

Abstract
Studies in animal models have shown that traumatic brain injury (TBI) induces the rapid accumulation of many of the same key proteins that form pathologic aggregates in neurodegenerative diseases. Here, we examined whether this rapid process also occurs in humans after TBI. Brain tissue from 18 cases who died after TBI and from 6 control cases was examined using immunohistochemistry. Following TBI, widespread axonal injury was persistently identified by the accumulation of neurofilament protein and amyloid precursor protein (APP) in axonal bulbs and varicosities. Axonal APP was found to co-accumulate with its cleavage enzymes, beta-site APP cleaving enzyme (BACE), presenilin-1 (PS1) and their product, amyloid-β (Aβ). In addition, extensive accumulation of α-synuclein (α-syn) was found in swollen axons and tau protein was found to accumulate in both axons and neuronal cell bodies. These data show rapid axonal accumulation of proteins implicated in neurodegenerative diseases including Alzheimer’s disease and the synucleinopathies. The cause of axonal pathology can be attributed to disruption of axons due to trauma, or as a secondary effect of raised intracranial pressure or hypoxia. Such axonal pathology in humans may provide a unique environment whereby co-accumulation of APP, BACE, and PS1 leads to intra-axonal production of Aβ as well as accumulation of α-syn and tau. This process may have important implications for survivors of TBI who have been shown to be at greater risk of developing neurodegenerative diseases.
Keywords: Traumatic brain injury, TBI, axonal injury, amyloid β, APP, BACE, PS-1, α-synuclein, tau
Introduction
It has become increasingly accepted that traumatic brain injury (TBI) results in pathophysiological changes similar to those seen in neurodegenerative diseases. Several investigations have suggested a link between a history of TBI and the subsequent development of Alzheimer’s disease (AD) (Mortimer et al. 1985; Rasmusson et al. 1995; Schofield et al. 1997; Nemetz et al. 1999; Guo et al. 2000; Lye and Shores 2000; Plassman et al. 2000). Likewise, TBI is an epidemiological risk factor for the development of sporadic Parkinson’s disease (PD) (Nayernouri 1985; Factor and Weiner 1991; Stern 1991; Ben-Shlomo 1997; Lees 1997; Goldman et al. 2006).
Pathologically, AD is characterized by Aβ-containing plaques and neurofibrillary tangles comprised of tau protein (Braak and Braak 1991; Selkoe 2001; Forman et al. 2004). To a lesser extent, both dystrophic neurites and Lewy bodies containing α-synuclein protein (α-syn) are also observed in AD. Lewy bodies and α-syn immunoreactivity are also hallmark pathological features of PD and other synucleinopathies such as dementia with Lewy bodies (DLB) and multi-system atrophy (MSA) (Smith et al. 2003; Norris et al. 2004). As with neurodegenerative diseases, protein accumulation is also a feature of TBI. Most notably, Aβ plaque formation and the accumulation of neurofilament proteins, tau and α-syn have been found in brain tissue of humans within hours to days following TBI (Grady et al. 1993; Roberts et al. 1994; Graham et al. 1995; Newell et al. 1999; Smith et al. 2003; Smith et al. 2003; Abrahamson et al. 2006). The mechanism underlying this rapid protein build-up after TBI remains unknown, as does its contribution to the later development of neurodegenerative disease.
Aβ peptide is generated via the trans-membrane cleavage of amyloid precursor protein (APP) by the β- and γ-secretases. More specifically, its anabolism is mediated by beta-site APP cleaving enzyme (BACE) and the catalytic component of β-secretase, presenilin-1 (PS1) (De Strooper et al. 1998; Vassar et al. 1999; Nunan and Small 2000; Selkoe and Wolfe 2000; Esler and Wolfe 2001). Mounting evidence suggests that this process may also occur within the axonal membrane compartment. Large accumulations of Aβ have been found in swollen axons after TBI in a pig model of head rotational acceleration (Smith et al. 1999; Chen et al. 2004), in rodent models of brain contusion (Iwata et al. 2002; Stone et al. 2002; Chen et al. 2004), and in humans (Roberts et al. 1994; Smith et al. 2003). Axonal accumulations of Aβ were frequently found near diffuse, extracellular AD-like Aβ plaques in both the pig and in humans at the earliest survival timepoints measured (3 days and 18 hours respectively). This suggests a potential link between axonal pathology and Aβ plaque formation. (Smith et al. 1999, 2003b). More recently, extensive co-accumulations of Aβ with APP, BACE, and PS-1 were identified at sites of axonal injury and disconnection after TBI in the pig (Chen et al. 2004). Thus, disruption of axonal transport after TBI may create an environment whereby large accumulations of APP are processed to form Aβ, potentially leading to subsequent neurodegneration. Indeed, other recent studies have demonstrated the intra-axonal generation of Aβ in both central and peripheral nerve axons (Kamal et al. 2000; Kamal et al. 2001). Similarly, in a transgenic mouse model of AD, interrupted axonal transport and axonal swelling was shown to promote Aβ generation (Stokin et al. 2005).
The other classic pathological findings in AD are neurofibrillary tangles (NFTs) and neuropil threads (Braak and Braak 1991; Selkoe 2001; Forman et al. 2004). These intracellular structures are found to contain abnormal forms of the microtubule associated protein tau. NFTs with similarly abnormal tau are found in the brains of patients with dementia pugilistica; a progressive dementing disorder resulting from repetitive head trauma (Schmidt et al. 2001). Following a single episode of TBI in humans, hyperphosphorolyated tau has been demonstrated in brain tissue as well as elevated levels of the protein in cerebrospinal fluid (Newman SJ 1995; Zemlan et al. 1999). Additionally, excessive tau protein accumulation has been found in swollen axons in a pig model of TBI (Smith et al. 1999).
α-syn is a small, highly soluble protein believed to play a role in synaptic maintenance (Norris et al. 2004). In the context of AD and the synucleinopathies, this protein is found as abnormal, highly insoluble, filamentous perikaryal aggregates (Trojanowski and Lee 2002; Forman et al. 2004). It appears that the α-syn found in disease states is pathologically altered due to ubiquitination, oxidation/nitration, phosphorylation and/or conformational modification (Giasson et al. 2000; Duda et al. 2002; Fujiwara et al. 2002). α-syn accumulation has also been demonstrated in neurons and axons following a single episode TBI in humans (Newell et al. 1999; Ikonomovic et al. 2004) as well as in patients with dementia pugilistica (Schmidt et al. 2001). Accumulation of nitrated and conformationally modified α-syn in axons has also recently been found after TBI in transgenic mice (Uryu et al. 2003).
