

Abstract
Interactions of cytochrome c (cyt c) with a unique mitochondrial glycerophospholipid cardiolipin (CL) are relevant for the protein’s function in oxidative phosphorylation and apoptosis. Binding to CL-containing membranes promotes cyt c unfolding and dramatically enhances the protein’s peroxidase activity, which is critical in early stages of apoptosis. We have employed a collection of seven dansyl variants of horse heart cyt c to probe the sequence of steps in this functional transformation. Kinetic measurements have unraveled four distinct processes during CL-induced cyt c unfolding: rapid protein binding to CL liposomes; rearrangements of protein substructures with small unfolding energies; partial insertion of the protein into the lipid bilayer; and extensive protein restructuring leading to “open” extended structures. While early rearrangements depend on a hierarchy of foldons in the native structure, the later process of large-scale unfolding is influenced by protein interactions with the membrane surface. The opening of the cyt c structure exposes the heme group, which enhances the protein’s peroxidase activity and also frees the C-terminal helix to aid in the translocation of the protein through CL membranes.
Keywords: protein folding, foldons, membrane, heme, fluorescence, apoptosis
Introduction
A small heme protein cytochrome c (cyt c) has become a true workhorse for studies of protein folding. Its investigations have not only uncovered intricacies of the cyt c folding mechanism but also shaped general concepts in the field and served as a valuable platform for the development of new methods for triggering and probing folding reactions.1-14 Among the key findings, hydrogen-deuterium amide exchange experiments revealed that the cyt c structure consists of five cooperative units with different thermodynamic stability (foldons), which undergo folding and unfolding in solution in a stepwise, sequential mechanism (Figure 1).15 The terminal N- and C- helices of cyt c are the regions of greatest stability and contact between them has been observed in early folding intermediates of cyt c.16 The loop carrying the heme ligand Met80 is the least stable substructure and folds last.15 The foldon model has been tested with experiments,8, 17, 18 as well as theoretical simulations,19 and both showed consistent results.
Figure 1.
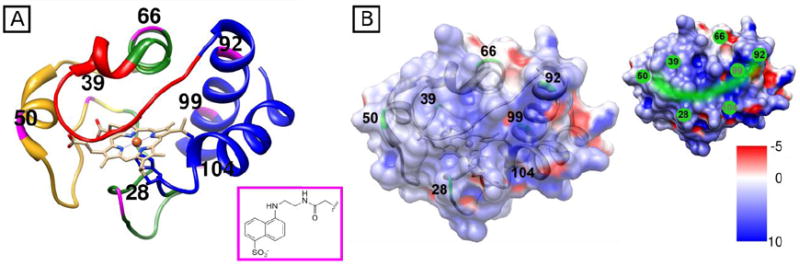
Structure of horse heart cyt c (PDB 1HRC)58 showing (A) color-coded foldons and positions of Dns labels (pink); and (B) electrostatic surface map as well as the likely site (green) for primary interactions with CL liposomes, the area that encompasses a prominent positively charged patch on the protein surface. Images created with Chimera 1.5.3.42
Unfolding of cyt c by acid, urea, and guanidine hydrochloride (GuHCl) has been extensively investigated.20-24 In addition to these common denaturants, cyt c binding to detergent micelles,25, 26 as well as anionic polymers and lipid membranes,27, 28 destabilizes the native protein structure. Since the prime function of cyt c is in mitochondria,29 studies of cyt c-membrane interactions not only allow one to address interesting biophysical questions but also have important biological implications.
The discovery of the role of altered cyt c conformations in apoptosis has renewed interest in lipid-induced unfolding reactions of this protein. Interactions of cyt c with the negatively-charged mitochondrial lipid cardiolipin (CL) convert the protein into a peroxidase capable of oxidizing CL itself.30, 31 This activity has been linked to the apoptotic release of cyt c into the cytosol.30 The topic of cyt c - CL interactions has become a vibrant area of research: structural features of CL-bound cyt c, the role of ionic strength and ATP, as well as effects of the interaction on the membrane morphology have been explored.32-38 Collectively, the studies have pointed to disruption of the protein tertiary structure and loss of Met80-heme coordination upon interaction with CL membrane surfaces, but the exact mechanism of the polypeptide transformation remained uncertain.
Recent fluorescence studies in our laboratory have identified primary surface sites on cyt c for interaction with CL liposomes (Figure 1B).35, 39 Furthermore, analyses of dye-to-heme distance distributions P(r) from time-resolved FRET (TR-FRET) experiments have revealed that CL-bound cyt c is a heterogeneous ensemble of interconverting species that vary in their degree of polypeptide compactness around the heme.35, 40 These studies have provided evidence for massive cyt c unfolding, with the breakup of critical stabilizing contacts between the N- and C-terminal helices, for at least a fraction of the polypeptide ensemble.
Herein, we employ a collection of seven dansyl (Dns)-labeled variants of horse heart cyt c (Figure 1A) to characterize the sequence of cyt c structural transitions induced by interactions with CL membranes. The labeling sites were chosen to explore the roles of the native-structure foldons as well as the effects of the protein establishing new interactions with the lipid surface. By monitoring Dns quenching by the heme, our kinetics experiments have uncovered distinct steps of cyt c unfolding that ultimately lead to the peroxidase-active species.
Materials and Methods
General
Distilled water was demineralized to a resistivity greater than 18.2 MΩ•cm. Compounds 4-(2-Hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) and guanidine hydrochloride (GuHCl, ultrapure) were purchased from Alfa Aesar and MP Biomedicals, respectively. Isolated from horse heart, wild-type cyt c was obtained from Sigma-Aldrich. All other chemicals were obtained from Fisher Scientific. HEPES buffer and GuHCl solutions were stored at 4 °C. Protein concentrations were determined spectrophotometrically using molar absorptivities of 106,000 M-1cm-1 at 409 nm and 129,000 M-1cm-1 at 416 nm for ferric and ferrous variants,41 respectively. Molecular graphics and data analyses were performed with Chimera (UCSF)42 and MATLAB (Mathworks). Chimera was developed by the UCSF Resource for Biocomputing, Visualization, and Informatics and funded by grants from the National Institutes of Health National Center for Research Resources (2P41RR001081) and National Institute of General Medical Sciences (9P41GM103311).42
CL Liposomes
Chloroform stock solutions of 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) and 1,1’,2,2’-tetraoleoylcardiolipin (TOCL) were obtained from Avanti Polar Lipids. DOPC and TOCL were mixed in 1:1 molar ratios in glass vials and dried under nitrogen stream with occasional manual rotation of the vial until a thin, dry lipid film was formed. Afterwards, the lipids were resuspended in a freshly prepared 25 mM HEPES buffer at pH 7.4 to a final concentration of 2.2 mM. The suspension was vortexed and then incubated for 30 min at 37 °C at 220 rpm. Bath sonication (Branson) followed for one hour to break up large liposomes into smaller vesicles. The milky solution was extruded eleven times through a 100 nm pore diameter polycarbonate membrane (SPI supplies). Vesicle solutions were stored at room temperature and used within five days. Dynamic light scattering (DLS, DynaPro Nanostar, Wyatt Technology) measurements confirmed the homogenous size distribution of the extruded vesicles. During the storage time, no evidence of vesicle aggregation was observed by DLS. Fluorescent liposomes were prepared following the same protocol except they were shielded from light after adding 0.35% of 1-Oleoyl-2-[12-[(7-nitro-2-1,3-benzoxadiazol-4-yl)amino]dodecanoyl]-sn-Glycero-3-Phosphocholine (18:1-12:0 NBD PC, Avanti Polar Lipids).
