

Abstract
Traumatic axonal injury (TAI) is the most common and important pathology of traumatic brain injury (TBI). However, little is known about potential indirect effects of TAI on dendrites. In this study, we used a well-established in vitro model of axonal stretch injury to investigate TAI-induced changes in dendrite morphology. Axons bridging two separated rat cortical neuron populations plated on a deformable substrate were used to create a zone of isolated stretch injury to axons. Following injury, we observed the formation of dendritic alterations or beading along the dendrite shaft. Dendritic beading formed within minutes after stretch then subsided over time. Pharmacological experiments revealed a sodium-dependent mechanism, while removing extracellular calcium exacerbated TAI's effect on dendrites. In addition, blocking ionotropic glutamate receptors with the N-methyl-D-aspartate (NMDA) receptor antagonist MK-801 prevented dendritic beading. These results demonstrate that axon mechanical injury directly affects dendrite morphology, highlighting an important bystander effect of TAI. The data also imply that TAI may alter dendrite structure and plasticity in vivo. An understanding of TAI's effect on dendrites is important since proper dendrite function is crucial for normal brain function and recovery after injury.
Keywords: Dendrite, Traumatic brain injury, Diffuse axonal injury, Cerebral cortex, In vitro
Introduction
Traumatic brain injury (TBI) is the leading cause of death and disability among people less than 45 years old. It afflicts up to two million people each year in the United States (US), and it is estimated that 2% of the US population lives with long-term TBI-related cognitive, behavioral and motor disability. TBI also may initiate insidious neurodegeneration that is associated with a greater risk of Alzheimer's disease (Rasmusson et al., 1995; Van Den Heuvel et al., 2007). Traumatic axonal injury (TAI), which results from damaged or dysfunctional axons throughout the white matter, is the most common pathology in TBI (Smith and Meaney, 2000), and is thought to contribute to the neurologic deficits found in most TBI survivors (Scheid et al., 2006; Kumar et al., 2009). However, how TAI affects cognitive outcome in humans is uncertain, particularly after mild or moderate TBI (Wallesch et al., 2001; Felmingham et al., 2004; Scheid et al., 2006), the most prevalent forms of TBI.
Dendrite diversity is a striking characteristic of the brain. In addition, dendrites can undergo rapid structural changes in response to a variety of stimuli (Cline, 2001; Jan and Jan, 2001), and this plasticity is essential to the cellular response of learning and memory (Comery et al., 1996; Lamprecht and Ledoux, 2004; Kozorovitskiy et al., 2005; Sjostrom et al., 2008). By contrast, any alteration in dendrite growth and/or structure can result in impaired brain function, e.g. cognitive dysfunction. Interestingly, impaired learning and memory are observed in many TBI survivors regardless of injury severity, and similar deficits are observed in animal models of TBI (Dixon et al., 1987; Lyeth et al., 1990; Winocur, 1990; Smith et al., 1991; Squire and Zola-Morgan, 1991; Smith et al., 1993). Additional evidence suggests that altered dendrite structure may underlie the cognitive deficits observed after TBI (Fineman et al., 2000; Zhu et al., 2000; Hoskison et al., 2009). For example, hippocampal long-term potentiation (LTP), that may account for many types of learning and is associated with dendritic plasticity, is suppressed for days to weeks following experimental TBI (Miyazaki et al., 1992). At the cellular level, changes in dendrite structural proteins such as microtubule-associated protein 2 (MAP2) and neurofilament proteins are prominent in animal models of TBI (Taft et al., 1992; Posmantur et al., 1994, 1996; Kanayama et al., 1996; Lewen et al., 1996; Folkerts et al., 1998; Saatman et al., 1998), and are also observed in the human brain of autopsy specimens after TBI (Castejon et al., 1997, 2004; Castejon and Arismendi, 2003). In addition, cerebrospinal fluid (CSF) MAP2 levels are increased in patients with severe TBI (Hayes et al., 2008). Together these data underline the importance of understanding how TBI affects dendrite structure and/or function, which may lead to novel insights about recovery after TBI.
There has been extensive research into the pathology and mechanisms of TAI (Povlishock, 1992; Smith and Meaney, 2000, Tang-Schomer et al., 2010). However, the effect of TAI on dendrite structure is less well understood. Therefore, we examined immediate and evolving dendritic changes using a well-established in vitro model of isolated axonal stretch injury (Smith et al., 1999; Wolf et al., 2001; Iwata et al., 2004), in which mechanical loading conditions replicate those occurring in head trauma (Meaney et al., 1995). We observed that stretch-induced axonal injury caused transient dendritic beading. This beading was sodium-dependent, exacerbated by extracellular calcium removal and blocked by N-methyl-D-aspartate receptor (NMDAR) antagonists. Our findings raise the possibility of a direct mechanical link between TAI and dendrite injury, and in particular that TAI may have a bystander effect on dendrites. Since dendrite structure and cognition are interconnected, understanding the mechanism(s) of dendrite injury after TBI may increase insight into TBI's pathobiology and lead to novel strategies to improve cognitive outcome.
Materials and methods
Animal welfare
All experiments were conducted in accordance with the Guide for the Care and Use of Laboratory Animals, U.S. Department of Health and Human Services, Pub No 85-23, 1985, and approved by the University of Pennsylvania Institutional Animal Care and Use Committee (IACUC). The minimum number of animals necessary for scientific validity was used and efforts were madeto minimize animal suffering.
Dissociated cerebral cortex neuronal cultures
Primary neuron cultures from E18 Sprague–Dawley rat embryos (Charles River Laboratories, Wilmington, MA) were prepared as previously described (Iwata et al., 2004). Briefly, neocortices were freed of meninges, dissociated and maintained in culture with Neurobasal medium (Invitrogen, Carlsbad, CA) supplemented with B-27 (Invitrogen) containing 0.5 mM glutamine and 5% fetal bovine serum. Cells were plated at a density of 3.75×105 cells/cm2 on a deformable membrane (Specialty Manufacturing, Saginaw, MI) coated with 1 mg/ml poly-L-lysine in custom-designed stainless steel wells (Smith et al., 1999). Two 2×10 mm silicone barriers (constructed from SYLGARD, Dow Corning Corporation, Midland, MI) with 0.4 mm wide micro-channels were placed on the deformable membrane at equal distance from the center of the well to create two cell-free gaps. These micro-channels were fabricated onto the surface of a stamp by casting polydimethylsiloxane (PDMS, Dow Corning) from patterned lithographic masters, as described previously (Tang et al., 2003). Using this system, we established 2 mm long axon tracks that span two neuronal populations. This micro-channel design offers several advantages over one-gap cultures (Iwata et al., 2004): 1) it provides directional cues for length-wise growth of axons, 2) parallel axon tracks are well separated and 3) as an essential requirement for anisotropic materials' elastic responses (Meaney et al., 1995), the longitudinal tracks insure a uniaxial stretch of most axons. We also used a novel method of water in the micro-channels to prevent cells from entering the gap (Tang-Schomer et al., 2010), instead of using pre-fabricated multi-layered channels (Taylor et al., 2005), so avoiding sophisticated lithography. Cells were grown for 11 days in vitro (DIV) when axon growth between the two cell populations separated by the gap is well-established. The barriers then were removed before axonal stretch injury was performed.
Axonal stretch injury
A specially designed axon stretch injury apparatus was used (Smith et al., 1999). To induce the injury, the culture well was placed in a device consisting of an aluminum cover block, a stainless steel plate with a machined 2×10 mm slit, and an air pulse-generating system. The culture well was inserted into the cover block and then placed on the slit plate so that the region containing the axon tracks coincided with the slit. The top plate was attached to the microscope stage, creating a sealed chamber. The top plate had a quartz viewing window in the center, an air inlet for compressed air and a dynamic pressure transducer (model EPX-V01-25P/16F-RF, Entran, Fairfield, NJ) to monitor internal chamber pressure. The introduction of compressed air into the chamber was gated by a solenoid (Parker General Valve, Elyria, OH). The solenoid and the pressure transducer were controlled and monitored by an analog-to-digital board (Metrabyte, Keithley Instruments, Cleveland, OH) integrated with a computer data acquisition system (Capital Equipment Corporation, Bellerica, MA). The device was mounted on the stage of a Nikon inverted microscope (Optical Apparatus, Ardmore, PA), to allow observation of the axon tracks throughout the experiment. To induce axonal stretch, a controlled air pulse was used to rapidly change the chamber pressure and deflect downward only the membrane containing the axon tracks within the gap. A strain rate of 20–50 s−1 was applied to the axons, well within the range for traumatic injury experienced by the human brain during rotational acceleration (Denny-Brown and Russell, 1940; Meaney et al., 1995). The nominal uniaxial strain (ε) was calculated by determining the centerline membrane deflection (δ) relative to the slit width (w) and substituting into the geometric relationship:
Experiments were conducted with a peak internal chamber pressure of 13–15 ψ to induce a transient uniaxial axonal strain calculated at 1.8–1.9 or 80–90% beyond axons' initial length.