Here, we examined whether the findings in animal TBI models of rapid axonal accumulation of proteins found in neurodegenerative diseases also occurs in human TBI. In particluar, we evaluated protein accumulation similar to that seen in AD and the synucleinopathies, including the accumulation of NF, APP, BACE, PS-1, Aβ, tau, and α-syn.
Materials and methods
This study was approved by the Ethics Committee of the Southern General Hospital, South Glasgow University Hospitals NHS Trust, UK.
Case Material and Preparation
Brain tissue from 18 cases following a single incident of fatal head injury was secured after full diagnostic autopsy using standardized techniques (Adams et al. 1980) by the Department of Neuropathology, Southern General Hospital, Glasgow, UK. Superficial and deep grey and white matter from the frontal lobe, temporal lobe, and brainstem was examined; however, the specific location of the tissue was unknown. None of the cases investigated had a prior history of TBI or other neurodegenerative disease. The mean +/- standard deviation age of TBI cases was 45.7 +/- 24.0 years. The survival time from TBI ranged from 4h to 5w and the post-mortem delay time was 50.2 +/- 33.6 hours.
The cause of injury was a fall in 8, a road traffic accident in 7 and assault in 3. A skull fracture was present in 14, contusions in 17 and there was an intracranial haematoma in 9. Diffuse axonal injury (Adams et al 1989) was identified in 11 (grade 3 in 4 cases; grade 2 in 2 and grade 1 in 5). Hypoxic damage was present in 15 (Graham et al. 1989) and graded as severe in 5, moderately severe in 3 and mild in 7. Brain swelling was present in 10 - unilateral in 5 and bilateral in 5, and there was internal herniation in 12 (Adams and Graham 1976). The cause of death was raised intracranial pressure in 11, pneumonia in 5, multiple injuries in 1 and systemic hypoxia in 1.
The brain of each case was collected and fixed in 10% neutral buffered formalin, then cut into slices 10mm thick and processed for paraffin embedding. Serial sections of 6 microns were cut on a Leitz rotary microtome and mounted on poly-L-lysine-coated slides for histological study.
Controls
Tissue was also secured from 6 control cases from the same institution. The mean +/- standard deviation age of control cases was 37.8 +/- 22.1 years and post-mortem delay time was 59.2 +/- 41.3 hours. These cases had no prior history of head injury or had any evidence of structural brain damage due to pre-existing disease or injury; the cause of death in 3 was septicemia and sudden unexpected death in epilepsy (Nashef 1997; Black and Graham 2002) in the remaining 3 cases.
Immunohistochemistry
Immunohistochemistry (IHC) was performed on serial paraffin-embedded sections with investigators blinded to the cases’ clinical history. Using a well-characterized panel of antibodies, we evaluated one antibody per slide for each case, resulting in the evaluation of a total of 15-20 slides per case. We chose to use several antibodies that targeted different binding sites of each protein of interest in order to provide a more comprehensive picture of the proteins present. Single-labeled IHC was carried out using the primary antibodies listed in Table 1, followed by incubation with the appropriate secondary antibody and the ABC kit (Vector Laboratories, Inc, Burlingame, CA). Visualization was achieved using DAB (Vector Laboratories, Inc, Burlingame, CA) and counterstaining with hematoxylin (Uryu et al. 2003). To evaluate the co-accumulation of proteins, we used double- or triple-labeled fluorescent immunohistochemistry (FIHC) as described elsewhere (Uryu et al. 2003; Chen et al. 2004). Briefly, tissue was incubated with a combination of primary antibodies (Table 1) followed by the appropriate fluorescent-conjugated secondary antibody. Omission of the primary antibody or application of control serum instead of the primary antibody was performed on selected sections of tissue to provide a negative control. Paraffin-embedded sections from pathologically confirmed human AD and Parkinson disease brain tissue served as positive control for tau, Aβ and α-syn staining.
Table 1.
Summary of antibodies used for immunohistochemical analysis.
| Protein | Antibody | Host | Recognition site | Dilution | Provider |
|---|---|---|---|---|---|
| Tau | 17026 | rabbit | Pan Tau | 1:10k | CNDR |
| PHF-1 | mouse | pS396/pS404 | 1:1000 | Davies* | |
| PHF-6 | mouse | p+ T231 | 1:1000 | CNDR | |
| α-Syn | syn202 | mouse | Pan synuclein | 1:20,000 | CNDR |
| syn303 | mouse | conformational a-syn | 1:5000 | CNDR | |
| NF | NF-L | rabbit | NF-L | 1:5000 | CNDR |
| RMO44 | mouse | Rod domains of NF-M | 1:500 | CNDR | |
| RMO217 | mouse | P+ NF-H side arm, C-terminus | 1:5 | CNDR | |
| APP | LN39 | mouse | APP | 1:50 | CNDR |
| Karen | donkey | APP/N-terminal | 1:800 | Greenberg** | |
| Aβ | BC05 | mouse | Aβ 1-42/43 | 1:10,000 | Suzuki*** |
| BAN27 | mouse | Aβ 1-40 | 1:10,000 | Suzuki | |
| 13335 | rabbit | Aβ 1-42 | 1:1000 | CNDR | |
| NAB288 | mouse | pan Aβ | 1:20,000 | CNDR | |
| Amy117 | mouse | Amyloid– 100kd protein present | 1:20,000 | CNDR | |
| BACE | BACE | rabbit | BACE | 1:500 | Alpha Diagnostics |
| BACE | rabbit | BACE/N-terminal | 1:1000 | CNDR | |
| PS1 | PS1 | goat | N-terminal | 1:100 | Chemicon |
CDNR = Center for Neurodegenerative Research (University of Pennsylvania), NF = neurofilament, APP = amyloid precursor protein, Aβ = amyloid beta, α-syn = alpha synuclein, BACE = beta amyloid cleaving enzyme, PS1 = presenilin-1.
Antibody courtesy of Dr. Peter Davies: Albert Einstein College of Medicine, New York, USA.
Antibody courtesy of Dr. B. Greenberg: Cephalon Inc., Frazer, Pennsylvania, USA.
Antibody courtesy of Dr. N. Suzuki: Takeda Pharmaceuticals North America Inc., Deerfiel, Illinois, USA.