Dns-Labeled Cyt c Derivatives
Protein expression, Dns-labeling, purification, and characterization of Dns-derivatives have been described previously.35, 39A new variant, Dns28, was prepared in this work; its spectra in native, CL-bound, and GuHCl-unfolded states, as well as equilibrium unfolding parameters are shown in Figure S1.
Centrifugation Assays
Centrifugation assays were conducted to compare the binding behavior of wild-type horse heart cyt c or Dns variants to liposomes (50 mol% CL). Solutions of liposomes ranging in total lipid concentrations from 0 μM to 300 μM and protein were mixed by pipetting in a 2.4:1 vol ratio. All samples were incubated for 45 min at room temperature and then centrifuged for 60 min using a Beckman Airfuge tabletop ultracentrifuge with a Beckman A-11 rotor at 120,000×g. After the centrifugation, the supernatants were removed immediately and their absorption spectra were measured in order to determine the concentration of unbound protein. The protein concentrations used for the assays were between 3.4 μM and 4.9 μM.
Spectroscopic Measurements
Absorption and fluorescence spectra were measured with an Agilent 8453 spectrophotometer and a Horiba Jobin Yvon Fluorolog 3 spectrofluorimeter, respectively. Signals from scattering of liposome solutions were removed using a blank spectrum. For steady-state and longer time-scale (60 min) fluorescence experiments (λex=336 nm, λem=400-600 nm), all samples were deoxygenated with argon prior to the measurements. Fluorescence lifetimes for samples at later stages (≥1 min) of unfolding were measured by time-correlated single photon counting (TCSPC) at 1,000 counts using a NanoLED-375L diode laser (λex=375 nm, <70 ps pulsewidth) as the excitation source and a TBX-04 detector for the detection of Dns emission at 500 nm. All fluorescence lifetime measurements were done under magic angle conditions and analyzed as previously described.35 The instrument response function (IRF) was measured by using a diluted LUDOX® AS-40 colloidal silica solution (Sigma Aldrich).
Stopped-Flow Fluorescence Experiments
Measurements were performed at room temperature with a BioLogic SF-300/S stopped-flow mixer. A 150 W Xenon-Mercury lamp was used as the excitation source. The excitation wavelengths (λex) of 365 and 436 nm for Dns and NBD, respectively, were selected with a Horiba Jobin Yvon H10-61 UV monochromator. Long-pass filters ≥400 nm and ≥500 nm were used to select the emission of Dns and NBD, respectively. A 0.8 mm (FC-08) cuvette was employed. The flow rate of 6 mL/s for all experiments resulted in an instrument dead-time of 5.1 ms. Between five and fifteen transients, each with 6000 data points, were collected and averaged for every kinetic trace reported. Multiple experiments conducted on different days with freshly purified proteins confirmed reproducibility of the reported data. The protein concentration in all experiments was 3 μM.
Results and Discussion
Dns-labeled Variants and their Interactions with CL Liposomes
Seven Dns variants of cyt c were employed in this work (Figure 1A). The labeling sites were all surface exposed in the native protein and probed different foldon units of cyt c (Table 1). Labeling of cyt c with a Dns chromophore at these sites introduced no detectable perturbations in the protein secondary structure or the environment of the heme group.35, 39 Moreover, the combined effects of Cys mutation and Dns labeling only minimally altered the stability of the protein. The midpoints of the unfolding transition [GuHCl]1/2 decreased by 0.1-0.3 M for all the variants in comparison to the value 2.7±0.1 M for the wild-type protein.35, 39 These findings show that the extent of destabilization was the same for all the variants. Centrifugation pelleting experiments were conducted in order to assess if the Dns label interferes with the protein binding to liposomes. For all the variants, the observed binding behavior was comparable to that of the wild-type protein (Figure S2).
Table 1.
Dns-labeled Variants, Foldons, and Characteristics of their Native, CL-bound and Unfolded States from Integrated Fluorescence and TR-FRET measurementsa
| Variant | Substructure Probed | Foldonb | Unit’s Unfolding ΔG (kcal/mol)b | CαD - FeA Distance (Å)c | Dns-Heme Distances (Å)d
|
λmax (nm)h
|
|||
|---|---|---|---|---|---|---|---|---|---|
| Ne | CLf | UGuHClg | Ne | CLf | |||||
| Dns28 | Loop A | Green | 10 | 8.4 | n.d.i | n.d.i | n.d.i | 490 | 500 |
| Dns39 | Loop B | Nested Yellow | 6 | 14.8 | 21 | 23 | 29 | 493 | 493 |
| Dns50 | 50’s helix / loop C | Nested Yellow | 6 | 15.1 | 23 | 33 | 41 | 493 | 484 |
| Dns66 | 60’s helix | Green | 10 | 13.8 | 20 | 30 | 51 | 488 | 489 |
| Dns92 | C-helix | Blue | 12.8 | 16.3 | 24 | 40 | 52 | 494 | 480 |
| Dns99 | C-helix | Blue | 12.8 | 15.2 | 23 | 29 | 50 | 479 | 490 |
| Dns104 | C-helix | Blue | 12.8 | 18.9 | 23 | 35 | 46 | 492 | 490 |
Measured at 2 hours after mixing.
From NMR NHX experiments.15
Calculated from the crystal structure, 1HRC.58 The Dns fluorophore adds about 6 to 8 Å to this distance.59
Given by the first moment of the experimental P(r) distributions from TR-FRET measurements. At distances longer than 1.5×R0=59 Å, energy transfer rate constants and D-A distances cannot be determined reliably; the structures with r ≥ 1.5 R0 are treated as having r=1.5×R0 and thus experimental distributions P(r) for more distant labeling sites yield apparent distances shorter than separation between Dns and the heme in the actual polypeptides.35, 59
In a 25 mM HEPES buffer at pH 7.4.