Drug treatment
Before axonal stretch, cells were washed once and incubated in control salt solution (CSS, 15 min at +37 °C) consisting of 120 mM NaCl, 5.4 mM KCl, 0.8 mM MgCl2, 1.8 mM CaCl2, 15 mM glucose, and 25 mM HEPES, pH 7.4. In some experiments, cells were pre-incubated (15 min) with tetrodotoxin (1 μM) or MK-801 (20 μM) by diluting drug stock solutions directly into CSS-containing cultures. Then, axon stretch was performed and cells were either fixed after 5 min and processed for immunocytochemistry or subjected to cell viability analysis. For time-course experiments, CSS was exchanged for culture medium immediately after stretch, and the cells returned to the incubator for 3 h and 5 h, until immunocytochemical or cell viability analyses were performed.
Calcium-free experiments were conducted in calcium-free CSS containing the calcium-chelating agent EGTA (1 mM). Cells were washed 3 times in calcium-free CSS, and incubated for 15 min at +37 °C before axonal stretch was performed. Sodium-free experiments were conducted by replacing sodium with N-methyl-D-glucamine and followed the same procedure described for calcium experiments.
Immunocytochemistry
Cells were fixed in cold 4% paraformaldehyde/4% sucrose in PBS for 20 min, thoroughly washed in PBS and permeabilized with 0.5% Triton X-100 for 5 min. Cells were pre-incubated in 5% goat serum (GS) in PBS for 30 min at room temperature, and then double-stained with a mouse monoclonal anti-MAP2 (1:600, Sigma-Aldrich, St Louis, MO) to label dendrites and a rabbit polyclonal anti-neurofilament protein 200 kDa (NF200; 1:900, Sigma-Aldrich) to label axons, in 2% GS in PBS overnight at +4 °C. After several washes in PBS, cells were incubated with Alexa Fluor 488-conjugated goat anti-mouse (1:700, Molecular Probes, Eugene, OR) and Alexa Fluor 546-conjugated goat anti-rabbit (1:1200, Molecular Probes) secondary antibodies, diluted in PBS/2% GS for 1 h at +37 °C. After rinsing in PBS, one drop of Fluoromount mounting medium (Southern Biotechnology, Birmingham, AL) was added to the center of each well and a coverslip was mounted onto the membrane. Neurons were visualized through 20× and 40× objectives and fluorescent images recorded with a CCD video camera (Dage-MTI, Michigan City, IN) mounted on a Nikon Optiphot microscope and analyzed with a Macintosh PowerMac (9500/200) and image processing program (NIH Image 1.59).
Morphological analysis
Control and stretched axon preparations were processed and analyzed in parallel and controls were subjected to the same number and type of solution changes as were stretched axon preparations. Dendritic alterations, i.e., beading, were examined in different, non-overlapping microscopic fields selected at random along both sides of the gap region (which contained the axon tracks) on a minimum of 3 wells for each condition, including controls. At least 3 experiments were performed for each condition, including controls. In both control and stretched axon preparations, a minimum of 30 dendrites were examined at a final image magnification of 400–800× per condition in each experiment. Dendrites from control preparations were smooth in appearance and displayed no pathological swellings or beads unlike altered dendrites from stretched axon preparations. Altered dendrites were scored as present if found in at least one dendrite/microscopic field. The percentage of beaded dendrites was determined by the ratio of beaded dendrite number/total number of dendrites within an area of approximately 130 μm width along each injured gap's side. For each condition, the percentage was evaluated for 3 different culture wells in at least 3 independent experiments, and the average percentage was calculated. To quantify the morphology of individual beads, dendrite swellings that are at least 3 times the diameter of the adjoining regions were analyzed using NIH Image. The total length of each beaded dendrite was measured, and bead number was expressed as the number of beads per dendrite Length Unit (LU), where LU is defined as 100 μm. Bead number was measured from at least 30 dendrites per condition in 3 independent experiments. Bead length (along the dendritic axis), diameter (normal to the dendritic axis), inter-bead distance and bead size were measured from the same beads used to generate bead number.
Determination of cell viability
Neuronal survival was assessed using fluorescein diacetate (FDA) to label viable cells, and propidium iodide (PI) to label dead or dying cells. Cells were washed once with pre-warmed CSS, and live and dead cells were determined by incubating with 15 μg/ml FDA and 4.5 μg/ ml PI for 15 min at +37 °C. Cells were counted in 24 adjacent microscopic fields along both sides ofthe gap region (which contained the axon tracks) at 20× magnification in each of 3 wells per condition. Experiments were repeated at least 3 times. Cell survival was expressed as percent of control values and represents a minimum of 4000 cells counted per condition in each experiment.
Statistical analysis
Statistical analysis was performed with StatView (SAS Institute, Cary, NC). Values are expressed as mean±S.E.M. Each dendritic segment analyzed was considered a single observation with several associated measured parameters (e.g., bead number, size, etc…). Each parameter was analyzed separately. Measurements from at least 3 independent experiments were combined (i.e., at least 100 dendritic segments/parameter were analyzed). Control and injured groups were compared byone-way analysisof variance (ANOVA) followed by Fisher's Protected Least Significant Difference (PLSD) post hoc test for pairwise comparisons. The significance level was p<0.05.
Results
Compartmentalization culture of axons and dendrites
To determine TAI's effect on dendrites, we used a modification of our well-established in vitro model of isolated axonal stretch injury (Smith et al., 1999; Wolf et al., 2001; Iwata et al., 2004; Yuen et al., 2009). The present experiments used two silicone barriers to create two cell-free gaps (rather than one as previously used) equidistant to the center of the steel well in which the cells are plated (Fig. 1A). Each barrier has micro-patterned channels of 2 mm long and 0.4 mm wide (Tang et al., 2003) small enough to confine cell bodies to the outside, while permissive to axon growth (Tang Schomer et al., 2010). This system creates well-defined 2 mm long axon tracts at regular intervals across each gap (Fig. 1B). In addition, stretch injury can be selectively induced on axons within one gap, whereas axons in the other gap remain uninjured, i.e. an internal control.
Fig. 1.
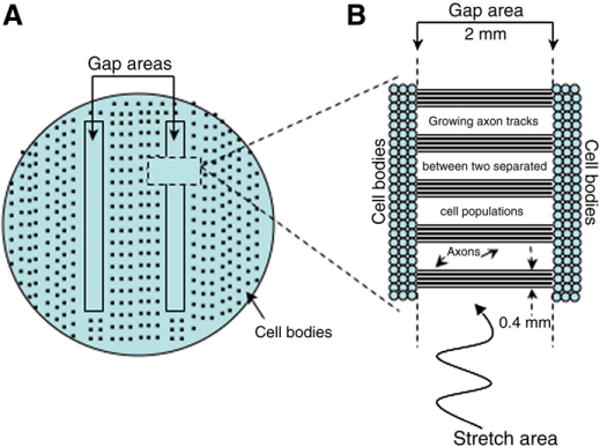
Axon stretch injury apparatus. Schematic representation of the steel well (A) in which two silicon barriers are placed over the elastic membrane to generate two cell-free gaps. Each barrier contains laddering micro-channels (B) that allow axon growth on 2 mm longitudinal tracks.
After 10–12 DIV, immunofluorescence analysis with anti-NF200 (to identify axons) and anti-MAP2 (to identify dendrites) confirmed that neuron processes crossing the gap were exclusively axonal (Fig. 2A). Axonal tracts were strictly confined to the micro-channels and showed a tendency to fasciculate. By contrast, MAP2 staining demonstrated that neuronal cell bodies and dendrites were located on each side of the gap, but were excluded from the gap region (Fig. 2B). Thus, clear boundaries between axonal and dendritic processes can be established in this system.
Fig. 2.