Semiquantitative Analysis
Examination of tissue was conducted by two individuals who were both blind to the clinical circumstances of the cases. Determination of the frequency of pathological profiles containing neurofilament (NF), APP, BACE, PS1, Aβ, tau, or α-syn proteins was achieved by reviewing microscopic sections stained with specific antibodies recognizing the respective molecules. We followed the pathological diagnostic criteria for AD as described previously by Mirra et al, 1993 (Mirra et al. 1993). This is an observational study where sections were examined at 100x magnification and profiles were ranked semi-quantitatively as no occurrence (-), low occurrence (+), moderate occurrence (++), or frequent occurrence (+++) in a 100μm2 field. The staining results for each protein of interest were in complete agreement, regardless of antibody recognition site; either all were positive for the protein or all were negative. Moreover, the number of profiles observed for each antibody was consistent across the tissue sections observed. Therefore, the findings presented in Table 2 are based on the consideration of all slides stained for a particular protein of interest.
Table 2.
Summary of immunohistochemistry findings following TBI in humans.
| Case | Age / sex | Survival | NF | APP | BACE | PS1 | AMY 117 | A-β (1-42) | α-syn | Tau |
|---|---|---|---|---|---|---|---|---|---|---|
| A | 56f | 4h | - | +++ | ++ | + | - | - | +++ | - |
| B | 47m | 12h | - | - | - | - | - | - | - | - |
| C | 51f | 12h | - | + | + | - | + | + | + | - |
| D | 5m | 13h | - | + | + | - | - | - | + | - |
| E | 57m | 15h | - | ++ | + | + | + | + | + | - |
| F | 75m | 16h | - | ++ | + | + | - | - | + | - |
| G | 28m | 18h | - | + | + | + | + | + | + | - |
| H | 60f | 24h | - | + | + | + | + | + | + | - |
| I | 65m | 27h | - | +++ | ++ | + | + | + | ++ | - |
| J | 47m | 36h | - | + | - | + | + | + | + | + |
| K | 16m | 7d | - | - | - | ++ | ++ | ++ | + | - |
| L | 15f | 8d | - | +++ | +++ | +++ | +++ | +++ | +++ | - |
| M | 79f | 8d | - | - | - | - | - | - | - | - |
| N | 31m | 27d | + | + | - | - | + | + | - | - |
| O | 18m | 4w | ++ | ++ | + | + | ++ | ++ | - | - |
| P | 23m | 4w | ++ | ++ | ++ | ++ | ++ | ++ | - | - |
| Q | 73f | 4w | - | +++ | +++ | ++ | +++ | +++ | +++ | ++ |
| R | 76m | 5w | + | + | - | - | + | + | - | - |
| S | 15m | Control | + | +++ | - | - | - | - | - | - |
| T | 26m | Control | - | - | - | - | - | - | - | - |
| U | 54m | Control | - | - | - | - | - | - | - | - |
| V | 21f | Control | - | - | - | - | - | - | - | - |
| W | 73f | Control | - | - | - | - | - | - | - | - |
| X | 38f | Control | - | - | - | - | - | - | - | - |
Cases S-X are control subjects. (-): no occurrence of profiles in a microscopic field (~100μm2); (+): low occurrence of profiles in a microscopic field; (++): moderate occurrence of profiles in a microscopic field; (+++): frequent occurrence of profiles in a microscopic field. NF = neurofilament, APP = amyloid precursor protein, BACE = beta amyloid cleaving enzyme, PS1 = presenilin-1, AMY117 = amyloid 100kd protein, Aβ = amyloid beta, α-syn = alpha synuclein,.
Results
Recognition of axonal injury
Axonal pathology, characterized by axonal bulbs and/or swellings, was identified using antibodies to NF protein and APP.
Axonal bulbs had the appearance of discrete spherical profiles surrounded by a halo and were morphologically distinct from the varicose swellings of the axon. The bulbs ranged in size from 5-100 μm. Only one subject in the uninjured group exhibited positive staining for NF and APP, while 15 of the 18 injured cases had extensive immunoreactivity to APP in the white matter. However, only about one-third of these cases showed positive staining for NF, an additional marker of axonal pathology (Table 2.) (Fig. 1a-d).
Figure 1.
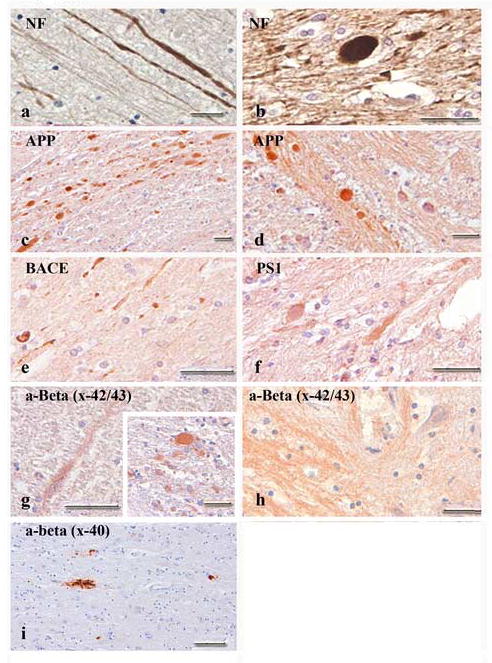
Bright-field photomicrographs showing pathological protein accumulation associated with axonal pathology in humans. Neurofilament and amyloid precursor proteins (APP) were both found in axonal swellings and bulbs following TBI (a-d). Enzymatic agents of APP cleavage (BACE and PS1) were also present, although to a lesser extent (e, f). Antibodies specific for Aβ revealed axonal swellings (g) and bulbs (g, inset) positive for Aβx-42/43 throughout the white matter of subjects and within the brainstem of several young cases (h). A limited number of swellings and bulbs stained positive for Aβx-40. Infrequent amyloid deposits in subjects over 70 years of age were seen with Aβx-40 (i). Scale bars = 50 μm.
Accumulation and Co-localization of APP, Aβ, BACE and PS1
In the control group, only one subject showed APP accumulation; however, no additional protein deposits were noted. As mentioned above, almost all cases subject to TBI displayed evidence of APP accumulation within axons. In addition, BACE and PS1 staining was observed within axonal bulbs in a majority of these cases. With the exception of 2 cases, the number of profiles appeared to increase in those cases survived between 8d and 4w (Fig. 1e,f). AβX-42/43 immunoreactivity within axons was noted in 13 of the total 18 injured cases (Fig 1g). Aβ IHC revealed axonal bulbs positive for AβX-42 but negative for AβX-40. Interestingly, brain tissue from four young cases (Cases K, L, N, O) indicated rather strong axonal Aβ staining in their brainstem (Fig. 1h). When detected, Aβ plaques were limited in number and restricted to cases aged 70 years or more (Fig. 1i).