With TOCL/DOPC liposomes (50 mol% CL, 750 μM total lipid) in a 25 mM HEPES buffer at pH 7.4 at 3 μM cyt c.
In 6 M GuHCl at pH 7.4.
Dns emission maxima λmax for all the variants in the GuHCl-denatured state were 500±2 nm.
High degree of Dns fluorescence quenching in Dns28 did not permit detection of TR-FRET decays with our current instrumentation.
Our prior studies examined the equilibrium CL-bound state of six of these seven variants (excluded Dns28).35, 39 The Dns label remained solvent-exposed (emission maximum λmax=500 nm) in CL-bound Dns28 (Figure S1), suggesting that this site is located away from the liposome surface. This new result is consistent with our previously proposed model of the primary protein surface area in contact with CL-liposomes (Figure 1B),39 where the protein regions near residues 50 and 92 are partially embeded into the membrane. The Dns flurescence is quenched by the heme in native variants owing to the proximity of the two groups (Table 1). Both cyt c binding to CL and protein unfolding in 6 M GuHCl increase Dns integrated fluorescence for all the variants, but the magnitude of the change is less for the former process.35, 39 Analyses of TR-FRET decays revealed the coexistence of compact and extended polypeptide species in the CL-bound cyt c ensemble, providing an explanation for only a partial increase in Dns fluorescence upon unfolding of cyt c induced by CL.35
Kinetics of Dns Fluorescence Change: Site Variations
The kinetics of the changes in Dns fluorescence upon mixing the variants with CL liposomes have been investigated on both short (60 s) and long (60 min) timescales. Measurements of integrated Dns fluorescence revealed distinct variations in the overall kinetics for the seven labeled proteins. The entire process can be described by a triexponential function. The site differences are particularly apparent on the short timescale in both the size of the “burst” amplitude and the amplitude a1 of the first observable phase (Table 2 and Figure 2). On the long timescale, when major increases in the Dns fluorescence intensities occur, two groups of sites with distinct behavior are observed: ones that unfold fast (Dns28, Dns39, Dns99, and Dns104) and others that unfold slower (Dns50, Dns66, and Dns92) (Figure 3). The rate constants k2 of the second phase are the same for all the variants; global fits of all seven kinetic traces yielded k2=0.04 s-1.
Table 2.
Normalized Intensities, Rates, and Relative Amplitudes of Different Phases for Changes in Dns Fluorescence during Cyt c Unfolding Induced by Interactions with CL-containing Liposomesa,b
| Variant | Relative Intensities
|
Phase 1
|
Phase 2c
|
Phase 3
|
||||
|---|---|---|---|---|---|---|---|---|
| Iburst/IU (%) | /IU (%) | /IU (%) | k1 (s-1) | a1, vscld (%) | a2, vscld (%) | k3×103 (s-1) | a3, vscld (%) | |
| Dns28 | 12.5 | 23.2 | 34.5 | 0.43 ± 0.27 | 21.4 | 48.5 | 3.3 ± 0.1 | 30.1 |
| Dns39 | 12.0 | 21.0 | 28.3 | 0.61 ± 0.09 | 31.0 | 40.2 | 4.4 ± 1.5 | 28.8 |
| Dns50 | 9.8 | 22.0 | 51.3 | 0.52 ± 0.02 | 23.8 | 17.7 | 0.9 ± 0.1 | 58.5 |
| Dns66 | 4.1 | 14.5 | 47.4 | 0.30 ± 0.01 | 11.8 | 16.0 | 0.9 ± 0.1 | 72.2 |
| Dns92 | 1.4 | 9.7 | 44.6 | 0.35 ± 0.01 | 7.3 | 9.7 | 1.2 ± 0.1 | 83.0 |
| Dns99 | 3.5 | 19.3 | 31.5 | 0.71 ± 0.03 | 16.0 | 37.1 | 5.7 ± 0.9 | 46.9 |
| Dns104 | 2.7 | 10.3 | 40.7 | 0.90 ± 0.05 | 3.1 | 7.1 | 3.2 ± 0.3 | 89.8 |
TOCL/DOPC liposomes (vscl, 50 mol% CL).
Determined with a combination of stopped-flow (Phases 1 and 2) and manual (Phase 3) mixing experiments in a 25 mM HEPES pH 7.4 at final concentrations of 3 μM of Dns-labeled cyt c and 750 μM of total lipid. All measurements were performed at 22±2 °C.
Kinetics progress curves for all the variants during Phase 2 could be fit to the process with the rate constant k2 having values between 0.03 and 0.04 s-1.
Percent of the observable amplitude for the Dns fuorescence signal during 60 min after mixing (a1, vscl + a2, vscl + a3, vscl=100%, the burst phase is not included).
Figure 2.
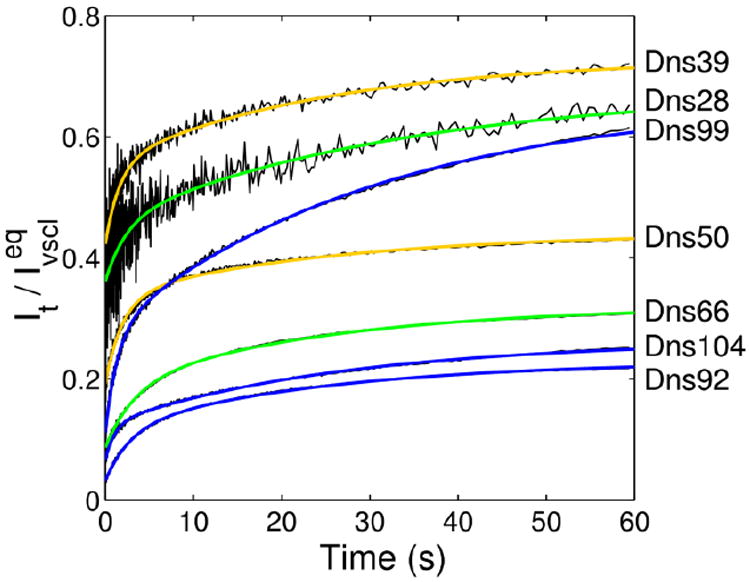
Changes in Dns fluorescence intensity (λex=365 nm, λem ≥ 400 nm) during the first 60 s after stopped-flow mixing of TOCL/DOPC liposomes (vscl, 50 mol% CL) with Dns-labeled cyt c in a 25 mM HEPES buffer at pH 7.4. Final concentrations after mixing were 3 μM cyt c and 750 μM total lipid. The Dns intensity of the native protein is subtracted from all the traces. Plotted are kinetics traces normalized to the respective intensities of the CL-bound proteins at equilibrium (measured 60 min after mixing). Kinetics traces normalized to the respective intensities of GuHCl-unfolded proteins are shown in Figure S3 in the Supporting Information. Colors of the fit lines correspond to accepted colors of the foldon units associated with these labeling sites.15
Figure 3.