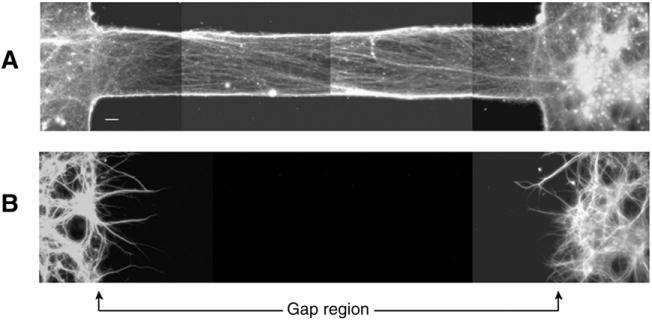
Composite fluorescent photomicrographs of neuronal processes double-stained for (A) NF200 and (B) MAP2, showing that axon tracts are devoid of cell bodies and dendritic processes. Instead, cell bodies and dendrites are restricted to each side of the gap region. Both panels represent the same microscopic field of double-labeled rat cortical neurons after 11 days in culture. Scale bar=50 μm.
Axonal stretch injury
We first confirmed tensile stretch selectively injured axon tracks within one gap and do not affect axons in the second gap. Only stretched axons displayed characteristic pathological changes similar to those we have observed in a single gap system (Smith et al., 1999; Wolf et al., 2001; Iwata et al., 2004; Tang-Schomer et al., 2010). These axonal pathologic changes were consistently observed when strain pulse duration was less than 50 ms, similar to conditions known to induce TAI during head rotational acceleration (Denny-Brown and Russell, 1940; Meaney et al., 1995). Following stretch, axons displayed “delayed elasticity”, characterized by multiple undulated regions, then gradually relaxed back to their pre-stretched length and straight morphology within an hour post-injury, similar to the one-gap well system (Smith et al., 1999; Wolf et al., 2001; Iwata et al., 2004; Tang-Schomer et al., 2010) (Fig. 3). In the two-gap system, no morphological changes were observed in axons that were not stretched. These results confirm the selectivity of our two-gap well injury model. While all the cultured axons were exposed to the same post-injury biochemical environment, only axons directly stretched showed mechanical injury. This indicates that the effect is not mediated by extracellular confounding factors. Subsequent experiments were conducted to examine the effect of mechanically injured axons on dendrites.
Fig. 3.
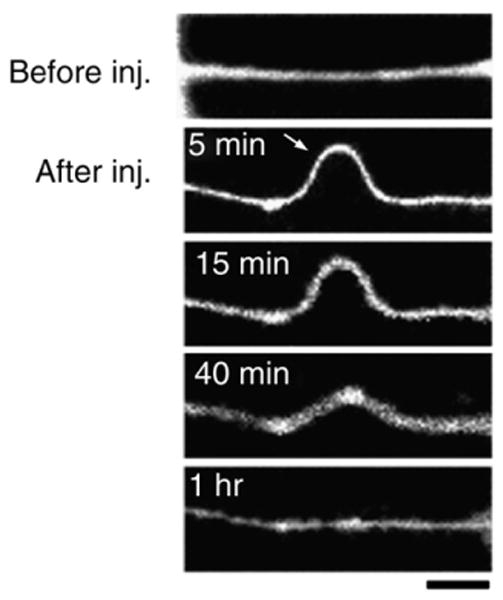
Representative fluorescent photomicrographs of NF200-labeled neuronal processes illustrating the temporal evolution of axons' delayed elastic response to dynamic stretch injury. Before stretch, axons are straight. Immediately after injury, they develop large undulations before gradually recovering their initial shape and length. Scale bar=10 μm.
Axonal stretch injury induces dendritic morphological alterations
Following axonal stretch, dendrite morphology was altered. Dendritic alterations manifested as periodic swellings along the dendritic shaft (hereafter referred to as “beading”). Beading also occurredon dendrites that projected away from the gap, i.e. where the dendrites were unlikely to be directly affected by the stretch, i.e. they had to be affected by the axon. Using a fixed strain of 80% level (i.e., axons were stretched up to 180% of their original lengths), we determined the time-course of dendritic morphological changes. Cells immunostained with anti-MAP2 demonstrated that dendritic beading formed within minutes after stretch and lasted for several hours (Fig. 4). Beading was maximal immediately after stretch and could affect up to 50% of dendritic processes within an area of approximately 130–160 μm width along each injured gap's side. This indicates that stretched axons originated from neurons located on both sides of the gap. In contrast, dendritic beading was absent along the other unstretched gap, consistent with our system's selective damage of axons growing within one gap. These results suggest that dendritic beading is dependent on direct mechanical damage of stretched axons.
Fig. 4.
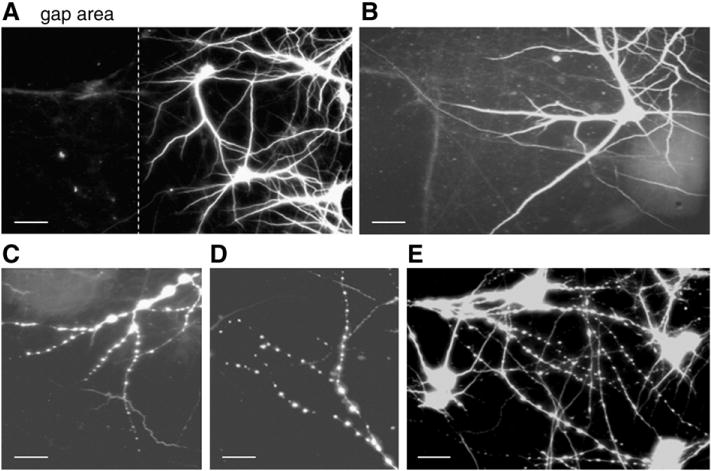
Fluorescent photomicrographs of control (uninjured) neurons (A and B) immunostained for the dendritic marker MAP2, and neurons displaying beaded dendrites 5 min after axonal stretch (C, D, and E). The dashed line in (A) indicates the boundary between the axon-only gap area (left) and the neuronal cell body region (right). Scale bar=50 μm (A) and 25 μm (B, C, D, and E).
The magnitude of dendrite beading decreased over time. Only 5–10% of dendritic processes displayed beading 5 h after axonal stretch injury, while beading was nearly absent when cells were examined 15 h post-stretch. Cell viability in the stretched gap area was similar to that in unstretched control wells 5 h after stretch (data not shown).
Morphological analysis of axonal stretch injury-induced dendritic beading
Next we characterized dendritic beading, including bead number, size and distribution along the dendritic shaft. Bead number per dendrite Length Unit (100 μm) was greatest 5 min after axonal stretch injury, then gradually decreased at 3 h and 5 h post-stretch (Fig. 5). By 15 h very few cells displayed dendritic beading. We therefore concentrated further analysis of dendrite morphology from neurons fixed during the first few hours after axonal stretch injury.
Fig. 5.
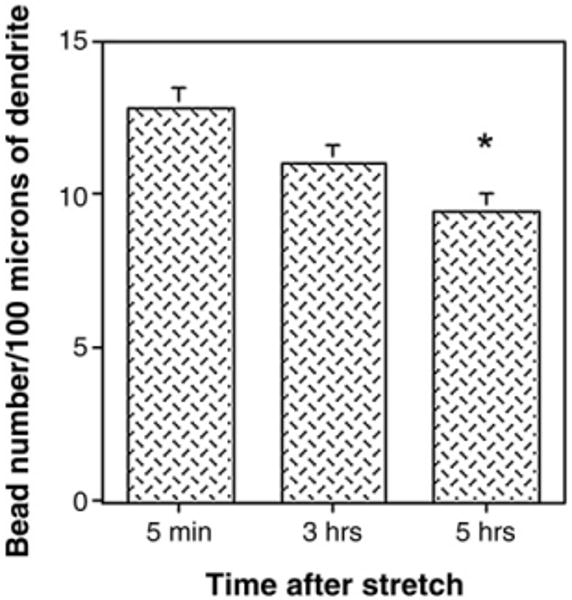
Histogram illustrating the time-course of dendritic bead formation. After dynamic stretch injury of axons, cultures were fixed at various time-points, and double-stained for MAP2 and NF200. The number of beads per dendrite Length Unit (100 μm) was determined at ×400 magnification. Values represent mean±S.E.M. from 3 independent experiments. *p<0.0005 vs. 5 min.
To examine bead distribution along the dendrite shaft, we measured inter-bead distances. We found that 70–90% of beads were separated by 0–10 μm regardless of which time-point was examined and that their distribution pattern along the dendrite shaft was similar between 5 min and 5 h (Fig. 6A). Greater than 60% of beads were separated by 5 μm or less at 5 min after stretch, and the percentage decreased to 35% at 5 h post-stretch. Conversely, an inter-bead distance of 11–16 μm was observed in less than 10% of beads at 5 min after axonal stretch injury, but in greater than 20% of beads after 5 h (Fig. 6B). These data indicate that over time beads were more spaced apart, consistent with the observation that there were fewer beads per dendrite Length Unit.