Using double- or triple-label fluorescence IHC, co-localization of Aβ and its precursor protein, APP, as well as co-factors of APP processing, BACE and PS1, was examined. APP was observed to co-localize with Aβ (Fig. 2a-c) or BACE and PS1 (Fig. 2d-g) within axonal bulbs following injury. Aβ was also noted to co-localize with BACE and PS1 in axonal bulbs (Fig. 2h-k).
Figure 2.
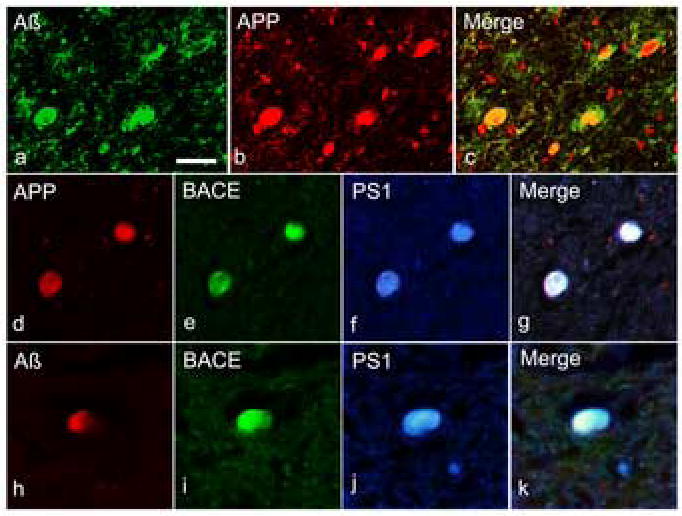
Multiple immunofluorescent staining showing co-accumulation of APP, BACE, PS1, and Aβ in axons following TBI. Double-labelling revealed co-accumulation of APP and Aβ1-42 in multiple axon bulbs (a-c). Further immunohistochemical staining showed co-accumulation of BACE and PS-1 with APP (d-g) and Aβ1-42 (h-k).Scale bars = 50 μm.
Tau and α-syn
None of the tissue from uninjured control cases stained positive for tau or α-syn proteins; and few of the injured cases stained positive for tau. However, a majority of the tissue from injured cases showed positive staining for α-syn. PHF-1 positive phosphorylated tau was noted in a small number of swollen axons and clusters of neuronal cell bodies in the cerebral cortex (Fig. 3a,b). In the same area, reactive astrocytes occasionally showed tau positive staining (Fig. 3c). Neuronal tau tended to more intensely stained than glial tau. α-syn protein was observed mostly in swollen, undulating axons as well as in bulbs, with little reactivity in the neuronal soma (Fig. 3d,e).
Figure 3.
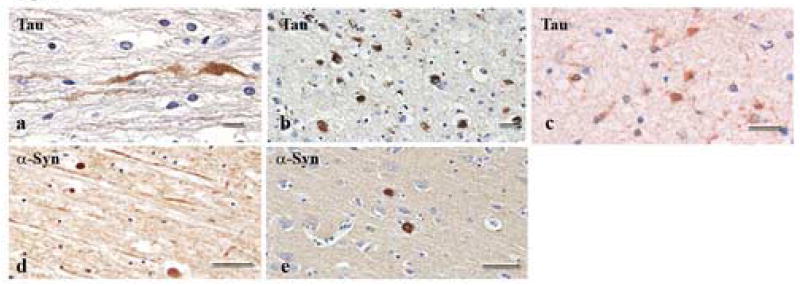
Bright-field photomicrographs showing accumulation of tau and α-syn proteins in axonal swellings and bulbs. Of the 18 TBI cases examined, only 2 stained for tau. Tau staining was observed in axonal swellings (a) and bulbs (b) following injury, and, to a lesser extent, in glial cells (c). α-syn was also present within axonal swellings (d) and bulbs (e) in a majority of the injured subjects. Scale bars = 50 um.
Summary of Principal Findings
Cases with a history of traumatic brain injury demonstrated axonal pathology in the majority of cases as shown by increased immunoreactivity to APP or NF. This compares to controls where just one case demonstrated minimal immunoreactivity. Immunoreactivity to BACE, PS-1 and Aβ were all found to be increased in TBI cases versus controls.
Co-immunoreacticity between BACE, PS-1 and APP was found within axonal bulbs. In addition, within these bulbs, Aβ was also found co-accumulating with APP, BACE and PS-1.
α-syn immunoreactivity was found in two thirds of the cases, predominantly in association with axonal pathology. Only 2 cases were positive for tau which was found in both neurons and nearby reactive astrocytes. No controls had immunoreactivity for either α-syn or tau.
It is important to note that while protein accumulation was seen in many of the injured cases we evaluated, the number and location of samples that can be evaluated in an autopsy study is limited. As such, some cases graded as negative may have actually had pathologic changes in areas not examined.
Discussion
There is increasing evidence that the brains of TBI cases display many of the same pathologies associated with several neurodegenerative diseases. TBI can also induce the rapid accumulation of several proteins that compose similar pathologic aggregates found in neurodegenerative diseases such as AD, of which the most widely studied include NF proteins, APP, Aβ, and α-syn. Here, the accumulation of multiple proteins and their anabolic agents implicated in neurodegenerative diseases were found within damaged axons up to 5 weeks after TBI in humans. This evidence suggests that damaged axons provide a key source of proteins that may play a role in neurodegenerative processes.
Traumatic axonal injury is a common and important pathology resulting from TBI in humans (Adams et al. 1989; Adams et al. 1991; Smith DH 2000) and is frequently observed after motor vehicle accidents, falls, and assaults (Adams et al. 1982; Pilz 1983; Adams et al. 1989; Gennarelli 1993). In DAI, the axoskeleton can be severely damaged, resulting in impaired axonal transport, build-up of transported proteins, axonal swellings and bulbs (Povlishock and Becker 1985; Maxwell et al. 1997).