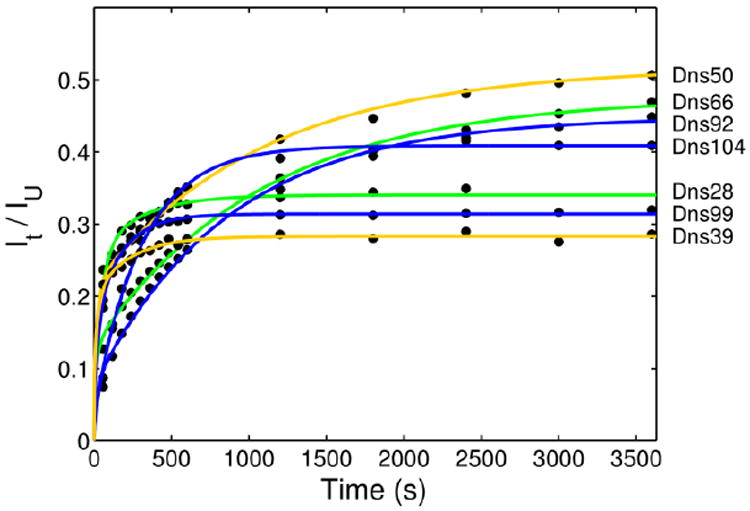
Changes in Dns fluorescence intensity (λex=336 nm, λem ≥ 400 nm) during the first 60 min after manual mixing of TOCL/DOPC liposomes (vscl, 50 mol% CL) with Dns-labeled cyt c in a 25 mM HEPES buffer at pH 7.4. Final concentrations after mixing were 3 μM cyt c and 750 μM total lipid. Both protein and liposome solutions were thoroughly deoxygenated with argon prior to mixing, and the cuvette was sealed during experiments to avoid photobleaching. The Dns intensity of the native protein is subtracted from all the traces. Plotted are kinetics traces normalized to the respective intensities of GuHCl-unfolded proteins. Colors of the fit lines correspond to accepted colors of the foldon units associated with these labeling sites.15
Protein-Liposome Binding and Early Rearrangements
The burst amplitudes in our kinetics fluorescence measurements indicate that some structural changes take place in cyt c during the stopped-flow dead-time (5.1 ms). We hypothesize that protein-liposome binding also occurs during this time and that the initial protein-liposome docking process precedes the protein conformational rearrangements. Two types of experiments provide support for this sequence. First, experiments with fluorescent NBD-labeled liposomes and wild-type cyt c have revealed dramatic quenching of the NBD-fluorescence by the heme within the stopped-flow dead-time (< 5.1 ms) (Figure 4). Second, the concentration dependence of the kinetics was investigated with one of the highly fluorescent mutants, Dns104, by varying the concentration of liposomes (Figure 5). The conditions yielded identical kinetics but varying magnitudes of the burst phase and observed fluorescent signal. These findings confirm that the binding event is fast and highlight the importance of the available large surface area for cyt c-liposome binding and unfolding, in accord with our previous report.40 The millisecond (or less) timescale of binding is consistent with the estimate of Pinheiro et al.27
Figure 4.
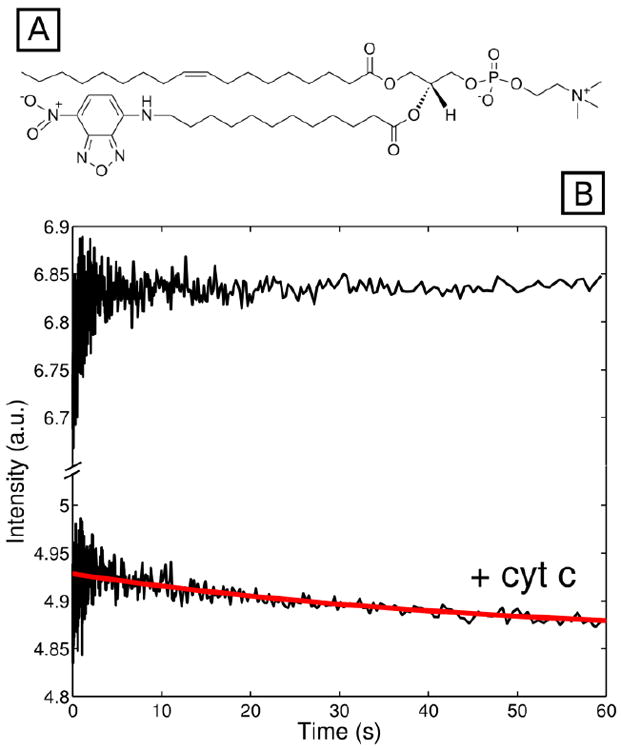
(A) Structure of the NBD-labeled PC used to prepare fluorescent liposomes. (B) NBD fluorescence signal from labeled TOCL/DOPC liposomes (λex=436 nm, λem ≥ 500 nm) in a 25 mM HEPES buffer at pH 7.4 after stopped-flow mixing with the same buffer (top) and wild-type cyt c (bottom). The final total lipid concentration in both experiments was 750 μM. The red line is a fit of the decrease in the NBD signal to a monoexponential decay function with a rate constant of 0.04 s-1.
Figure 5.
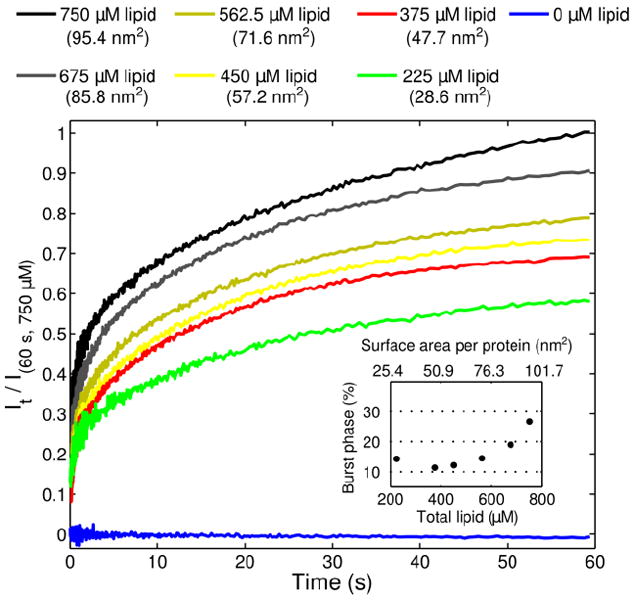
Changes in Dns fluorescence intensity (λex=365 nm, λem ≥ 400 nm) during the first 60 s after stopped-flow mixing of TOCL/DOPC liposomes (50 mol% CL) with the Dns104 cyt c variant in a 25 mM HEPES buffer at pH 7.4. Final concentration of cyt c after mixing was 3 μM and concentrations of total lipid were varied as indicated. All kinetics traces are normalized to the signal at 60 s observed with final concentration of the total lipid of 750 μM. Every trace (except for 0 μM lipid) can be fit to a biexponential function with the same rate constants and amplitudes as with 750 μM total lipid (Table 2). Inset: the increase in the burst phase at higher lipid concentrations; correlation with the surface area is also provided.