Fig. 6.
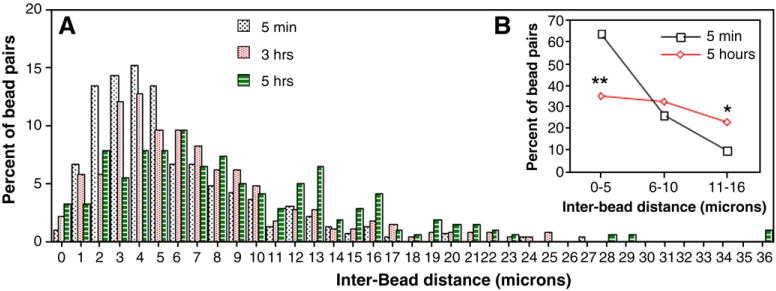
Histogram (A) and graph (B) showing inter-bead distances expressed as a percentage of bead pairs. After dynamic stretch injury of axons, cultures were fixed at various time-points and double-stained for MAP2 and NF200. Inter-bead distances were measured along dendritic processes at ×800 magnification, and were graphed as bead pairs. Values represent mean±S.E.M. from 3 independent experiments. *p<0.005 and **p<0.0001 vs. 5 min.
Bead size (perimeter) was similar when examined between 5 min and 5 h after axonal stretch injury (Fig. 7B). There was a large range in bead perimeter (Fig. 7A); greater than 60% of beads had a perimeter between 3 and 7 μm, regardless of the time-point examined. Large beads (>15 μm in perimeter) represented less than 10% of the bead population at any time-point. Other measures of bead size varied greatly; bead length varied from less than 1 μm to greater than 10 μm, and bead diameter from less than 1 μm to 6 μm. Dendritic beads were generally spherical, although some displayed a more irregular appearance, particularly large beads (greater than 5 μm in length). When beads formed, the dendritic segment between beads shrank in diameter and occasionally was undetectable, i.e. a fragmented MAP2 staining. Bead size generally decreased proximal to distal along the dendritic shaft, although smaller beads could precede bigger ones on a particular dendrite segment.
Fig. 7.
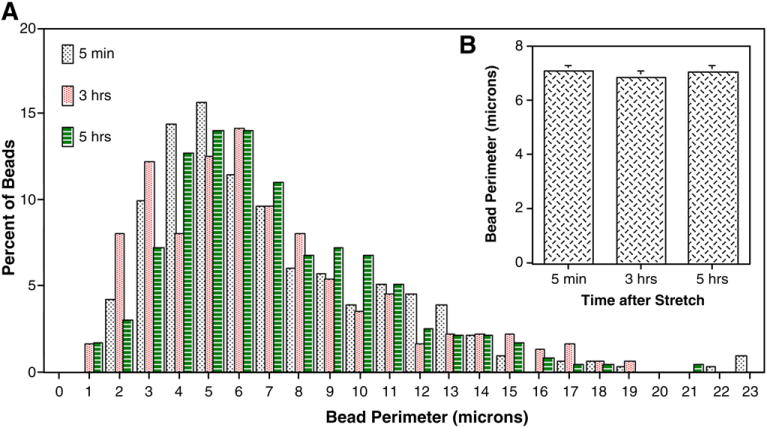
Histogram (A) and graph (B) of bead perimeter distribution. Cells were axon-stretched, then fixed at various time-points and double-labeled with MAP2 and NF200. Bead perimeter was determined at ×800 magnification. Values represent mean±S.E.M. from 3 independent experiments.
Pharmacology of axonal stretch injury-induced dendritic beading
Previous studies have implicated sodium channels in dendritic bead formation after glutamate exposure or oxygen–glucose deprivation (Hasbani et al., 1998; Al-Noori and Swann, 2000; Oliva et al., 2002). Therefore, we examined sodium channels' involvement in axonal stretch injury-induced dendritic beading. Cultures were pre-incubated with the voltage-gated sodium channel blocker (VGSC) tetrodotoxin (TTX; 1 μM) before axonal stretch injury. This resulted in nearly complete inhibition of dendritic beading (Fig. 8B). Similarly, when sodium in the CSS was replaced with equimolar N-methyl-D-glucamine, dendritic beading in response to axonal stretch injury also was attenuated (Fig. 8C). Dendritic beading was not observed in unstretched cultures pre-treated with TTX, or when extracellular sodium was removed. These data indicate that sodium influx is involved in dendritic beading after axonal stretch injury.
Fig. 8.
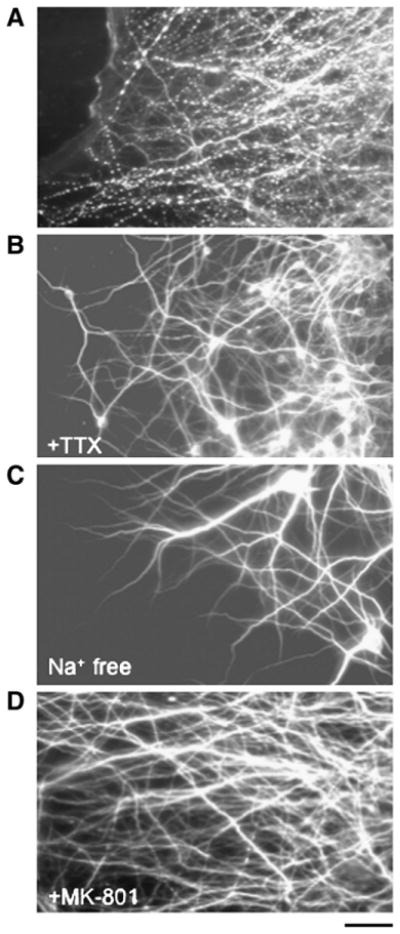
Pharmacological modulation of axonal stretch injury-induced dendritic beading. Fluorescence photomicrographs of dendritic processes immunostained with the dendritic marker MAP2. Cultures were fixed 5 min after dynamic stretch injury of axons. Compared with untreated cultures (A), pre-treatment with the VGSC inhibitor tetrodotoxin (TTX, 1 μM) (B), removal of extracellular sodium (C), or pre-incubation with the NMDA receptor antagonist MK-801 (20 μM) (D), prevented dendritic beading. Scale bar=25 μm.
N-methyl-D-aspartate receptors (NMDARs) are found on dendrites and not axons. These NMDARs have been implicated in excitotoxicity-induced dendritic beading (Park et al., 1996; Faddis et al., 1997; Hasbani et al., 1998), therefore we examined whether these receptors played a role, i.e., we could potentially compartmentalize the effect of stretch. Pre-treatment with the selective NMDAR blocker MK-801 (20 μM) before axonal stretch injury, suppressed dendritic beading (Fig. 8D). Since NMDARs are highly permeable to calcium ions, we examined whether calcium influx played a role in axonal stretch injury-induced dendritic beading. Cells were placed in calcium-free CSS before axonal stretch injury. This removal of extracellular calcium significantly increased the number of beaded dendrites (>90% in calcium-free CSS versus 40–50% in calcium-containing medium). However, this beading was qualitatively different from cultures in which calcium was present during axonal stretch injury since the majority of beads appeared larger and were spaced further apart along the dendritic shafts. Quantitative analysis confirmed this observation (Fig. 9). When cells were incubated in calcium-free CSS but axons were not stretched, dendrite beading did not occur.
Fig. 9.