Axonal pathology in humans has been identified by the accumulation of NF in damaged axons from 6 hours onwards after head trauma in both animals and humans (Grady et al. 1993; Christman et al. 1994). In cases of death very shortly following injury, accumulation of NF is very limited, if present at all, in our experience. This is likely due to the fact that NF is slowly transported and accumulation is insufficient for detection in this early phase. In addition, we also note that in humans, NF immunoreactivity has a tendency to increase as survival time post-trauma elapses. This explains why in this sample we see axonal pathology as identified by NF accumulation in both fewer cases and specifically those with a longer survival period. As such, immunohistochemical detection of the fast transport APP has become a standard method of diagnosing axonal injury in human brain tissue, where axonal swellings can be identified within one hour of injury (Adams et al. 1980; Adams et al. 1989; Otsuka et al. 1991; Sherriff et al. 1994; Lambri et al. 2001; Gorrie et al. 2002; Reichard et al. 2003). The presence of acute axonal pathology is confirmed here. However, we note that within our heterogeneous population, there are cases who ultimately died secondary to raised intracranial pressure (RICP) and the vascular complications of internal herniation. Therefore, in many cases of fatal TBI it is possible to identify axonal pathology that is both traumatic and non-traumatic (infarction, related to hematomas and contusions) in origin. Axonal swellings are also seen in many other non-traumatic conditions and as part of the aging process. As so, it is appropriate to suggest that such pressure / vascular complications may have independently contributed to the neuropathological findings described (Geddes et al. 2000). Our findings also demonstrate that APP accumulation may be far more than a simple marker of axonal pathology in humans; it also may be the primary substrate for posttraumatic Aβ formation. In particular, in damaged axons, we observed APP accumulation along with its catalytic enzymes, BACE and PS1.To a lesser extent, other AD-associated proteins, tau and α-syn, were observed. Thus, extensive axonal damage may serve as a key reservoir of proteins implicated in neuropathologic processes. Lysis or release of accumulate proteins from damaged axons may lead to plaque formation or toxicity.
The present histopathologic findings in human TBI are consistent with findings in previous animal studies. Extensive co-accumulation of APP with Aβ has been found in swollen axons in a swine model of diffuse axonal injury induced by rotational acceleration (Smith et al. 1999). More recently, in the same model, axonal Aβ has been found to co-localize with its precursor protein, APP, along with the catalytic enzymes BACE and PS1 necessary to cleave Aβ from APP (Chen et al. 2004).
A number of studies suggest that axons and their terminals may be a critical site of Aβ production. Firstly, Aβ plaques have been shown to develop in close relationship with axonal terminals in AD brains (Van Hoesen and Hyman 1990; Kamal et al. 2000; Schonheit et al. 2004). It has also been shown that APP is transported along axons by direct binding to kinesin in mouse sciatic nerves (Kamal et al. 2000). This study demonstrates that APP operates as a kinesin-1 receptor and mediates the transport of its cleavage enzymes PS-1 and β-secretease. This in turn permits the intra-axonal generation of Aβ. However, this mechanism remains controversial and is contradicted by other studies (Lazarov et al. 2005). If such were the case, it is reasonable to assume that disruption of axonal transport may lead to abnormal Aβ generation intraxonally and potentially deposition. Indeed, Stokin et al, using an APP transgenic mouse model, demonstrated that both Aβ levels and deposition after disruption of axonal transport (Stokin et al. 2005). In addition, inter-axonal Aβ deposits formed in APP transgenic mouse brain could be reduced by anti-Aβ antibody therapy (Brendza et al. 2005). These studies suggest that axons can play an important role in APP processing and Aβ formation both intraxonally and in the generation of extracellular Aβ plaques. Thus, the extensive axonal damage found in TBI may provide a unique environment in which unusually concentrated co-accumulation of APP, BACE, and PS1 occurs, providing the tools to produce Aβ. In turn, this intra-axonal process may play a critical role in rapid Aβ plaque formation.
Accumulation of both tau and α-syn proteins were also found in the cases we examined; although tau was observed in fewer cases than α-syn. Based on the antibodies used, tau protein appeared to be abnormally phosphorylated and the α-syn protein was conformationally changed. These findings are consistent with the pathologies that are observed in AD brain lesions and other diseases characterized by α-syn accumulation (Fujiwara et al. 2002; Norris et al. 2004). Thus, it seems as though TBI may initiate similar processes leading to the pathological modification of these proteins that occurs in neurodegenerative disease. It is also interesting to note that both tau and α-syn are observed in the grey matter in AD brains. (Forman et al. 2004); yet here, we observed both proteins within the axons and axonal bulbs. Additionally, tau protein appeared in far fewer injured cases than did α-syn. If damaged axons provide a source of this protein, the pathological accumulation may occur over a more protracted time course than was observed here.
The present study illustrates the potential contribution of axonal injury to creating pathological protein accumulation in human brain within 4 hours – 5 weeks following TBI. Thus, it is possible that axonal injury associated with pathological protein accumulation may contribute to AD-related pathogenesis. A further understanding of the mechanistic aspects of the long-term pathophysiology of AD-related proteins in the injured brain may aid the development of interventions to halt possible TBI induced neurodegeneration.