The burst phase varies appreciably for the seven variants (Figure 2). The variants Dns28, Dns39, and Dns50, with reporter labels at the lower stability nested-yellow and green foldons, show the largest signal increase (I/Ivscl) within this short time. A recent study suggests a possible trigger for this change: disruption of His26-Pro44 hydrogen bonding initiates rearrangements in the residue 37-61 loop and Met80 dissociation.36
The rate constants and their amplitudes for the first observable process, k1 and a1, also reflect site variations (Table 2 and Figure 2). The relative amplitudes of this phase a1 are small, particularly for the variants with labels at the end of the protein sequence, arguing against major protein unfolding at this stage. Also being independent of the liposome concentration and thus, available surface area (Figure 5), this phase likely represents an overall loosening of the native protein structure.
The higher amplitudes of this phase for Dns39 and Dns50, with labels at the nested-yellow foldon, suggest that loosening is most prominent for the loop containing residue 39 and the adjacent 50’s helix, in accord with the low ΔG of unfolding for this region (Table 1 and Figure 1A). The rapid fluorescence changes of Dns28, with a label at the more stable green foldon, may reflect a ruptured His26-Pro44 hydrogen bond at the early stages of cyt c interactions with CL liposomes. Probed by Dns66 (Figure 1A), the 60’s helix, another green-foldon structure, loosens slower and this transformation contributes less to the overall kinetics.
Since Dns92, Dns99, and Dns104 have probe labels within the blue foldon with the highest ΔG of unfolding (Table 1 and Figure 1A), the larger k1 values for Dns99 and Dns104 seem to be counterintuitive. This finding perhaps is related to the ease of rearrangement of the corresponding polypeptide units as they do not establish prominent interactions with the liposome surface (Figure 1B).39 On the other hand, changes during this phase for Dns92 are slow, suggesting that the protein has likely established interactions with the liposome next to this labeling site. Because the lowest-stability red foldon is directly upstream of the C-terminal helix, the observed decrease in Dns quenching by the heme and thus the increase in separation between the two chromophores for Dns92, Dns99, and Dns104, may largely reflect structural changes in the adjacent red foldon rather than rearrangements of the C-terminal helix itself. Small amplitudes for a1, particularly with respect to the cyt c GuHCl-unfolded state UGuHCl (Table 2 and Figure S3), are consistent with this hypothesis. The large burst phase for Dns39 and Dns50 variants (Figure 2) and the rapid disruption of the Met80-heme ligation linked to structural changes in the yellow foldon36, 43 provide additional support for this assignment. The reported rate constants of 0.7 s-1 and 1.8 s-1 for Met80-dissociation from the heme induced by anionic liposomes and CL,27, 44 respectively, are consistent with our proposal that changes in the red foldon occur before (burst phase) or during the first observable kinetic phase.
The behavior of the Dns99 variant is intriguing: compared to other C-terminal variants, its CL-bound state is the least fluorescent, yet structural rearrangements involving the Dns label at this site occur faster (Figures 2 and 3). The red shift of the Dns emisson maximum λmax is characteristic of a solvent exposure of this label in the CL-bound structures. This result suggests that rearrangement of this polypeptide fragment may not be impeded by protein interactions with liposomes (Table 1), offering an explanation for faster kinetics. However, it cannot be ruled out that labeling at this site may also have affected the CL-induced unfolding dynamics of this variant. The blue-shifted Dns λmax for the native protein variant suggests that the Dns label is located in the hydrophobic environment before CL binding. The native-state hydrophobic interaction, perhaps with Phe36 in the absence of the Lys99-Glu61 salt bridge for this variant, could be readily disrupted upon CL binding, before other changes in the C-terminal helix occur. Folding kinetics studies of the Dns99 variant of yeast iso-1 cyt c suggested that modification at this site does not change the folding dynamics of cyt c in solution,45 but there may be additional factors when CL liposomes are involved. Nevertheless, this variant provides information about the mechanism of CL-induced unfolding, especially at longer times when massive structural rearrangements of cyt c take place.
Site-Independent Phase: Cyt c Interactions with Lipid Membranes
The k2 rate constants of 0.03-0.04 s-1 for the second phase are independent of the Dns site location, suggesting a common process that limits the observed structural rearrangements. The independence of this phase on the liposome concentration (Figure 5) suggests that the available liposome surface is not critical for this process. This finding, as well as small a2 amplitudes for variants with the Dns label at the C-terminal helix (Table 2), argue against major protein unfolding at this stage.
Interestingly, a continuing decrease in the fluorescent signal of NBD-labeled vesicles following addition of cyt c on the 60 s timescale (Figure 4) can be fitted to a monoexponential decay with the same rate constant of 0.04 s-1. This finding suggests that the heme (and thus, the protein) gets closer to the lipid bilayer. This observation cannot be attributed to photobleaching since there is no corresponding decrease of the signal for fluorescent vesicles without adding the protein.
The prominent blue shifts of the emission maxima λmax in Dns50 and Dns92 (Table 1), indicative of the chromophore in the hydrophobic environment, can already be observed at 60 s (our earliest full-spectra measurement) after protein mixing with liposomes and these λmax positions do not change in subsequent measurements (Figure S4). These findings suggest that cyt c insertion into the lipid bilayer occurs during the first 60 s of the cyt c-CL interactions, consistent with our assignment of k2 to this process. This timescale also accords with results from studies of other protein-membrane interactions.46, 47
Major Protein Unfolding
Over the time span of an hour, a third phase for CL-induced structural changes in cyt c is observed (Figure 3). For many variants, particularly with labels at the end of the protein sequence, this is a major kinetic phase (Table 2). The progress curves (Figure 3) clearly illustrate that there are two groups of variants. The variants Dns50 and Dns92, with labels that partially insert into the lipid bilayer, and Dns66, with the labeling site close to residues 50 and 92 (the primary interaction site of cyt c with CL liposomes, Figure 1B), unfold slower than the other four variants. These results suggest that structural changes of the regions probed by Dns50, Dns66, and Dns92 are impeded by already established cyt c interactions with liposomes.