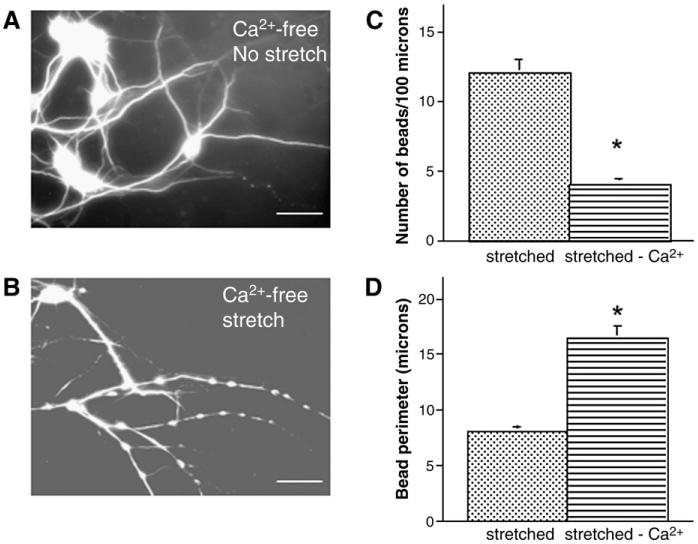
Effect of extracellular calcium on axon stretch injury-induced dendritic beading. Fluorescent photomicrographs of dendritic processes immunostained for MAP2 demonstrate smooth processes of uninjured cultures pre-incubated in calcium-free medium (A). However, 5 min after dynamic stretch injury of axons, dendrites displayed extensive beading (B). Histograms (C) and (D) illustrate quantitative analysis of dendritic beading and show a decreased number of beads (C) and an increased bead perimeter (D) in calcium-free medium after stretch, compared tocalcium-containing medium. Bead number and perimeter were determined at ×400 and ×800 magnification, respectively. Values represent mean±S.E.M. from 3 independent experiments. *p<0.0001 vs. calcium-containing medium. Scale bar=25 μm in (A and B).
Discussion
When we induced isolated TAI in culture, we observed that axonal stretch injury alters dendrite morphology by causing periodical swellings, i.e. dendritic beading, a phenomenon that most probably involves changes in ionic homeostasis. This suggests a ‘bystander’ effect, whereby dynamic mechanical deformation of isolated axons induces indirect pathologic changes of associated dendrites. The dynamic uniaxial axon stretch used in this study, replicates the mechanical loading conditions experienced by axons during inertial brain injury (Smith et al., 1999) and induces many of the morphological and ultrastructural changes found in TAI in vivo (Povlishock, 1992; Sherriff et al., 1994; Maxwell and Graham, 1997; Smith and Meaney, 2000; Tang-Schomer et al., 2010). This raises the possibility that a similar relationship between TAI and dendrite structure and plasticity may exist in vivo. Indeed, loss of MAP2 staining and changes in dendritic morphology have been observed in rodent models of TBI and in humans (Taft et al., 1992; Posmantur et al., 1996; Lewen et al., 1996; Castejon et al., 1997, 2004; Folkerts et al., 1998; Castejon and Arismendi, 2003). Our results further suggest a sodium- and NMDAR-dependent mechanism for TAI-induced changes in dendrite morphology. Understanding the mechanism(s) of TAI-induced dendrite injury may be important since dendrite plasticity is postulated to be the structural cellular correlate of cognition. Indeed, cognitive and behavioral dysfunction among TBI survivors is a frequent finding (Scheid et al., 2006; Kumar et al., 2009).
There are several advantages to our in vitro system of TAI: 1) dendrite and axon growth occurs in two different areas that allows separate examination of both processes, 2) the system is highly reliable, with a precisely controlled biochemical and mechanical environment, and 3) the model produces an axon-only injury with mechanical conditions similar to that observed in vivo. This provides the unique opportunity to study dendritic alterations that result from axonal trauma. Indeed, the key findings of this study is that selective, direct mechanical axonal injury triggers changes in dendrites that are highly selective spatially, fast-forming and transient, and provides evidence for a direct link between axon pathology and dendrite injury.
In our model, axonal stretch injury-induced dendritic beading was restricted to a narrow area along the gap's sides (approximately 130– 160 μm in width). Only axons are found in the gap, suggesting that most of the axons crossing the gap that are injured, originate from neurons growing immediately adjacent to it. Our data suggest a bystander effect of TAI on dendrites and establish a direct link between the injured axons and their associated dendrites. Since dendritic beading was restricted to a narrow area contiguous to the gap region where axon stretch occurs, it is unlikely that a stretch-induced release of soluble factor(s) mediates this phenomenon. Since there is high density and interconnectivity of neurites in our culture, it is difficult to confirm whether this bystander effect is from postsynaptic neurons or only from neurons whose axons were mechanically injured. Therefore, two possibilities emerge: 1) axonal stretch injury disrupts synaptic transmission. Consistent with this, previous experiments demonstrate that disruption of the axon connection between a presynaptic neuron and a postsynaptic neuron's dendrites can affect dendrite structure (Valverde, 1968; Deitch and Rubel, 1984; Mizrahi and Libersat, 2002). It also is possible that impaired axonal transport and accumulation of axonal proteins following axonal stretch (Sherriff et al., 1994; Maxwell et al., 1997; Povlishock et al., 1997; Smith and Meaney, 2000), may lead to abnormal synaptic activity and so affect dendrite structure and integrity. 2) Axon injury sends a biochemical signal from the cell body and leads to dendrite injury from the same neuron. Further studies using techniques to isolate individual processes of single neurons will provide further insights. Nevertheless, this study provides the first evidence for a link between mechanical axon deformation and dendritic alterations in TAI.
The morphological changes in dendrites in our TAI model appear similar to that observed following excitotoxic stimulation of ionotropic glutamate receptors in vitro (Park et al., 1996; Hasbani et al., 1998; Al-Noori and Swann, 2000; Oliva et al., 2002; Greenwood et al., 2007). In both circumstances, beading occurs rapidly and is transient, i.e., this change represents a robust response to the initial trigger.In addition, the pattern of dendritic constriction and swelling (“string of beads”) is common to other forms of neuronal injury in vivo (Sloviter and Dempster, 1985; Stewart et al., 1991).In this dynamic process, we found that ionic fluxes similar to excitotoxicity play an important role: first, extracellular sodium is involved in the beading process since specific sodium channel blockade and sodium removal prevented dendritic beading. This finding is similar to the need for sodium influx to trigger glutamate- or glutamate agonist-induced dendritic beading (Hasbani et al., 1998; Al-Noori and Swann, 2000; Oliva et al., 2002; Greenwood et al., 2007). Second, calcium seems to modulate TAI-induced dendritic beading since removing extracellular calcium exacerbated dendritic beading. A role for calcium in dendritic beading also is observed when cultured neurons are exposed to glutamate receptor agonists. These studies suggest calcium may influence the extent of beading since beads are larger and form more rapidly when induced in calcium-free buffer (Hasbani et al., 1998). Consistent with this, Oliva et al. (2002) reported an increase in average bead length when voltage-gated calcium channels (VGCCs) are blocked during kainate receptor stimulation of mouse organotypic hippocampal slice cultures.
The effect of extracellular calcium elimination, however, may be linked to increased sodium influx as a consequence of calcium removal (Hasbani et al., 1998). Third, NMDARs appear to be involved. This is an important indication of the specificity of axon-induced dendritic injury. We observed that NMDAR blockade prevented TAI-induced dendritic beading. Since NMDARs are absent from axons, this suggests a direct link between dendritic receptor activation and axon-induced dendrite injury. A role for ionotropic glutamate receptors also is observed in excitotoxicity-induced bead formation in vitro (Park et al., 1996; Hasbani et al., 1998; Al-Noori and Swann, 2000; Oliva et al., 2002; Greenwood et al., 2007). In addition, several studies show rapid, mechanically-induced NMDAR activation after injury (Zhang et al., 1996; Lusardi et al., 2004; DeRidder et al., 2006; Geddes-Klein et al., 2006). Thus, it is possible that once NMDARs are activated, dendritic beading occurs. Alternatively, injury-induced glutamate release, (Kao et al., 2004; Geddes-Klein et al., 2006) may contribute to NMDAR-mediated bead formation.
How dendritic beads form is unclear. It has been proposed that water influx that accompanies sodium and chloride ion influx may contribute to glutamate-induced beading (Hasbani et al., 1998; Al-Noori and Swann, 2000; Greenwood et al., 2007). Our data suggest a similar mechanism may exist in axon injury-induced dendritic beading. For example, sodium influx is known to be triggered by axon stretch through VGSCs (Wolf et al., 2001). Proteolysis of VGSCs' subunits also can occur after axonal stretch injury (Iwata et al., 2004; von Reyn et al., 2009). It is conceivable then that a sodium wave may propagate over a long distance and so affect dendrites similar to long-range calcium wave propagation. It is possible that dysregulation of sodium movement also may affect dendrite integrity. However, our experiments using TTX did not distinguish between axonal vs. dendritic VGSCs. Bead formation also may result from a disrupted neuronal cytoskeleton. For example, microtubule depolymerization induced by NMDAR activation, leads to dendritic alterations in CNS neurons (Goldberg and Bateman, 1993; Emery and Lucas, 1995; Greenwood et al., 2007). This perturbation in the microtubule cytoskeleton may explain the disrupted MAP2 staining we observed. Indeed, microtubule breakage and disintegration recently was demonstrated in our axonal stretch injury model (Tang-Schomer et al., 2010). Whether this effect propagates further than the axon or whether altered dendrite structure occurs independently, will need further study. Alternatively, protease activation including calpain after axonal stretch injury (Pike et al., 2000; Iwata et al., 2004; DeRidder et al., 2006; von Reyn et al., 2009), may lead to cytoskeletal damage, and in particular MAP2 degradation. Thus, the existence of a biochemical cross-talk between the directly injured axons and the dendrites may account for the dendritic morphological changes after axonal stretch injury.