Acknowledgments
We thank the families of patients whose generosity made this research possible. This work was supported by grants from the National Institutes of Health including the National Institute on Aging (AG10124 and AG11542) and the National Institute of Neurological Disorders and Stroke (NS38104, AG12527 and NS08803). V.M.Y.L. is the John H. Ware Third Chair of Alzheimer’s disease research; and J.Q.T. is the William Maul Measey-Truman G. Schnabel, Jr., Professor of Geriatric Medicine and Gerontology.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abrahamson EE, Ikonomovic MD, Ciallella JR, Hope CE, Paljug WR, Isanski BA, Flood DG, Clark RS, DeKosky ST. Caspase inhibition therapy abolishes brain trauma-induced increases in Abeta peptide: implications for clinical outcome. Exp Neurol. 2006;197(2):437–50. doi: 10.1016/j.expneurol.2005.10.011. [DOI] [PubMed] [Google Scholar]
- Adams JH, Doyle D, Ford I, Gennarelli TA, Graham DI, McLellan DR. Diffuse axonal injury in head injury: definition, diagnosis and grading. Histopathology. 1989;15(1):49–59. doi: 10.1111/j.1365-2559.1989.tb03040.x. [DOI] [PubMed] [Google Scholar]
- Adams JH, Graham DI. The relationship between ventricular fluid pressure and the neuropathology of raised intracranial pressure. Neuropath Appl Neurobiol. 1976;2:323–332. [Google Scholar]
- Adams JH, Graham DI, Gennarelli TA, Maxwell WL. Diffuse axonal injury in non-missile head injury. J Neurol Neurosurg Psychiatry. 1991;54(6):481–3. doi: 10.1136/jnnp.54.6.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams JH, Graham DI, Murray LS, Scott G. Diffuse axonal injury due to nonmissile head injury in humans: an analysis of 45 cases. Ann Neurol. 1982;12(6):557–63. doi: 10.1002/ana.410120610. [DOI] [PubMed] [Google Scholar]
- Adams JH, Graham DI, Scott G, Parker LS, Doyle D. Brain damage in fatal non-missile head injury. J Clin Pathol. 1980;33(12):1132–45. doi: 10.1136/jcp.33.12.1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Shlomo Y. The epidemiology of Parkinson’s disease. Baillieres Clin Neurol. 1997;6(1):55–68. [PubMed] [Google Scholar]
- Black M, Graham DI. Sudden unexplained death in adults caused by intracranial pathology. J Clin Pathol. 2002;55(1):44–50. doi: 10.1136/jcp.55.1.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol (Berl) 1991;82(4):239–59. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- Brendza RP, Bacskai BJ, Cirrito JR, Simmons KA, Skoch JM, Klunk WE, Mathis CA, Bales KR, Paul SM, Hyman BT, Holtzman DM. Anti-Abeta antibody treatment promotes the rapid recovery of amyloid-associated neuritic dystrophy in PDAPP transgenic mice. J Clin Invest. 2005;115(2):428–33. doi: 10.1172/JCI23269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen XH, Siman R, Iwata A, Meaney DF, Trojanowski JQ, Smith DH. Long-term accumulation of amyloid-beta, beta-secretase, presenilin-1, and caspase-3 in damaged axons following brain trauma. Am J Pathol. 2004;165(2):357–71. doi: 10.1016/s0002-9440(10)63303-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christman CW, Grady MS, Walker SA, Holloway KL, Povlishock JT. Ultrastructural studies of diffuse axonal injury in humans. J Neurotrauma. 1994;11(2):173–86. doi: 10.1089/neu.1994.11.173. [DOI] [PubMed] [Google Scholar]
- De Strooper B, Saftig P, Craessaerts K, Vanderstichele H, Guhde G, Annaert W, Von Figura K, Van Leuven F. Deficiency of presenilin-1 inhibits the normal cleavage of amyloid precursor protein. Nature. 1998;391(6665):387–90. doi: 10.1038/34910. [DOI] [PubMed] [Google Scholar]
- Duda JE, Giasson BI, Mabon ME, Lee VM, Trojanowski JQ. Novel antibodies to synuclein show abundant striatal pathology in Lewy body diseases. Ann Neurol. 2002;52(2):205–10. doi: 10.1002/ana.10279. [DOI] [PubMed] [Google Scholar]
- Esler WP, Wolfe MS. A portrait of Alzheimer secretases--new features and familiar faces. Science. 2001;293(5534):1449–54. doi: 10.1126/science.1064638. [DOI] [PubMed] [Google Scholar]
- Factor SA, Weiner WJ. Prior history of head trauma in Parkinson’s disease. Mov Disord. 1991;6(3):225–9. doi: 10.1002/mds.870060306. [DOI] [PubMed] [Google Scholar]
- Forman MS, Trojanowski JQ, Lee VM. Neurodegenerative diseases: a decade of discoveries paves the way for therapeutic breakthroughs. Nat Med. 2004;10(10):1055–63. doi: 10.1038/nm1113. [DOI] [PubMed] [Google Scholar]
- Fujiwara H, Hasegawa M, Dohmae N, Kawashima A, Masliah E, Goldberg MS, Shen J, Takio K, Iwatsubo T. alpha-Synuclein is phosphorylated in synucleinopathy lesions. Nat Cell Biol. 2002;4(2):160–4. doi: 10.1038/ncb748. [DOI] [PubMed] [Google Scholar]
- Geddes JF, Whitwell HL, Graham DI. Traumatic axonal injury: practical issues for diagnosis in medicolegal cases. Neuropathol Appl Neurobiol. 2000;26(2):105–16. doi: 10.1046/j.1365-2990.2000.026002105.x. [DOI] [PubMed] [Google Scholar]
- Gennarelli TA. Mechanisms of brain injury. J Emerg Med. 1993;11(Suppl 1):5–11. [PubMed] [Google Scholar]
- Giasson BI, Duda JE, Murray IV, Chen Q, Souza JM, Hurtig HI, Ischiropoulos H, Trojanowski JQ, Lee VM. Oxidative damage linked to neurodegeneration by selective alpha-synuclein nitration in synucleinopathy lesions. Science. 2000;290(5493):985–9. doi: 10.1126/science.290.5493.985. [DOI] [PubMed] [Google Scholar]
- Goldman SM, Tanner CM, Oakes D, Bhudhikanok GS, Gupta A, Langston JW. Head injury and Parkinson’s disease risk in twins. Ann Neurol. 2006;60(1):65–72. doi: 10.1002/ana.20882. [DOI] [PubMed] [Google Scholar]
- Gorrie C, Oakes S, Duflou J, Blumbergs P, Waite PM. Axonal injury in children after motor vehicle crashes: extent, distribution, and size of axonal swellings using beta-APP immunohistochemistry. J Neurotrauma. 2002;19(10):1171–82. doi: 10.1089/08977150260337976. [DOI] [PubMed] [Google Scholar]
