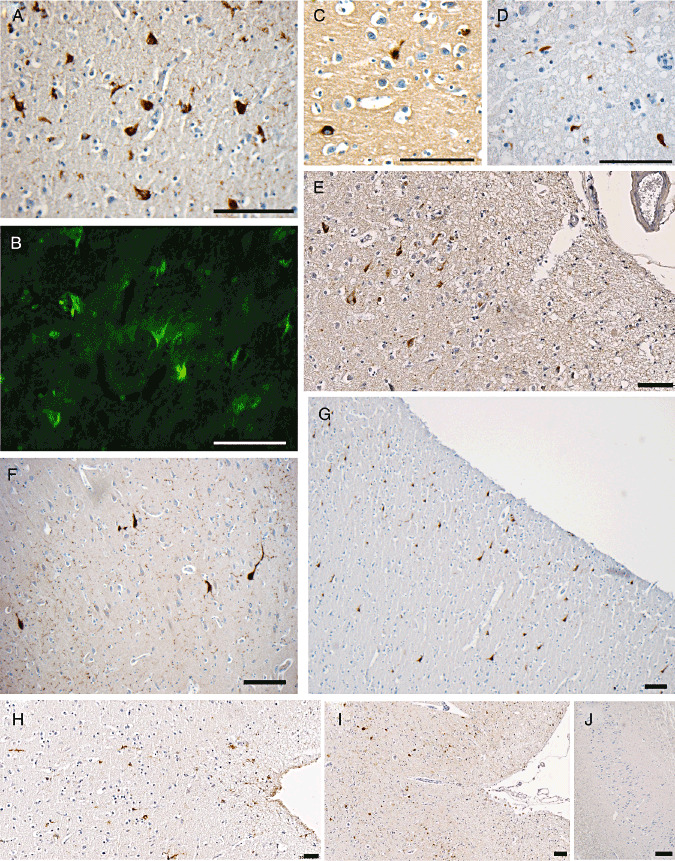
Representative immunohistochemical and thioflavine‐S staining for neurofibrillary tangles (NFTs). A. NFTs in the parahippocampal gyrus of a 49‐year‐old male 1 year post‐traumatic brain injury (TBI). B. Representative thioflavine‐S‐positive staining in the same case as A. C. NFTs in the fusiform gyrus of a 27‐year‐old male 1.5 years after TBI. D. NFTs in the frontal lobe of a case of advanced Alzheimer's disease (positive control). E, G. Representative images showing prevalent NFTs in the superficial layers of the cortex of the medial temporal lobe. F. Extensive neuropil threads and occasional NFTs in a 53‐year‐old individual who died 8 years following TBI. H, I. Representative images showing isolated clusters of NFTs within the depth of sulci. J. Uninjured control case displaying no neurons positive for tau immunostaining in the hippocampal region CA1. All scale bars approximately 100 µm.
Figure 2.
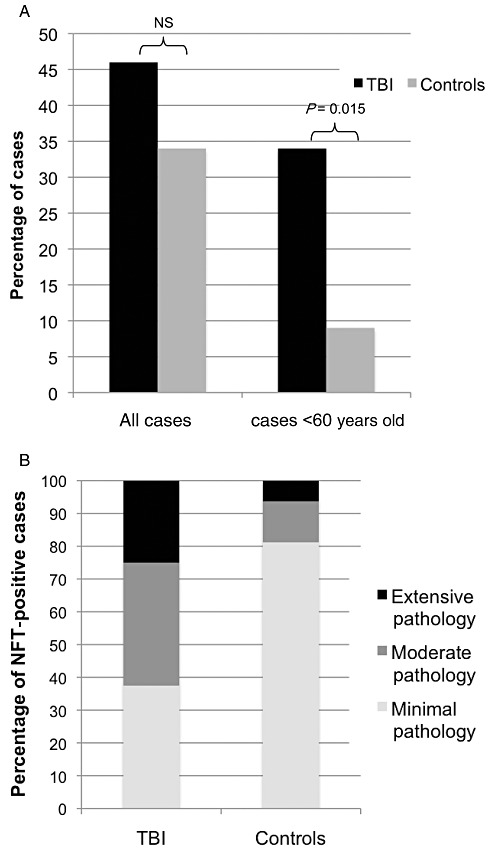
Graphical representation of findings regarding neurofibrillary tangles (NFTs). A. Percentage of cases with NFTs in entire cohort/cohort with those aged >60 excluded: traumatic brain injury (TBI) vs. controls. B. Extent and distribution of NFTs in TBI positive cases vs. controls by pathological group. Group 1 (minimal pathology): NFTs of sparse to moderate density in the transentorhinal cortex with or without small numbers in the CA1 sector of the hippocampus. Group 2 (moderate pathology): as described in Group 1, together with NFTs extending into the fusiform gyrus (lateral to entorhinal cortex) and sparse tangles in the isocortex (cingulate/insular blocks). Group 3 (extensive pathology): NFTs in more widespread sectors of the hippocampus and subiculum and with extensive isocortical involvement.