The implications of TAI-induced changes in dendrite morphology remain unclear. Potentially, altered dendrite morphology is an early sign of excitotoxic injury that occurs in several acute neurologic and chronic neurodegenerative diseases. In addition, several lines of evidence show that TBI alters dendrite structure. For example, reduced immunoreactivity and increased MAP2 and spectrin degradation and changes in neurofilament proteins in dendrites occur in animal TBI models (Taft et al., 1992; Posmantur et al., 1994, 1996, 1997, 2000; Kanayama et al., 1996; Lewen et al., 1996; Saatman et al., 1996, 1998). In human, ultrastructural studies demonstrate dendritic changes in brains obtained from dead TBI patients (Castejon et al., 1997, 2004; Castejon and Arismendi, 2003), and increased CSF MAP2 levels are observed in patients with severe TBI (Hayes et al., 2008). TBI also can blunt experience-dependent dendrite growth and neural plasticity (Fineman et al., 2000; Ip et al., 2002), whereas increased MAP2 expression or dendrite growth, appears to be important in TBI recovery (Jones et al., 1996; Kanayama et al., 1996; Rowntree and Kolb, 1997; Kozlowski et al., 2004; Zepeda et al., 2004; Briones et al., 2006). This phenomenon of dendrite growth and recovery may be particularly important since proper dendrite growth is a key determinant of neuron and so CNS function. Dendrites are highly plastic and undergo structural changes, reacting to various stimuli, and this may represent the cellular response to learning and memory (Comery et al., 1996; Lamprecht and Ledoux, 2004; Kozorovitskiy et al., 2005; Sjostrom et al., 2008). Cognitive and behavioral dysfunction is common after clinical and experimental TBI regardless of injury severity, and so emphasizes the need to better define TBI-induced dendropathology. Thus, understanding mechanism(s) of TAI-induced dendrite pathology may provide insights to develop novel therapies to improve learning, memory and behavioral dysfunctions after TBI. The model described in this report may represent a useful method to study this important question. Together our data identify dendritic alterations as the direct result of mechanically injured axons. Although the signaling mechanism is still unclear, this study represents a first step towards understanding cognitive dysfunction after TBI at the cellular level.
Acknowledgments
This work was supported by a grant (021.BIR1) to H. A. K. and P. D. L. from the New Jersey Commission on Brain Injury Research, and NIH grants NS038104, NS048949 and NS056202 to D. H. S.
References
- Al-Noori S, Swann JW. A role for sodium and chloride in kainic acid-induced beading of inhibitory interneuron dendrites. Neuroscience. 2000;101:337–348. doi: 10.1016/s0306-4522(00)00384-5. [DOI] [PubMed] [Google Scholar]
- Briones TL, Woods J, Wadowska M, Rogozinska M. Amelioration of cognitive impairment and changes in microtubule-associated protein 2 after transient global cerebral ischemia are influenced bycomplex environment experience. Behav Brain Res. 2006;168:261–271. doi: 10.1016/j.bbr.2005.11.015. [DOI] [PubMed] [Google Scholar]
- Castejon OJ, Arismendi GJ. Morphological changes of dendrites in the human edematous cerebral cortex. A transmission electron microscopic study. J Submicrosc Cytol Pathol. 2003;35:395–413. [PubMed] [Google Scholar]
- Castejon OJ, Valero C, Diaz M. Light and electron microscopy study of nerve cells in traumatic oedematous human cerebral cortex. Brain Inj. 1997;11:363–388. doi: 10.1080/026990597123520. [DOI] [PubMed] [Google Scholar]
- Castejon OJ, Castellano A, Arismendi G. Transmission electron microscopy of cortical dendritic spines in the human oedematous cerebral cortex. J Submicrosc Cytol Pathol. 2004;36:181–191. [PubMed] [Google Scholar]
- Cline HT. Dendritic arbor development and synaptogenesis. Curr Opin Neurobiol. 2001;11:118–126. doi: 10.1016/s0959-4388(00)00182-3. [DOI] [PubMed] [Google Scholar]
- Comery TA, Stamoudis CX, Irwin SA, Greenough WT. Increased density of multiple-head dendritic spines on medium-sized spiny neurons of the striatum in rats reared in a complex environment. Neurobiol Learn Mem. 1996;66:93–96. doi: 10.1006/nlme.1996.0049. [DOI] [PubMed] [Google Scholar]
- Deitch JS, Rubel EW. Afferent influences on brain stem auditory nuclei of the chicken: time-course and specificity of dendritic atrophy following deafferentation. J Comp Neurol. 1984;229:66–79. doi: 10.1002/cne.902290106. [DOI] [PubMed] [Google Scholar]
- Denny-Brown D, Russell RW. Experimental cerebral concussion. J Physiol. 1940;99:153. doi: 10.1113/jphysiol.1940.sp003887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeRidder MN, Simon MJ, Siman R, Auberson YP, Raghupathi R, Meaney DF. Traumatic mechanical injury to the hippocampus in vitro causes regional caspase-3 and calpain activation that is influenced by NMDA receptor subunit composition. Neurobiol Dis. 2006;22:165–176. doi: 10.1016/j.nbd.2005.10.011. [DOI] [PubMed] [Google Scholar]
- Dixon CE, Lyeth BG, Povlishock JT, Findling RL, Hamm RJ, Marmarou A, Young HF, Hayes RL. A fluid percussion model of experimental brain injury in the rat. J Neurosurg. 1987;67:110–119. doi: 10.3171/jns.1987.67.1.0110. [DOI] [PubMed] [Google Scholar]
- Emery DG, Lucas JH. Ultrastructural damage and neuritic beading in cold-stressed spinal neurons with comparisons to NMDA and A23187 toxicity. Brain Res. 1995;692:161–173. doi: 10.1016/0006-8993(95)00726-7. [DOI] [PubMed] [Google Scholar]
- Faddis BT, Hasbani MJ, Goldberg MP. Calpain activation contributes to dendritic remodeling after brief excitotoxic injury in vitro. J Neurosci. 1997;17:951–959. doi: 10.1523/JNEUROSCI.17-03-00951.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felmingham KL, Baguley IJ, Green AM. Effects of diffuse axonal injury on speed of information processing following severe traumatic brain injury. Neuropsychology. 2004;18:564–571. doi: 10.1037/0894-4105.18.3.564. [DOI] [PubMed] [Google Scholar]
- Fineman I, Giza CC, Nahed BV, Lee SM, Hovda DA. Inhibition of neocortical plasticity during development by a moderate concussive brain injury. J Neurotrauma. 2000;17:739–749. doi: 10.1089/neu.2000.17.739. [DOI] [PubMed] [Google Scholar]
- Folkerts MM, Berman RF, Muizelaar JP, Rafols JA. Disruption of MAP2 immunostaining in rat hippocampus after traumatic brain injury. J Neurotrauma. 1998;15:349–363. doi: 10.1089/neu.1998.15.349. [DOI] [PubMed] [Google Scholar]
- Geddes-Klein DM, Serbest G, Mesfin MN, Cohen AS, Meaney DF. Pharmacologically induced calcium oscillations protect neurons from increases in cytosolic calcium after trauma. J Neurochem. 2006;97:462–474. doi: 10.1111/j.1471-4159.2006.03761.x. [DOI] [PubMed] [Google Scholar]
- Goldberg MP, Bateman MC. Taxol attenuates glutamate receptor-mediated neurite swelling in vitro. Soc Neurosci Abstr. 1993;19:26. [Google Scholar]
- Greenwood SM, Mizielinska SM, Frenguelli BG, Harvey J, Connolly CN. Mitochondrial dysfunction and dendritic beading during neuronal toxicity. J Biol Chem. 2007;282:26235–26244. doi: 10.1074/jbc.M704488200. [DOI] [PubMed] [Google Scholar]