- Grady MS, McLaughlin MR, Christman CW, Valadka AB, Fligner CL, Povlishock JT. The use of antibodies targeted against the neurofilament subunits for the detection of diffuse axonal injury in humans. J Neuropathol Exp Neurol. 1993;52(2):143–52. doi: 10.1097/00005072-199303000-00007. [DOI] [PubMed] [Google Scholar]
- Graham DI, Ford I, Adams JH, Doyle D, Teasdale GM, Lawrence AE, McLellan DR. Ischaemic brain damage is still common in fatal non-missile head injury. J Neurol Neurosurg Psychiatry. 1989;52(3):346–50. doi: 10.1136/jnnp.52.3.346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham DI, Gentleman SM, Lynch A, Roberts GW. Distribution of beta-amyloid protein in the brain following severe head injury. Neuropathol Appl Neurobiol. 1995;21(1):27–34. doi: 10.1111/j.1365-2990.1995.tb01025.x. [DOI] [PubMed] [Google Scholar]
- Guo Z, Cupples LA, Kurz A, Auerbach SH, Volicer L, Chui H, Green RC, Sadovnick AD, Duara R, DeCarli C, Johnson K, Go RC, Growdon JH, Haines JL, Kukull WA, Farrer LA. Head injury and the risk of AD in the MIRAGE study. Neurology. 2000;54(6):1316–23. doi: 10.1212/wnl.54.6.1316. [DOI] [PubMed] [Google Scholar]
- Ikonomovic MD, Uryu K, Abrahamson EE, Ciallella JR, Trojanowski JQ, Lee VM, Clark RS, Marion DW, Wisniewski SR, DeKosky ST. Alzheimer’s pathology in human temporal cortex surgically excised after severe brain injury. Exp Neurol. 2004;190(1):192–203. doi: 10.1016/j.expneurol.2004.06.011. [DOI] [PubMed] [Google Scholar]
- Iwata A, Chen XH, McIntosh TK, Browne KD, Smith DH. Long-term accumulation of amyloid-beta in axons following brain trauma without persistent upregulation of amyloid precursor protein genes. J Neuropathol Exp Neurol. 2002;61(12):1056–68. doi: 10.1093/jnen/61.12.1056. [DOI] [PubMed] [Google Scholar]
- Kamal A, Almenar-Queralt A, LeBlanc JF, Roberts EA, Goldstein LS. Kinesin-mediated axonal transport of a membrane compartment containing beta-secretase and presenilin-1 requires APP. Nature. 2001;414(6864):643–8. doi: 10.1038/414643a. [DOI] [PubMed] [Google Scholar]
- Kamal A, Stokin GB, Yang Z, Xia CH, Goldstein LS. Axonal transport of amyloid precursor protein is mediated by direct binding to the kinesin light chain subunit of kinesin-I. Neuron. 2000;28(2):449–59. doi: 10.1016/s0896-6273(00)00124-0. [DOI] [PubMed] [Google Scholar]
- Lambri M, Djurovic V, Kibble M, Cairns N, Al-Sarraj S. Specificity and sensitivity of betaAPP in head injury. Clin Neuropathol. 2001;20(6):263–71. [PubMed] [Google Scholar]
- Lazarov O, Morfini GA, Lee EB, Farah MH, Szodorai A, DeBoer SR, Koliatsos VE, Kins S, Lee VM, Wong PC, Price DL, Brady ST, Sisodia SS. Axonal transport, amyloid precursor protein, kinesin-1, and the processing apparatus: revisited. J Neurosci. 2005;25(9):2386–95. doi: 10.1523/JNEUROSCI.3089-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lees AJ. Trauma and Parkinson disease. Rev Neurol (Paris) 1997;153(10):541–6. [PubMed] [Google Scholar]
- Lye TC, Shores EA. Traumatic brain injury as a risk factor for Alzheimer’s disease: a review. Neuropsychol Rev. 2000;10(2):115–29. doi: 10.1023/a:1009068804787. [DOI] [PubMed] [Google Scholar]
- Maxwell WL, Povlishock JT, Graham DL. A mechanistic analysis of nondisruptive axonal injury: a review. J Neurotrauma. 1997;14(7):419–40. doi: 10.1089/neu.1997.14.419. [DOI] [PubMed] [Google Scholar]
- Mirra SS, Hart MN, Terry RD. Making the diagnosis of Alzheimer’s disease. A primer for practicing pathologists. Arch Pathol Lab Med. 1993;117(2):132–44. [PubMed] [Google Scholar]
- Mortimer JA, French LR, Hutton JT, Schuman LM. Head injury as a risk factor for Alzheimer’s disease. Neurology. 1985;35(2):264–7. doi: 10.1212/wnl.35.2.264. [DOI] [PubMed] [Google Scholar]
- Nashef L. Unexpected death in epilepsy: terminology and definitions. Epilepsia. 1997;38(Suppl.11):S6–8. doi: 10.1111/j.1528-1157.1997.tb06130.x. [DOI] [PubMed] [Google Scholar]
- Nayernouri T. Posttraumatic parkinsonism. Surg Neurol. 1985;24(3):263–4. doi: 10.1016/0090-3019(85)90035-7. [DOI] [PubMed] [Google Scholar]
- Nemetz PN, Leibson C, Naessens JM, Beard M, Kokmen E, Annegers JF, Kurland LT. Traumatic brain injury and time to onset of Alzheimer’s disease: a population-based study. Am J Epidemiol. 1999;149(1):32–40. doi: 10.1093/oxfordjournals.aje.a009724. [DOI] [PubMed] [Google Scholar]
- Newell KL, Boyer P, Gomez-Tortosa E, Hobbs W, Hedley-Whyte ET, Vonsattel JP, Hyman BT. Alpha-synuclein immunoreactivity is present in axonal swellings in neuroaxonal dystrophy and acute traumatic brain injury. J Neuropathol Exp Neurol. 1999;58(12):1263–8. doi: 10.1097/00005072-199912000-00007. [DOI] [PubMed] [Google Scholar]
- Newman SJ, G S, Graham DI, Brown F, Roberts GW. Tissue distribution and cellular localisation of hyperphosphorylated tau in human head injury and agematched controls. Chichester: Wiley; 1995. [Google Scholar]
- Norris EH, Giasson BI, Lee VM. Alpha-synuclein: normal function and role in neurodegenerative diseases. Curr Top Dev Biol. 2004;60:17–54. doi: 10.1016/S0070-2153(04)60002-0. [DOI] [PubMed] [Google Scholar]
- Nunan J, Small DH. Regulation of APP cleavage by alpha-, beta- and gamma-secretases. FEBS Lett. 2000;483(1):6–10. doi: 10.1016/s0014-5793(00)02076-7. [DOI] [PubMed] [Google Scholar]
- Otsuka N, Tomonaga M, Ikeda K. Rapid appearance of beta-amyloid precursor protein immunoreactivity in damaged axons and reactive glial cells in rat brain following needle stab injury. Brain Res. 1991;568(1-2):335–8. doi: 10.1016/0006-8993(91)91422-w. [DOI] [PubMed] [Google Scholar]
- Pilz P. Axonal injury in head injury. Acta Neurochir Suppl (Wien) 1983;32:119–23. doi: 10.1007/978-3-7091-4147-2_17. [DOI] [PubMed] [Google Scholar]