Notably, following TBI, NFTs were commonly observed in the superficial cortical layers, with clustering of NFTs among the depths of the sulci and at points of geometric inflection. Indeed, following TBI, well‐defined clusters of NFTs could be found in the cingulate gyrus extending through the superior frontal gyrus and also within the insular cortex. This pattern was not observed in controls who rarely displayed pathology outside the transentorhinal cortex and CA1. In addition, in those cases with NFTs, glia immunoreactive for tau could also be observed following TBI and, to a lesser extent, in control cases, although these were not independently quantified.
The distribution of NFTs in both TBI cases and controls permitted classification using a semiquantitative protocol reflecting the density and distribution of tau pathology. Following TBI, cases displayed more extensive pathology than controls (Figure 2B). Specifically six of 18 (33%) cases met the criteria for Group 1, with tangles in the transentorhinal cortex and CA1 of hippocampus; eight of 18 (44%) were Group 2, with NFTs in the same regions as Group 1, but with extension into the fusiform gyrus; the remaining four of 18 (22%) were Group 3, with NFTs more widespread in the hippocampus and subiculum and with more extensive isocortical involvement. In comparison, the distribution of NFTs in controls was consistent with the published literature for so called “normal aging”34, 44, with 13 of the 16 (81%) controls with NFTs displaying Group 1 pathology, two cases (13%) with Group 2 pathology and just a single example displaying Group 3 pathology. Thus, in addition to being more prevalent at an earlier age, the extent and distribution of NFTs, as assessed by this semiquantitative protocol, is significantly greater in material from individuals surviving a year or more post‐TBI than in controls (P = 0.019; chi‐square).
Aβ plaque pathology
Aβ plaques were observed in 13 of 47 (28%) controls vs. 11 of 39 (28%) TBI cases (Figure 3). While the incidence of plaque pathology was not statistically different between controls and TBI cases, the extent of plaques differed greatly. Specifically, following TBI, age‐adjusted plaque density was moderate (27%; three of 11 cases) or, more commonly, high (73%; eight of 11 cases), whereas in controls 31% (four of 13 cases) had sparse plaque, 31% (four of 13 cases) moderate and just 38% (five of 13 cases) high (Figure 4A). While this observation failed to reach statistical significance (P = 0.095; chi‐square), there is a clear trend to a wider distribution and a higher density of Aβ plaques (either diffuse or thioflavine‐S‐positive) following survival from TBI.
Figure 3.
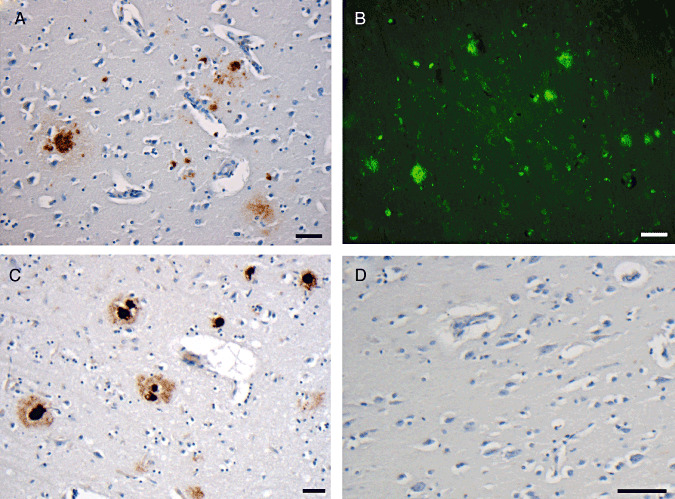
Representative immunohistochemical and thioflavine‐S staining for amyloid‐beta (Aβ) plaques. A. Plaques in the inferior temporal gyrus of a 55‐year‐old female 47 years post‐ traumatic brain injury. B. Representative thioflavine‐S‐positive staining in the same cases as A. C. Plaques in the frontal lobe of a case of advanced Alzheimer's disease. D. Uninjured control displaying no Aβ immunostaining. All scale bars approximately 50 µm.