- Hasbani MJ, Hyrc KL, Faddis BT, Romano C, Goldberg MP. Distinct roles for sodium, chloride, and calcium in excitotoxic dendritic injury and recovery. Exp Neurol. 1998;154:241–258. doi: 10.1006/exnr.1998.6929. [DOI] [PubMed] [Google Scholar]
- Hayes RL, Gabrielli A, Buki A, Robinson G, Robicsek S, Tepas J, Pineda J, Robertson C, Oli M, Akinyl L, Mo JX, Scharf D, Liu MC, Zheng WR, Wang K. Increased CSF levels of microtubule-associated protein-2 (MAP-2) in human subjects with severe traumatic brain injury (TBI) J Neurotrauma. 2008;25:863. [Google Scholar]
- Hoskison MM, Moore AN, Hu B, Orsi S, Koboric N, Dash PK. Persistent working memory dysfunction following traumatic brain injury: evidence for a time-dependent mechanism. Neuroscience. 2009;159:483–491. doi: 10.1016/j.neuroscience.2008.12.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ip EY, Giza CC, Griesbach GS, Hovda DA. Effects of enriched environment and fluid percussion injury on dendritic arborization within the cerebral cortex of the developing rat. J Neurotrauma. 2002;19:573–585. doi: 10.1089/089771502753754055. [DOI] [PubMed] [Google Scholar]
- Iwata A, Stys PK, Wolf JA, Chen XH, Taylor AG, Meaney DF, Smith DH. Traumatic axonal injury induces proteolytic cleavage of the voltage-gated sodium channels modulated by tetrodotoxin and protease inhibitors. J Neurosci. 2004;24:4605–4613. doi: 10.1523/JNEUROSCI.0515-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jan YN, Jan LY. Dendrites Genes Dev. 2001;15:2627–2641. doi: 10.1101/gad.916501. [DOI] [PubMed] [Google Scholar]
- Jones TA, Kleim JA, Greenough WT. Synaptogenesis and dendritic growth in the cortex opposite unilateral sensorimotor cortex damage in adult rats: a quantitative electron microscopic examination. Brain Res. 1996;733:142–148. doi: 10.1016/0006-8993(96)00792-5. [DOI] [PubMed] [Google Scholar]
- Kao CQ, Goforth PB, Ellis EF, Satin LS. Potentiation of GABAA currents after mechanical injury of cortical neurons. J Neurotrauma. 2004;21:259–270. doi: 10.1089/089771504322972059. [DOI] [PubMed] [Google Scholar]
- Kanayama G, Takeda M, Niigawa H, Ikura Y, Tamii H, Taniguchi N, Kudo T, Miyamae Y, Morihara T, Nishimura T. The effects of repetitive mild brain injury on cytoskeletal protein and behavior. Methods Find Exp Clin Pharmacol. 1996;18:105–115. [PubMed] [Google Scholar]
- Kozlowski DA, Nahed BV, Hovda DA, Lee SM. Paradoxical effects of cortical impact injury on environmentally enriched rats. J Neurotrauma. 2004;21:513–519. doi: 10.1089/089771504774129856. [DOI] [PubMed] [Google Scholar]
- Kozorovitskiy Y, Gross CG, Kopil C, Battaglia L, McBreen M, Stranahan AM, Gould E. Experience induces structural and biochemical changes in the adult primate brain. Proc Natl Acad Sci U S A. 2005;102:17478–17482. doi: 10.1073/pnas.0508817102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar R, Husain M, Gupta RK, Hasan KM, Haris M, Agarwal AK, Pandey CM, Narayana PA. Serial changes in the white matter diffusion tensor imaging metrics in moderate traumatic brain injury and correlation with neuro-cognitive function. J Neurotrauma. 2009;26:481–495. doi: 10.1089/neu.2008.0461. [DOI] [PubMed] [Google Scholar]
- Lamprecht R, LeDoux J. Structural plasticity and memory. Nat Rev Neurosci. 2004;5:45–54. doi: 10.1038/nrn1301. [DOI] [PubMed] [Google Scholar]
- Lewen A, Li GL, Olsson Y, Hillered L. Changes in microtubule-associated protein 2 and amyloid precursor protein immunoreactivity following traumatic brain injury in rat: influence of MK-801 treatment. Brain Res. 1996;719:161–171. doi: 10.1016/0006-8993(96)00081-9. [DOI] [PubMed] [Google Scholar]
- Lusardi TA, Wolf JA, Putt ME, Smith DH, Meaney DF. Effect of acute calcium influx after mechanical stretch injury in vitro on the viability of hippocampal neurons. J Neurotrauma. 2004;21:61–72. doi: 10.1089/089771504772695959. [DOI] [PubMed] [Google Scholar]
- Lyeth LG, Jenkins LW, Hamm RJ, Dison CE, Phillips LL, Clifton GL, Young HF, Hayes RL. Prolonged memory impairment in the absence of hippocampal cell-death following traumatic brain injury in the rat. Brain Res. 1990;526:249–258. doi: 10.1016/0006-8993(90)91229-a. [DOI] [PubMed] [Google Scholar]
- Maxwell WL, Graham DI. Loss of axonal microtubules and neurofilaments after stretch-injury to guinea pig optic nerve fibers. J Neurotrauma. 1997;14:603–614. doi: 10.1089/neu.1997.14.603. [DOI] [PubMed] [Google Scholar]
- Maxwell WL, Povlishock JT, Graham DI. A mechanistic analysis of non-disruptive axonal injury: a review. J Neurotrauma. 1997;14:419–440. doi: 10.1089/neu.1997.14.419. [DOI] [PubMed] [Google Scholar]
- Meaney DF, Smith DH, Shreiber DI, Bain AC, Miller RT, Ross DT, Gennarelli TA. Biomechanical analysis of experimental diffuse axonal injury. J Neurotrauma. 1995;12:689–694. doi: 10.1089/neu.1995.12.689. [DOI] [PubMed] [Google Scholar]
- Mizrahi A, Libersat F. Afferent input regulates the formation of distal dendritic branches. J Comp Neurol. 2002;452:1–10. doi: 10.1002/cne.10275. [DOI] [PubMed] [Google Scholar]
- Miyazaki Y, Katayama Y, Lyeth BG, Jenkins LW, Dewitt DS, Goldberg SJ, Newlon PG. Enduring suppression of hippocampal long-term potentiation following traumatic brain injury in rat. Brain Res. 1992;585:335–339. doi: 10.1016/0006-8993(92)91232-4. [DOI] [PubMed] [Google Scholar]
- Oliva AA, Lam TT, Swann JW. Distally directed dendrotoxicity induced by kainic acid in hippocampal interneurons of green fluorescent protein-expressing transgenic mice. J Neurosci. 2002;22:8052–8062. doi: 10.1523/JNEUROSCI.22-18-08052.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JS, Bateman MC, Goldberg MP. Rapid alterations in dendrite morphology during sublethal hypoxia or glutamate receptor activation. Neurobiol Dis. 1996;3:215–227. doi: 10.1006/nbdi.1996.0022. [DOI] [PubMed] [Google Scholar]
- Pike BR, Zhao X, Newcomb JK, Glenn CC, Anderson DK, Hayes RL. Stretch injury causes calpain and caspase-3 activation and necrotic and apoptotic cell death in septo-hippocampal cell cultures. J Neurotrauma. 2000;17:283–298. doi: 10.1089/neu.2000.17.283. [DOI] [PubMed] [Google Scholar]
- Posmantur R, Hayes RL, Dixon E, Taft WC. Neurofilament 68 and neurofilament 200 protein levels decrease after traumatic brain injury. J Neurotrauma. 1994;11:533–545. doi: 10.1089/neu.1994.11.533. [DOI] [PubMed] [Google Scholar]
- Posmantur RM, Kampfl A, Taft WC, Bhattacharjee M, Dixon CE, Bao J, Hayes RL. Diminished microtubule-associated protein 2 (MAP2) immunoreactivity following cortical impact brain injury. J Neurotrauma. 1996;13:125–137. doi: 10.1089/neu.1996.13.125. [DOI] [PubMed] [Google Scholar]
- Posmantur R, Kampfl A, Siman R, Liu SJ, Zhao X, Clifton GL, Hayes RL. A calpain inhibitor attenuates cortical cytoskeletal protein loss after experimental traumatic brain injury in the rat. Neuroscience. 1997;77:875–888. doi: 10.1016/s0306-4522(96)00483-6. [DOI] [PubMed] [Google Scholar]