No additional blue shifts of the Dns emission maxima λmax can be observed during this phase (Figure S4) suggesting that no further protein insertion into the lipid bilayer occurs after the first 60 s. The Dns λmax position in the Dns99 variant, however, red-shifts (Figure S4), indicating an increase in the solvent exposure of this site. This change in the probe environment is consistent with protein unfolding into more “open” extended (E) structures as inferred from previous TR-FRET studies at equilibrium. A major increase in the Dns fluorescence signal for many of the variants supports a dramatic increase in Dns-to-heme distances during this phase.
Measurements of TR-FRET with highly-fluorescent Dns92 allowed us to examine changes in Dns-to-heme distance distributions P(r) during later stages of CL-induced unfolding (Figure 6). Results of these experiments clearly indicate that the population of E structures is indeed increasing during this phase (from 29% at ~60 s to 57% at ~20 min for Dns92). The sizable population of E conformers in Dns92 at 60 s as well as the red shift of the λmax position from that of the native protein in Dns99 (Figure S4) suggest that some of the large-scale protein unfolding has already occurred at this time.
Figure 6.
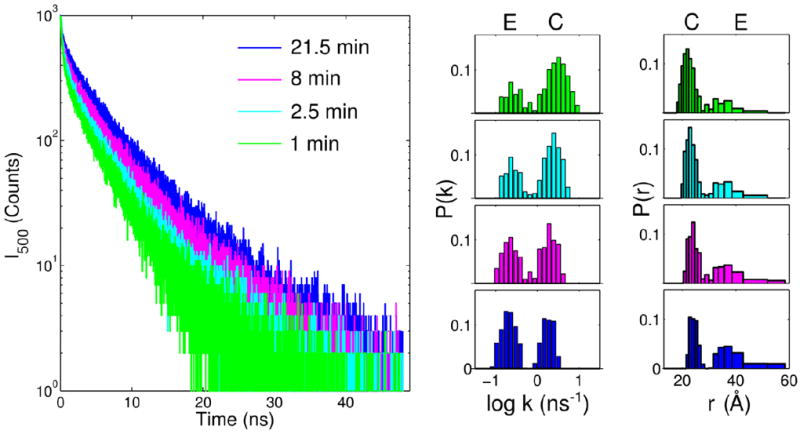
TR-FRET and extracted distributions of rate constants P(k) and Dns-heme distances P(r) for Dns92 at indicated times following manual mixing with TOCL/DOPC liposomes in a 25 mM HEPES buffer at pH 7.4. Final concentrations after mixing were 3 μM cyt c and 750 μM total lipid. Populations associated with extended (E) and compact (C) conformers are indicated.
In contrast to k1 and k2, the rate constant k3 shows a prominent dependence on the liposome concentration (Figure S5). The increase in k3 with increasing liposome concentrations suggests that a large liposome surface area may be important for mediating large-scale protein unfolding during this phase. With broken contacts between N- and C-terminal helices, the polypeptide is likely able to stretch out on the membrane surface. The recent observation of the two-dimensional cyt c “refolding” on solid surfaces provides a precedent for these types of protein structures.48 Coulombic interactions between the many positively charged residues in cyt c and the negatively-charged lipid surface provide plentiful opportunities for stabilizing unfolded cyt c. Our observation of partial protein insertion into CL liposomes preceding the formation of extended cyt c structures suggests that restriction of protein mobility by anchoring may facilitate dramatic polypeptide unfolding and favor new interactions with the liposome surface.
Multiple replicates of mixing experiments with different batches of liposome vesicles and freshly purified proteins confirmed the reproducibility of kinetic traces (Figures 2 and 3). At later times (>60 min), however, we have occasionally noticed small variations in the kinetics progress curves (Figure S6). Changes in the membrane morphology37 or mere photobleaching of the Dns dye could potentially affect the kinetics on this timescale. At this point, the exact origin of these deviations remains unknown, and we have not included these later time points in our analysis.
Mechanism of Cyt c Unfolding
Site-dependent kinetics described in this report together with known triggers of cyt c structural rearrangements and findings at equilibrium suggest the following scenario for CL-induced unfolding of cyt c (Figure 7). Electrostatic attraction between positively-charged cyt c and negatively-charged CL liposomes guides protein docking to the membrane.40, 49 This initial interaction occurs rapidly, during the dead time of the stopped-flow instrument (< 5.1 ms). A large patch of basic residues spans the surface of folded cyt c from Asp50 to the beginning of the C-terminal helix (Figure 1B). This region contains Lys residues 72, 73, 86, and 87, previously implicated in binding to CL liposomes,50, 51 as well as residues 50, 66, and 92, identified in our recent work.35, 39
Figure 7.
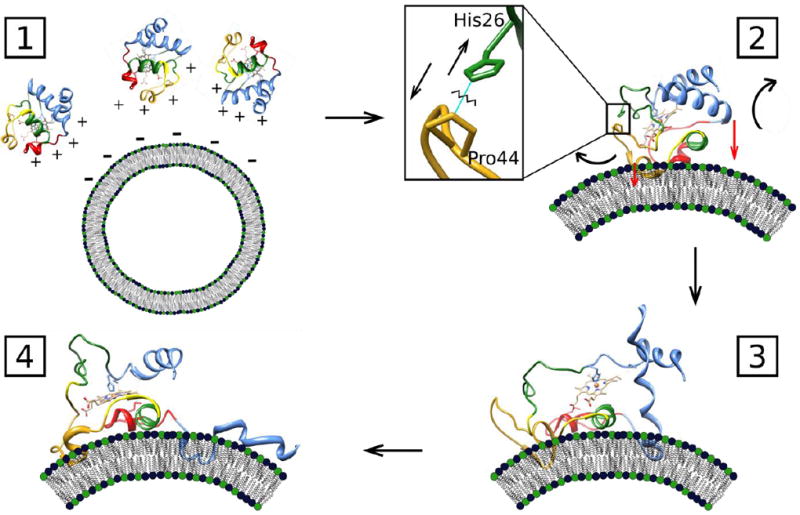
A cartoon depicting the proposed mechanism of cyt c unfolding upon contact with CL-containing liposomes. (1) Positively-charged cyt c and negatively-charged liposomes are attracted electrostatically, leading to binding of cyt c to the membrane within milliseconds. (2) A protein surface region near Lys72, Lys73, Lys86, and Lys87 as well as Asp50, Glu66, and Glu92 is the likely contact site for binding to the membrane. The His26-Pro44 hydrogen bond breaks upon binding of cyt c to the surface. (3) The low-stability loop containing Met80 rearranges and the weak Met80-heme coordination is broken. In addition, unfolding of the green and yellow foldon units loosens the protein tertiary structure. (4) The 50’s helix and the beginning of the C-terminal helix insert partially into the membrane and anchor the protein, while the 60’s helix interacts flat with the membrane surface. The 40’s Ω loop is pulled closer to the membrane, while staying in relatively close proximity to the heme group. The end of the C-terminal helix largely unfolds and N- and C-terminal helix contacts are broken, leading to an extended protein conformer E with an easily accessible heme group, turning cyt c into a peroxidase.