Figure 4.
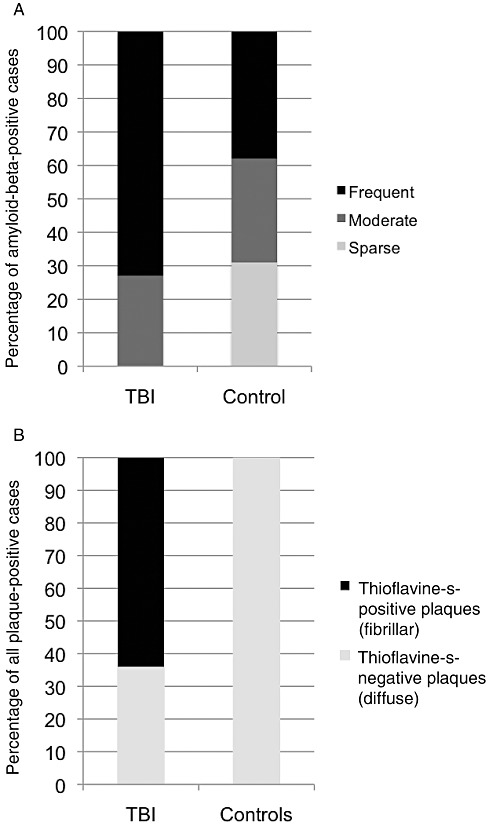
Graphical representation of findings regarding amyloid‐beta plaques. A. Age‐adjusted plaque density in plaque‐positive traumatic brain injury (TBI) cases (11 of 39; 28%) vs. controls (13 of 47; 28%). B. Percentage of plaque‐positive cases displaying thioflavine‐S‐positive staining in TBI cases vs. controls.
Typically cases could broadly be described as having either (i) widespread and relatively evenly distributed plaques dispersed throughout all layers of the cortex vs. (ii) isolated clusters of plaques scattered throughout the cortex with interspersed, plaque‐free regions. Typically, following TBI, cases were less likely to display smaller clustered regions of plaques and more likely to have widespread plaques across the entire cortical region examined. This was particularly true extending from the cingulate gyrus through the superior frontal gyrus, which frequently displayed extensive and widespread pathology after TBI.
However, while plaques could often be observed in the entorhinal cortex, throughout the fusiform gyrus and the inferior temporal gyrus, there were comparatively minimal plaques in the hippocampus and subiculum. Cases demonstrating involvement of the hippocampus and subiculum universally displayed extensive plaque pathology throughout all other regions analyzed, potentially indicating a relative sparing of this region in early disease.
In addition to an increased density of plaques revealed by immunohistochemistry, thioflavine‐S staining revealed that the nature of Aβ plaques following TBI also differed from controls. Specifically, while all 13 of the controls with Aβ plaques displayed predominantly diffuse plaques with very occasional, isolated fibrillar plaques, in seven of the 11 (64%) plaque‐positive TBI cases, plaques were predominantly fibrillar in nature or displayed a mixed diffuse/fibrillar pattern (P = 0.003; chi‐square) (Figure 4B).
DISCUSSION
This study demonstrates that NFTs are present at a higher density and in a wider distribution in brains of patients surviving greater than 1 year following just a single TBI when compared with age‐matched controls from the same archive. Similarly, there is evidence of more extensive Aβ plaque pathology, often fibrillary in nature, in survivors of TBI. These observations raise the intriguing possibility that the pathological mechanisms leading to neurodegenerative disease may be initiated or accelerated as part of the chronic pathological milieu following survival post‐TBI, with the pathology emerging even in young adults.