- Plassman BL, Havlik RJ, Steffens DC, Helms MJ, Newman TN, Drosdick D, Phillips C, Gau BA, Welsh-Bohmer KA, Burke JR, Guralnik JM, Breitner JC. Documented head injury in early adulthood and risk of Alzheimer’s disease and other dementias. Neurology. 2000;55(8):1158–66. doi: 10.1212/wnl.55.8.1158. [DOI] [PubMed] [Google Scholar]
- Povlishock JT, Becker DP. Fate of reactive axonal swellings induced by head injury. Lab Invest. 1985;52(5):540–52. [PubMed] [Google Scholar]
- Rasmusson DX, Brandt J, Martin DB, Folstein MF. Head injury as a risk factor in Alzheimer’s disease. Brain Inj. 1995;9(3):213–9. doi: 10.3109/02699059509008194. [DOI] [PubMed] [Google Scholar]
- Reichard RR, White CL, 3rd, Hladik CL, Dolinak D. Beta-amyloid precursor protein staining of nonaccidental central nervous system injury in pediatric autopsies. J Neurotrauma. 2003;20(4):347–55. doi: 10.1089/089771503765172309. [DOI] [PubMed] [Google Scholar]
- Roberts GW, Gentleman SM, Lynch A, Murray L, Landon M, Graham DI. Beta amyloid protein deposition in the brain after severe head injury: implications for the pathogenesis of Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 1994;57(4):419–25. doi: 10.1136/jnnp.57.4.419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt ML, Zhukareva V, Newell KL, Lee VM, Trojanowski JQ. Tau isoform profile and phosphorylation state in dementia pugilistica recapitulate Alzheimer’s disease. Acta Neuropathol (Berl) 2001;101(5):518–24. doi: 10.1007/s004010000330. [DOI] [PubMed] [Google Scholar]
- Schofield PW, Tang M, Marder K, Bell K, Dooneief G, Chun M, Sano M, Stern Y, Mayeux R. Alzheimer’s disease after remote head injury: an incidence study. J Neurol Neurosurg Psychiatry. 1997;62(2):119–24. doi: 10.1136/jnnp.62.2.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schonheit B, Zarski R, Ohm TG. Spatial and temporal relationships between plaques and tangles in Alzheimer-pathology. Neurobiol Aging. 2004;25(6):697–711. doi: 10.1016/j.neurobiolaging.2003.09.009. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer’s disease: genes, proteins, and therapy. Physiol Rev. 2001;81(2):741–66. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ, Wolfe MS. In search of gamma-secretase: presenilin at the cutting edge. Proc Natl Acad Sci U S A. 2000;97(11):5690–2. doi: 10.1073/pnas.97.11.5690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherriff FE, Bridges LR, Sivaloganathan S. Early detection of axonal injury after human head trauma using immunocytochemistry for beta-amyloid precursor protein. Acta Neuropathol (Berl) 1994;87(1):55–62. doi: 10.1007/BF00386254. [DOI] [PubMed] [Google Scholar]
- Smith C, Graham DI, Murray LS, Nicoll JA. Tau immunohistochemistry in acute brain injury. Neuropathol Appl Neurobiol. 2003;29(5):496–502. doi: 10.1046/j.1365-2990.2003.00488.x. [DOI] [PubMed] [Google Scholar]
- Smith DH, Chen XH, Iwata A, Graham DI. Amyloid beta accumulation in axons after traumatic brain injury in humans. J Neurosurg. 2003;98(5):1072–7. doi: 10.3171/jns.2003.98.5.1072. [DOI] [PubMed] [Google Scholar]
- Smith DH, Chen XH, Nonaka M, Trojanowski JQ, Lee VM, Saatman KE, Leoni MJ, Xu BN, Wolf JA, Meaney DF. Accumulation of amyloid beta and tau and the formation of neurofilament inclusions following diffuse brain injury in the pig. J Neuropathol Exp Neurol. 1999;58(9):982–92. doi: 10.1097/00005072-199909000-00008. [DOI] [PubMed] [Google Scholar]
- Smith DH, M D. Axonal damage in traumatic brain injury. The neuroscientist. 2000;6:483–495. [Google Scholar]
- Smith DH, Uryu K, Saatman KE, Trojanowski JQ, McIntosh TK. Protein accumulation in traumatic brain injury. Neuromolecular Med. 2003;4(1-2):59–72. doi: 10.1385/NMM:4:1-2:59. [DOI] [PubMed] [Google Scholar]
- Stern MB. Head trauma as a risk factor for Parkinson’s disease. Mov Disord. 1991;6(2):95–7. doi: 10.1002/mds.870060202. [DOI] [PubMed] [Google Scholar]
- Stokin GB, Lillo C, Falzone TL, Brusch RG, Rockenstein E, Mount SL, Raman R, Davies P, Masliah E, Williams DS, Goldstein LS. Axonopathy and transport deficits early in the pathogenesis of Alzheimer’s disease. Science. 2005;307(5713):1282–8. doi: 10.1126/science.1105681. [DOI] [PubMed] [Google Scholar]
- Stone JR, Okonkwo DO, Singleton RH, Mutlu LK, Helm GA, Povlishock JT. Caspase-3-mediated cleavage of amyloid precursor protein and formation of amyloid Beta peptide in traumatic axonal injury. J Neurotrauma. 2002;19(5):601–14. doi: 10.1089/089771502753754073. [DOI] [PubMed] [Google Scholar]
- Trojanowski JQ, Lee VM. Parkinson’s disease and related synucleinopathies are a new class of nervous system amyloidoses. Neurotoxicology. 2002;23(4-5):457–60. doi: 10.1016/s0161-813x(02)00065-7. [DOI] [PubMed] [Google Scholar]
- Uryu K, Giasson BI, Longhi L, Martinez D, Murray I, Conte V, Nakamura M, Saatman K, Talbot K, Horiguchi T, McIntosh T, Lee VM, Trojanowski JQ. Age-dependent synuclein pathology following traumatic brain injury in mice. Exp Neurol. 2003;184(1):214–24. doi: 10.1016/s0014-4886(03)00245-0. [DOI] [PubMed] [Google Scholar]
- Van Hoesen GW, Hyman BT. Hippocampal formation: anatomy and the patterns of pathology in Alzheimer’s disease. Prog Brain Res. 1990;83:445–57. doi: 10.1016/s0079-6123(08)61268-6. [DOI] [PubMed] [Google Scholar]
- Vassar R, Bennett BD, Babu-Khan S, Kahn S, Mendiaz EA, Denis P, Teplow DB, Ross S, Amarante P, Loeloff R, Luo Y, Fisher S, Fuller J, Edenson S, Lile J, Jarosinski MA, Biere AL, Curran E, Burgess T, Louis JC, Collins F, Treanor J, Rogers G, Citron M. Beta-secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science. 1999;286(5440):735–41. doi: 10.1126/science.286.5440.735. [DOI] [PubMed] [Google Scholar]
- Zemlan FP, Rosenberg WS, Luebbe PA, Campbell TA, Dean GE, Weiner NE, Cohen JA, Rudick RA, Woo D. Quantification of axonal damage in traumatic brain injury: affinity purification and characterization of cerebrospinal fluid tau proteins. J Neurochem. 1999;72(2):741–50. doi: 10.1046/j.1471-4159.1999.0720741.x. [DOI] [PubMed] [Google Scholar]