The residue Pro44 is situated in the vicinity of this patch and, as suggested by Spiro and coworkers,36 the breakup of the His26-Pro44 hydrogen-bonding interaction is a likely consequence of protein binding to the CL membrane surface. T-jump/UV resonance Raman experiments at pH 3 have revealed that the loss of this critical hydrogen bond initiates rapid formation of the β-sheet structure in the 40s Ω loop of cyt c, followed by extension of the sheet into the adjacent 60s and 70s helices.36 These changes can trigger rearrangements of the low-stability Met80-containing loop, disrupting the Met80-heme ligation and loosening the protein structure. Indeed, the His26-Pro44 contact plays an important role in controlling the dynamics of opening of the Met80 loop in yeast cyt c.52 Furthermore, protonation and mutations of His26 are known to perturb the protein stability and have been suggested to uncouple the green 20-30s Ω loop from the 60s helix.53
The burst phases and prominent unfolding of the nested-yellow and green foldons at the early stages of cyt c-CL interaction are consistent with the unfolding course initially targeting low-stability substructures in cyt c. Previous analysis of cyt c unfolding reactions suggested that dissociation of the Met80 ligand is necessary for further unfolding to proceed27, 54 and we anticipate that this important change is among the early events in CL-induced cyt c unfolding.
Upon contact with the liposome surface, the 50’s helix and the beginning of the C-terminal helix move away from the heme, a process likely aided by the unfolding of the Met80-containing loop, and these protein regions partially insert into the lipid bilayer. The 60’s helix and the rest of the C-terminal helix are left to interact with the liposome surface. Owing to the swiping motion of the 50’s helix, the 40s Ω loop is also pulled closer to the membrane surface, but does not move as far away as the 50’s, 60’s, and C-terminal helices. Misligation of the heme by His33 or His 2655 is a possible factor in maintaining the heme proximity to the 40s Ω loop as well as to the region probed by Dns28. Heme misligation by a Lys residue rather than a His for CL-bound cyt c has been suggested by the recent MCD study.33 One of the Lys residues next to His26 (e.g. Lys22, Lys25, or Lys27) could possibly become a heme ligand and thus play a role in keeping the protein region containing residues 28 and 39 close to the heme.
Anchoring the protein to the membrane facilitates large-scale cyt c unfolding as the polypeptide establishes new contacts with the liposome surface. Breakup of the N- and C-terminal contacts leads to extended protein structures E with the exposed heme, transforming cyt c into a powerful peroxidase. These structures, although lacking many of the native tertiary interactions, retain a high degree of α-helicity and have a propensity to refold into compact conformers C (C ⇄ E)40.
Besides its effects on enhancing the protein peroxidase activity, the liberation of the C-terminal helix during CL-induced cyt c unfolding may have an additional role in the apoptotic mechanism. Peptides with sequences derived from that of the C-terminal helix penetrate through membranes,56 and the recent study by Groves and coworkers proposes that insertion of the C-helix may contribute to cyt c-induced pore formation in CL liposomes.57
Conclusions
Upon binding to CL-containing liposomes, cyt c undergoes a dramatic unfolding process that yields a mixture of extended and compact non-native conformers. General loosening of the cyt c tertiary structure and initial unfolding of low-stability foldons is followed by a partial insertion of the loosened structure into the lipid bilayer. After protein anchoring to the membrane, major rearrangements of the protein structure lead to a stretching of the polypeptide on the liposome surface. The opening of the cyt c structure exposes the heme group, which enhances the protein’s peroxidase activity and also frees the C-terminal helix to aid in the translocation of the protein through CL membranes. The derived sequence of cyt c transformations provides a rationale for the protein gaining new functions and may help in the development of drugs that interfere with the apoptotic activity of cyt c.
Supplementary Material
Acknowledgments
We thank Jay R. Winkler for sharing his MATLAB codes, Kara L. Bren for the pBTR plasmid, Anna M. Morenz for her work on thermodynamic characterization of Dns28, and Erik J. Snider for his help in preparation of some of the protein samples used in this study.
Funding Sources
This work was supported by NIH RO1-GM098502 (E.V.P.). The DAAD RISE program provided a summer undergraduate fellowship to J.M.
Footnotes
Supporting Information Available. Six figures showing fluorescence spectra and equilibrium unfolding parameters for Dns28; results of ultracentrifugation pelleting experiments; kinetics of Dns fluorescence changes for seven variants relative to the corresponding Dns intensities in the GuHCl-unfolded proteins; shifts of λmax for Dns50, Dns92, and Dns99; dependence of the slow kinetics phase on the liposome concentration; and variability in the kinetics traces at longer times (6 pages). This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interests.