Although a previous study failed to find NFTs following a single TBI, analysis was limited to within 4 weeks of injury (40). By extending the examination to years after injury, the present data demonstrate that NFTs can indeed emerge following just a single TBI. Appearing in over 30% of individuals aged less than 60 years, NFTs were often regionally widespread and extensive. This finding is in contrast to the minimal pathology observed in controls. Indeed, NFTs observed in the control group were almost exclusively observed in cases aged 60 or greater at a frequency highly consistent with a large cohort of 2661 nonselected autopsy cases reported by Braak and Braak (3). Specifically, according to this large data set, 91.6% of cases over the age of 60 displayed NFT pathology to some extent. For the control group presented here, this number is 91.2% indicating a high level of consistency with the largest series in the literature. Only one control case aged less than 60 (age 55) was determined to have NFTs, which again is consistent with the comprehensive data by Braak and Braak (3). In contrast, following survival of a year or more from a moderate to severe TBI, NFTs were observed in autopsy‐derived brain material from individuals dying in their third to fourth decade.
The apparent delayed appearance of NFTs after trauma suggests that even a single TBI may, in the long term, be associated with a neurodegenerative process. Moreover, this finding provides a potential pathological substrate for the epidemiological observation of an increased risk of developing syndromes of cognitive impairment, such as AD, following TBI 14, 16, 26, 27, 28, 30, 33, 38, 39. A similar tau pathology has been observed in autopsy‐acquired brains from athletes involved in contact sports, thought to be a consequence of repetitive TBI 7, 8, 10, 23, 31, 32, 35, 43. NFTs observed in the current, relatively large cohort of patients following a single moderate/severe TBI suggest tau pathology may not be exclusive to situations where there is a history of repetitive injury. Of note, although cases with a known history of repetitive injury, such as boxing, were excluded, it is difficult to determine with complete certainty whether any of the cases within this study experienced repeat head injury because of the retrospective nature of the archive and available clinical information. However, a large age and demographically matched control group for which tissue was accrued at the same institution was included for comparison and failed to display pathology at the same frequency or extent.
The observation of NFTs in relatively young individuals long after just a single TBI supports epidemiological studies suggesting that TBI may act to accelerate the clinical onset of neurodegenerative disease 11, 29, 39, 42. Notably, analysis of over 1200 TBI survivors demonstrated that the time to onset of AD was significantly reduced in those who sustained TBI (29). The median time to onset from TBI to the development of AD in that cohort was 10 years—a finding that is consistent with the NFTs encountered post‐TBI in this study.
The regional distribution of NFTs observed post‐trauma follows a similar hierarchical progression to that of AD 1, 2. However, in AD, the deep cortical layers are predominantly affected (2), whereas in CTE it is the superficial layers (17). While NFT pathology in our cohort was not uniformly superficial, this was a common finding, possibly indicating a pattern of tau pathology specific to trauma and common to both single and repetitive head injury. Furthermore, at points of geometric inflection, such as the depths of sulci, clustering of NFTs was frequently found. Although not typical of AD, similar clustering has been demonstrated in CTE 5, 23. Potentially unique to TBI, this may arise as the brain is rapidly deformed, producing concentrations of mechanical stress at inflection points that, ultimately, may lead to more severe cellular pathology, such as the formation of NFTs. Interestingly, glial cells immunoreactive for tau were also observed more commonly following TBI, typically in regions where NFTs were observed. Although not directly quantified, this is a finding worthy of further exploration with regards to delineating the mechanisms of tau accumulation and, possibly, clearance.
Previous investigations of the link between TBI and neurodegenerative disease primarily focused on Aβ plaques as these were found in approximately 30% of cases in the immediate period after injury (20). However, the long‐term Aβ plaque pathology following TBI had been in doubt as a consequence of the observation that, in contrast to the acute phase, few plaques are seen in the months following TBI (6). Plaque regression mediated by the Aβ‐clearing enzyme neprilysin has been suggested as a potential mechanism that may drive plaque clearance in the weeks following TBI 6, 21.
In this study we observed Aβ plaques in long‐term TBI survivors at a greater density than in age‐matched controls. In contrast to NFTs, plaques were evident in older cases, a feature previously observed with plaques forming in the acute phase post‐injury 12, 37. Furthermore, unlike Aβ plaques encountered in both normal aging and acutely post‐TBI, which are almost universally diffuse in nature in individuals aged less than 75 12, 34, 37, 44, the plaques observed in this cohort of survivors of a year or more post‐TBI were more often fibrillary.