- Posmantur R, Newcomb JK, Kampfl A, Hayes RL. Light and confocal microscopic studies of evolutionary changes in neurofilament proteins following cortical impact injury in the rat. Exp Neurol. 2000;161:15–26. doi: 10.1006/exnr.1999.7244. [DOI] [PubMed] [Google Scholar]
- Povlishock JT. Traumatically induced axonal injury: pathogenesis and pathobiological implications. Brain Pathol. 1992;2:1–12. [PubMed] [Google Scholar]
- Povlishock JT, Marmarou A, McIntosh TK, Trojanowski JQ, Moroi J. Impact acceleration injury in the rat: evidence for focal axolemmal change and related neurofilament sidearm alteration. J Neuropathol Exp Neurol. 1997;56:347–359. [PubMed] [Google Scholar]
- Rasmusson DX, Brandt J, Martin DB. Head injury as a risk factor in Alzheimer's disease. Brain Inj. 1995;3:213–219. doi: 10.3109/02699059509008194. [DOI] [PubMed] [Google Scholar]
- Rowntree S, Kolb B. Blockade of basic fibroblast growth factor retards recovery from motor cortex injury in rats. Eur J Neurosci. 1997;9:2432–2441. doi: 10.1111/j.1460-9568.1997.tb01660.x. [DOI] [PubMed] [Google Scholar]
- Saatman KE, Bozyczko-Coyne D, Marcy V, Siman R, McIntosh TK. Prolonged calpain-mediated spectrin breakdown occurs regionally following experimental brain injury in the rat. J Neuropathol Exp Neurol. 1996;55:850–860. doi: 10.1097/00005072-199607000-00010. [DOI] [PubMed] [Google Scholar]
- Saatman KE, Graham DI, McIntosh TK. The neuronal cytoskeleton is at risk after mild and moderate brain injury. J Neurotrauma. 1998;15:1047–1058. doi: 10.1089/neu.1998.15.1047. [DOI] [PubMed] [Google Scholar]
- Scheid R, Walther K, Guthke T, Preul C, von Cramon DY. Cognitive sequelae of diffuse axonal injury. Arch Neurol. 2006;63:418–424. doi: 10.1001/archneur.63.3.418. [DOI] [PubMed] [Google Scholar]
- Sherriff FE, Bridges LR, Sivaloganathan S. Early detection of axonal injury after human head trauma using immunocytochemistry for beta-amyloid precursor protein. Acta Neuropathol. 1994;87:55–62. doi: 10.1007/BF00386254. [DOI] [PubMed] [Google Scholar]
- Sjostrom PJ, Rancz EA, Roth A, Hausser M. Dendritic excitability and synaptic plasticity. Physiol Rev. 2008;88:769–840. doi: 10.1152/physrev.00016.2007. [DOI] [PubMed] [Google Scholar]
- Sloviter RS, Dempster DW. Epileptic brain damage is replicated qualitatively in the rat hippocampus by central injection of glutamate or aspartate but not by GABA or acetylcholine. Brain Res Bull. 1985;15:39–60. doi: 10.1016/0361-9230(85)90059-0. [DOI] [PubMed] [Google Scholar]
- Smith DH, Meaney DF. Axonal damage in traumatic brain injury. Neuroscientist. 2000;6:483–495. [Google Scholar]
- Smith DH, Okiyama K, Thomas MJ, Claussen B, McIntosh TK. Evaluation of memory dysfunction following experimental brain injury using the Morris water maze. J Neurotrauma. 1991;8:259–269. doi: 10.1089/neu.1991.8.259. [DOI] [PubMed] [Google Scholar]
- Smith DH, Okiyama K, Gennarelli TA, McIntosh TK. Magnesium and ketamine attenuate cognitive dysfunction following experimental brain injury. Neurosci Lett. 1993;157:211–214. doi: 10.1016/0304-3940(93)90739-8. [DOI] [PubMed] [Google Scholar]
- Smith DH, Wolf JA, Lusardi TA, Lee VMY, Meaney DF. High tolerance and delayed elastic response of cultured axons to dynamic stretch injury. J Neurosci. 1999;19:4263–4269. doi: 10.1523/JNEUROSCI.19-11-04263.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Squire LR, Zola-Morgan S. The medial temporal-lobe memory system. Science. 1991;253:1380–1386. doi: 10.1126/science.1896849. [DOI] [PubMed] [Google Scholar]
- Stewart GR, Olney JW, Pathikonda M, Snider WD. Excitotoxicity in the embryonic chick spinal cord. Ann Neurol. 1991;30:758–760. doi: 10.1002/ana.410300604. [DOI] [PubMed] [Google Scholar]
- Taft WC, Yang K, Dixon CE, Hayes RL. Microtubule-associated protein 2 levels decrease in hippocampus following traumatic brain injury. J Neurotrauma. 1992;9:281–290. doi: 10.1089/neu.1992.9.281. [DOI] [PubMed] [Google Scholar]
- Tang MD, Golden AP, Tien J. Molding of three-dimensional microstructures of gels. J Am Chem Soc. 2003;125:12988–12989. doi: 10.1021/ja037677h. [DOI] [PubMed] [Google Scholar]
- Tang-Schomer MD, Patel AR, Baas PW, Smith DH. Mechanical breaking of microtubules in axons during dynamic stretch injury underlies delayed elasticity, microtubule dissassembly, and axon degeneration. FASEB J. 2010;24:1401–1410. doi: 10.1096/fj.09-142844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor AM, Blurton-Jones M, Rhee SW, Cribbs DH, Cotman CW, Jeon NL. A microfluidic culture platform for CNS axonal injury, regeneration and transport. Nat Methods. 2005;2:599–605. doi: 10.1038/nmeth777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valverde F. Structural changes in the area striata of the mouse after enucleation. Exp Brain Res. 1968;5:274–292. doi: 10.1007/BF00235903. [DOI] [PubMed] [Google Scholar]
- Van Den Heuvel C, Thornton E, Vink R. Neurotrauma: new insights into pathology and treatment. Prog Brain Res. 2007;161:303–316. doi: 10.1016/S0079-6123(06)61021-2. [DOI] [PubMed] [Google Scholar]
- Von Reyn CR, Spaethling JM, Mesfin MN, Ma M, Neumar RW, Smith DH, Siman R, Meaney DF. Calpain mediates proteolysis of the voltage-gated sodium channel α-subunit. J Neurosci. 2009;29:10350–10356. doi: 10.1523/JNEUROSCI.2339-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallesch CW, Curio N, Kutz S, Jost S, Bartels C, Synowitz H. Outcome after mild-to-moderate blunt head injury: effects of focal lesions and diffuse axonal injury. Brain Inj. 2001;15:401–412. doi: 10.1080/02699050010005959. [DOI] [PubMed] [Google Scholar]
- Winocur G. Anterograde and retrograde-amnesia in rats with dorsal hippocampal or dorsomedial thalamic lesions. Behav Brain Res. 1990;38:145–154. doi: 10.1016/0166-4328(90)90012-4. [DOI] [PubMed] [Google Scholar]
- Wolf JA, Stys PK, Lusardi T, Meaney D, Smith DH. Traumatic axonal injury induces calcium influx modulated by tetrodotoxin-sensitive sodium channels. J Neurosci. 2001;21:1923–1930. doi: 10.1523/JNEUROSCI.21-06-01923.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuen TJ, Browne KD, Iwata A, Smith DH. Sodium channelopathy induced by mild axonal trauma worsens outcome after a repeat injury. J Neurosci Res. 2009;87:3620–3625. doi: 10.1002/jnr.22161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zepeda A, Sengpiel F, Guagnelli MA, Vaca L, Arias C. Functional reorganization of visual cortex maps after ischemic lesions is accompanied by changes in expression of cytoskeletal proteins and NMDA and GABAA receptor subunits. J Neurosci. 2004;24:1812–1821. doi: 10.1523/JNEUROSCI.3213-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Rzigalinski BA, Ellis EF, Satin LS. Reduction of voltage-dependent Mg2+ blockage of NMDA current in mechanically injured neurons. Science. 1996;274:1921–1923. doi: 10.1126/science.274.5294.1921. [DOI] [PubMed] [Google Scholar]
- Zhu J, Hamm RJ, Reeves TM, Povlishock JT, Phillips LL. Postinjury administration of L-deprenyl improves cognitive function and enhances neuroplasticity after traumatic brain injury. Exp Neurol. 2000;166:136–152. doi: 10.1006/exnr.2000.7484. [DOI] [PubMed] [Google Scholar]