While this work indicates a clear association between a history of a single moderate to severe TBI and later development of Aβ and tau pathologies, the limitations inherent in a retrospective study such as this based on archival tissue did not permit extensive regional analyses or subanalyses with regard to correlating the specific TBI‐induced pathologies, mechanism of injury or therapeutic intervention with observed pathological findings. However, the results do support the establishment of prospective studies with suitably sampled brain tissue to explore these specific points. In addition, detailed analysis of lesion location in relation to pathology may also be of interest. Here, a standardized analysis over three regions was performed to determine whether trauma is capable of inducing a widespread pathology that might be associated with neurodegenerative disease. Specific regions were sampled because of their known vulnerability in both TBI and neurodegenerative disorders, as well as reflecting previous observations of the acute pathology following TBI, such as plaques 6, 18, 19, 36, 37. Notably, previous data examining plaques following acute TBI suggest the location of any focal lesion (e.g., cerebral contusion) does not influence the regional distribution of plaques, in other words, it would appear it is the diffuse consequences of TBI that may be mechanistically important (6). Again, however, larger scale prospective studies may permit direct comparisons with the pathologies identified here and those of both AD and following repetitive TBI.
Collectively, these data demonstrating the presence of widespread and extensive tau and Aβ pathology many years after a single moderate to severe TBI may, in part, represent a substrate for the long‐term development of neurodegenerative disease, such as AD, known to be at increased incidence in long‐term survivors of TBI. There is undoubtedly a need for direct quantitation and exploration of the mechanistic basis underpinning these observations, including potential genetic determinants such as apolipoprotein E genotype. However, these findings may indicate important potential therapeutic considerations for survivors of TBI, such as emerging anti‐tau and anti‐Aβ drugs 4, 22.
ACKNOWLEDGMENTS
This work was supported by NIH grants NS038104 and NS056202. We would like to express our gratitude to Ms Janice E. Stewart who assisted with the tissue preparation and immunohistochemical techniques described in this study.
REFERENCES
- 1. Braak H, Braak E (1990) Neurofibrillary changes confined to the entorhinal region and an abundance of cortical amyloid in cases of presenile and senile dementia. Acta Neuropathol 80:479–486. [DOI] [PubMed] [Google Scholar]
- 2. Braak H, Braak E (1991) Neuropathological stageing of Alzheimer‐related changes. Acta Neuropathol 82:239–259. [DOI] [PubMed] [Google Scholar]
- 3. Braak H, Braak E (1997) Frequency of stages of Alzheimer‐related lesions in different age categories. Neurobiol Aging 18:351–357. [DOI] [PubMed] [Google Scholar]
- 4. Brunden KR, Trojanowski JQ, Lee VM (2009) Advances in tau‐focused drug discovery for Alzheimer's disease and related tauopathies. Nat Rev Drug Discov 8:783–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Buee L, Hof PR, Bouras C, Delacourte A, Perl DP, Morrison JH, Fillit HM (1994) Pathological alterations of the cerebral microvasculature in Alzheimer's disease and related dementing disorders. Acta Neuropathol 87:469–480. [DOI] [PubMed] [Google Scholar]
- 6. Chen XH, Johnson VE, Uryu K, Trojanowski JQ, Smith DH (2009) A lack of amyloid beta plaques despite persistent accumulation of amyloid beta in axons of long‐term survivors of traumatic brain injury. Brain Pathol 19:214–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Corsellis JA, Bruton CJ, Freeman‐Browne D (1973) The aftermath of boxing. Psychol Med 3:270–303. [DOI] [PubMed] [Google Scholar]
- 8. Dale GE, Leigh PN, Luthert P, Anderton BH, Roberts GW (1991) Neurofibrillary tangles in dementia pugilistica are ubiquitinated. J Neurol Neurosurg Psychiatry 54:116–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Faul M, Xu L, Wald MM, Coronado VG (2010) Traumatic Brain Injury in the United States: Emergency Department Visits, Hospitalizations, and Deaths. Centers for Disease Control and Prevention, National Center for Injury Prevention and Control: Atlanta. [Google Scholar]
- 10. Geddes JF, Vowles GH, Nicoll JA, Revesz T (1999) Neuronal cytoskeletal changes are an early consequence of repetitive head injury. Acta Neuropathol 98:171–178. [DOI] [PubMed] [Google Scholar]
- 11. Gedye A, Beattie BL, Tuokko H, Horton A, Korsarek E (1989) Severe head injury hastens age of onset of Alzheimer's disease. J Am Geriatr Soc 37:970–973. [DOI] [PubMed] [Google Scholar]
- 12. Gentleman SM, Greenberg BD, Savage MJ, Noori M, Newman SJ, Roberts GW et al (1997) A beta 42 is the predominant form of amyloid beta‐protein in the brains of short‐term survivors of head injury. Neuroreport 8:1519–1522. [DOI] [PubMed] [Google Scholar]
- 13. Graham DI, Gentleman SM, Lynch A, Roberts GW (1995) Distribution of beta‐amyloid protein in the brain following severe head injury. Neuropathol Appl Neurobiol 21:27–34. [DOI] [PubMed] [Google Scholar]
- 14. Graves AB, White E, Koepsell TD, Reifler BV, van Belle G, Larson EB, Raskind M (1990) The association between head trauma and Alzheimer's disease. Am J Epidemiol 131:491–501. [DOI] [PubMed] [Google Scholar]
- 15. Guntern R, Bouras C, Hof PR, Vallet PG (1992) An improved thioflavine S method for staining neurofibrillary tangles and senile plaques in Alzheimer's disease. Experientia 48:8–10. [DOI] [PubMed] [Google Scholar]
- 16. Guo Z, Cupples LA, Kurz A, Auerbach SH, Volicer L, Chui H et al (2000) Head injury and the risk of AD in the MIRAGE study. Neurology 54:1316–1323. [DOI] [PubMed] [Google Scholar]
- 17. Hof PR, Bouras C, Buee L, Delacourte A, Perl DP, Morrison JH (1992) Differential distribution of neurofibrillary tangles in the cerebral cortex of dementia pugilistica and Alzheimer's disease cases. Acta Neuropathol 85:23–30. [DOI] [PubMed] [Google Scholar]
- 18. Huber AGK, Kelemen J, Cervod‐Navarro J (1993) Density of amyloid plaques in brains after head trauma. J Neurotrauma 10(Suppl.):S180. [Google Scholar]
- 19. Ikonomovic MD, Uryu K, Abrahamson EE, Ciallella JR, Trojanowski JQ, Lee VM et al (2004) Alzheimer's pathology in human temporal cortex surgically excised after severe brain injury. Exp Neurol 190:192–203. [DOI] [PubMed] [Google Scholar]
- 20. Johnson VE, Stewart W, Smith DH (2010) Traumatic brain injury and amyloid‐beta pathology: a link to Alzheimer's disease? Nat Rev Neurosci 11:361–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Johnson VE, Stewart W, Stewart JE, Graham DI, Praestgaard AH, Smith DH (2009) A neprilysin polymorphism and amyloid‐beta plaques following traumatic brain injury. J Neurotrauma 26:1197–1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Loane DJ, Pocivavsek A, Moussa CE, Thompson R, Matsuoka Y, Faden AI et al (2009) Amyloid precursor protein secretases as therapeutic targets for traumatic brain injury. Nat Med 15:377–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. McKee AC, Cantu RC, Nowinski CJ, Hedley‐Whyte ET, Gavett BE, Budson AE et al (2009) Chronic traumatic encephalopathy in athletes: progressive tauopathy after repetitive head injury. J Neuropathol Exp Neurol 68:709–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. McKee AC, Gavett BE, Stern RA, Nowinski CJ, Cantu RC, Kowall NW et al (2010) TDP‐43 proteinopathy and motor neuron disease in chronic traumatic encephalopathy. J Neuropathol Exp Neurol 69:918–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM et al (1991) The Consortium to Establish a Registry for Alzheimer's Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer's disease. Neurology 41:479–486. [DOI] [PubMed] [Google Scholar]
- 26. Molgaard CA, Stanford EP, Morton DJ, Ryden LA, Schubert KR, Golbeck AL (1990) Epidemiology of head trauma and neurocognitive impairment in a multi‐ethnic population. Neuroepidemiology 9:233–242. [DOI] [PubMed] [Google Scholar]
- 27. Mortimer JA, French LR, Hutton JT, Schuman LM (1985) Head injury as a risk factor for Alzheimer's disease. Neurology 35:264–267. [DOI] [PubMed] [Google Scholar]
- 28. Mortimer JA, van Duijn CM, Chandra V, Fratiglioni L, Graves AB, Heyman A et al (1991) Head trauma as a risk factor for Alzheimer's disease: a collaborative re‐analysis of case‐control studies. EURODEM Risk Factors Research Group. Int J Epidemiol 20(Suppl. 2):S28–S35. [DOI] [PubMed] [Google Scholar]
- 29. Nemetz PN, Leibson C, Naessens JM, Beard M, Kokmen E, Annegers JF, Kurland LT (1999) Traumatic brain injury and time to onset of Alzheimer's disease: a population‐based study. Am J Epidemiol 149:32–40. [DOI] [PubMed] [Google Scholar]
- 30. O'Meara ES, Kukull WA, Sheppard L, Bowen JD, McCormick WC, Teri L et al (1997) Head injury and risk of Alzheimer's disease by apolipoprotein E genotype. Am J Epidemiol 146:373–384. [DOI] [PubMed] [Google Scholar]
- 31. Omalu BI, DeKosky ST, Minster RL, Kamboh MI, Hamilton RL, Wecht CH (2005) Chronic traumatic encephalopathy in a National Football League player. Neurosurgery 57:128–134; discussion ‐34. [DOI] [PubMed] [Google Scholar]
- 32. Omalu BI, DeKosky ST, Hamilton RL, Minster RL, Kamboh MI, Shakir AM, Wecht CH (2006) Chronic traumatic encephalopathy in a national football league player: part II. Neurosurgery 59:1086–1092; discussion 92‐3. [DOI] [PubMed] [Google Scholar]
- 33. Plassman BL, Havlik RJ, Steffens DC, Helms MJ, Newman TN, Drosdick D et al (2000) Documented head injury in early adulthood and risk of Alzheimer's disease and other dementias. Neurology 55:1158–1166. [DOI] [PubMed] [Google Scholar]
- 34. Price JL, Morris JC (1999) Tangles and plaques in nondemented aging and “preclinical” Alzheimer's Disease. Ann Neurol 45:358–368. [DOI] [PubMed] [Google Scholar]
- 35. Roberts GW, Allsop D, Bruton C (1990) The occult aftermath of boxing. J Neurol Neurosurg Psychiatry 53:373–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Roberts GW, Gentleman SM, Lynch A, Graham DI (1991) beta A4 amyloid protein deposition in brain after head trauma. Lancet 338:1422–1423. [DOI] [PubMed] [Google Scholar]
- 37. Roberts GW, Gentleman SM, Lynch A, Murray L, Landon M, Graham DI (1994) Beta amyloid protein deposition in the brain after severe head injury: implications for the pathogenesis of Alzheimer's disease. J Neurol Neurosurg Psychiatry 57:419–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Salib E, Hillier V (1997) Head injury and the risk of Alzheimer's disease: a case control study. Int J Geriatr Psychiatry 12:363–368. [DOI] [PubMed] [Google Scholar]
- 39. Schofield PW, Tang M, Marder K, Bell K, Dooneief G, Chun M et al (1997) Alzheimer's disease after remote head injury: an incidence study. J Neurol Neurosurg Psychiatry 62:119–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Smith C, Graham DI, Murray LS, Nicoll JA (2003) Tau immunohistochemistry in acute brain injury. Neuropathol Appl Neurobiol 29:496–502. [DOI] [PubMed] [Google Scholar]
- 41. Smith DH, Chen XH, Iwata A, Graham DI (2003) Amyloid beta accumulation in axons after traumatic brain injury in humans. J Neurosurg 98:1072–1077. [DOI] [PubMed] [Google Scholar]
- 42. Sullivan P, Petitti D, Barbaccia J (1987) Head trauma and age of onset of dementia of the Alzheimer type. JAMA 257:2289–2290. [DOI] [PubMed] [Google Scholar]
- 43. Tokuda T, Ikeda S, Yanagisawa N, Ihara Y, Glenner GG (1991) Re‐examination of ex‐boxers' brains using immunohistochemistry with antibodies to amyloid beta‐protein and tau protein. Acta Neuropathol 82:280–285. [DOI] [PubMed] [Google Scholar]
- 44. Troncoso JC, Martin LJ, Dal Forno G, Kawas CH (1996) Neuropathology in controls and demented subjects from the Baltimore longditudinal study of aging. Neurobiol Aging 17:365–371. [DOI] [PubMed] [Google Scholar]
- 45. Uryu K, Chen XH, Martinez D, Browne KD, Johnson VE, Graham DI et al (2007) Multiple proteins implicated in neurodegenerative diseases accumulate in axons after brain trauma in humans. Exp Neurol 208:185–192. [DOI] [PMC free article] [PubMed] [Google Scholar]