References
- 1.Winkler JR. Cytochrome c Folding Dynamics. Curr Opin Chem Biol. 2004;8:169–174. doi: 10.1016/j.cbpa.2004.02.009. [DOI] [PubMed] [Google Scholar]
- 2.Lyubovitsky JG, Gray HB, Winkler JR. Mapping the Cytochrome c Folding Landscape. J Am Chem Soc. 2002;124:5481–5485. doi: 10.1021/ja017399r. [DOI] [PubMed] [Google Scholar]
- 3.Pascher T, Chesick JP, Winkler JR, Gray HB. Protein Folding Triggered by Electron Transfer. Science. 1996;271:1558–1560. doi: 10.1126/science.271.5255.1558. [DOI] [PubMed] [Google Scholar]
- 4.Bandi S, Bowler BE. Probing the Bottom of a Folding Funnel Using Conformationally Gated Electron Transfer Reactions. J Am Chem Soc. 2008;130:7540–7541. doi: 10.1021/ja801941r. [DOI] [PubMed] [Google Scholar]
- 5.Jones CM, Henry ER, Hu Y, Chan C-K, Luck SD, Bhuyan A, Roder H, Hofrichter J, Eaton WA. Fast Events in Protein Folding Initiated by Nanosecond Laser Photolysis. Proc Natl Acad Sci USA. 1993;90:11860–11864. doi: 10.1073/pnas.90.24.11860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chan CK, Hu Y, Takahashi S, Rousseau DL, Eaton WA, Hofrichter J. Submillisecond Protein-Folding Kinetics Studied by Ultrarapid Mixing. Proc Natl Acad Sci USA. 1997;94:1779–1784. doi: 10.1073/pnas.94.5.1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Takahashi S, Yeh S-R, Das TK, Chan C-K, Gottfried DS, Rousseau D. Folding of Cytochrome c Initiated by Submillisecond Mixing. Nature Struct Biol. 1997;4:44–50. doi: 10.1038/nsb0197-44. [DOI] [PubMed] [Google Scholar]
- 8.Maity H, Maity M, Krishna MM, Mayne L, Englander SW. Protein Folding: The Stepwise Assembly of Foldon Units. Proc Natl Acad of Sci USA. 2005;102:4741–4746. doi: 10.1073/pnas.0501043102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hoang L, Bédard S, Krishna MMG, Lin Y, Englander SW. Cytochrome c Folding Pathway: Kinetic Native-State Hydrogen Exchange. Proc Natl Acad Sci USA. 2002;99:12173–12178. doi: 10.1073/pnas.152439199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shastry MCR, Roder H. Evidence for Barrier-Limited Protein-Folding Kinetics on the Microsecond Time-Scale. Nature Struct Biol. 1998;5:385–392. doi: 10.1038/nsb0598-385. [DOI] [PubMed] [Google Scholar]
- 11.Chen E, Goldbeck RA, Kliger DS. Probing Early Events in Ferrous Cytochrome c Folding with Time-Resolved Natural and Magnetic Circular Dichroism Spectroscopies. Curr Protein Pept Sci. 2009;10:464–475. doi: 10.2174/138920309789352001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Abel CJ, Goldbeck RA, Latypov RF, Roder H, Kliger DS. Conformational Equilibration Time of Unfolded Protein Chains and the Folding Speed Limit. Biochemistry. 2007;46:4090–4099. doi: 10.1021/bi0622930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yu W, Dawson PE, Zimmermann J, Romesberg FE. Carbon-Deuterium Bonds as Probes of Protein Thermal Unfolding. J Phys Chem B. 2012;116:6397–6403. doi: 10.1021/jp303521t. [DOI] [PubMed] [Google Scholar]
- 14.Weinkam P, Zimmermann J, Romesberg FE, Wolynes PG. The Folding Energy Landscape and Free Energy Excitations of Cytochrome c. Acc Chem Res. 2010;43:652–660. doi: 10.1021/ar9002703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maity H, Maity M, Englander SW. How Cytochrome c Folds, and Why: Submolecular Foldon Units and Their Stepwise Sequential Stabilization. J Mol Biol. 2004;343:223–233. doi: 10.1016/j.jmb.2004.08.005. [DOI] [PubMed] [Google Scholar]
- 16.Krishna MMG, Lin Y, Mayne L, Englander SW. Intimate View of a Kinetic Protein Folding Intermediate: Residue-Resolved Structure, Interactions, Stability, Folding and Unfolding Rates, Homogeneity. J Mol Biol. 2003;334:501–513. doi: 10.1016/j.jmb.2003.09.070. [DOI] [PubMed] [Google Scholar]
- 17.Duncan MG, Williams MD, Bowler BE. Compressing the Free Energy Range of Substructure Stabilities in Iso-1-Cytochrome c. Prot Sci. 2009;18:1155–1164. doi: 10.1002/pro.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Krishna MM, Maity H, Rumbley JN, Lin Y, Englander SW. Order of Steps in the Cytochrome c Folding Pathway: Evidence for a Sequential Stabilization Mechanism. J Mol Biol. 2006;359:1410–1419. doi: 10.1016/j.jmb.2006.04.035. [DOI] [PubMed] [Google Scholar]
- 19.Weinkam P, Zong C, Wolynes PG. A Funneled Energy Landscape for Cytochrome c Directly Predicts the Sequential Folding Route Inferred from Hydrogen Exchange Experiments. Proc Natl Acad Sci USA. 2005;102:12401–12406. doi: 10.1073/pnas.0505274102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Russell BS, Melenkivitz R, Bren KL. Nmr Investigation of Ferricytochrome c Unfolding: Detection of an Equilibrium Unfolding Intermediate and Residual Structure in the Denatured State. Proc Natl Acad Sci USA. 2000;97:8312–8317. doi: 10.1073/pnas.150239397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ensign AA, Jo I, Yildirim I, Krauss TD, Bren KL. Zinc Porphyrin: A Fluorescent Acceptor in Studies of Zn-Cytochrome c Unfolding by Fluorescence Resonance Energy Transfer. Proc Natl Acad of Sci USA. 2008;105:10779–10784. doi: 10.1073/pnas.0802737105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Latypov RF, Cheng H, Roder NA, Zhang J, Roder H. Structural Characterization of an Equilibrium Unfolding Intermediate in Cytochrome c. J Mol Biol. 2006;357:1009–1025. doi: 10.1016/j.jmb.2006.01.055. [DOI] [PubMed] [Google Scholar]
- 23.Drew HR, Dickerson RE. The Unfolding of Cytochromes c in Methanol and Acid. J Mol Biol. 1978;253:8420–8427. [PubMed] [Google Scholar]
- 24.Goto Y, Hagihara Y, Hamada D, Hoshino M, Nishii I. Acid-Induced Unfolding and Refolding Transitions of Cytochrome c: A Three-State Mechanism in H2o and D2o. Biochemistry. 1993;32:11878–11885. doi: 10.1021/bi00095a017. [DOI] [PubMed] [Google Scholar]
- 25.Bertini I, Turano P, Vasos PR, Bondon A, Chevance S, Simonneaux G. Cytochrome c and SDS: A Molten Globule Protein with Altered Axial Ligation. J Mol Biol. 2004;336:489–496. doi: 10.1016/j.jmb.2003.12.045. [DOI] [PubMed] [Google Scholar]
- 26.Bhuyan AK. On the Mechanism of Sds-Induced Protein Denaturation. Biopolymers. 2010;93:186–199. doi: 10.1002/bip.21318. [DOI] [PubMed] [Google Scholar]
- 27.Pinheiro TJ, Elöve GA, Watts A, Roder H. Structural and Kinetic Description of Cytochrome c Unfolding Induced by the Interaction with Lipid Vesicles. Biochemistry. 1997;36:13122–13132. doi: 10.1021/bi971235z. [DOI] [PubMed] [Google Scholar]
- 28.Sun J, Ruchmann J, Pallier A, Jullien L, Desmadril M, Tribet C. Unfolding of Cytochrome c Upon Interaction with Azobenzene-Modified Copolymers. Biomacromolecules. 2012;13:3736–3746. doi: 10.1021/bm301200p. [DOI] [PubMed] [Google Scholar]
- 29.Moore GR, Pettigrew GW. Cytochromes c: Evolutionary, Structural, and Physicochemical Aspects. Springer-Verlag; New York: 1990. [Google Scholar]
- 30.Kagan VE, Tyurin VA, Jiang J, Tyurina YY, Ritov VB, Amoscato AA, Osipov AN, Belikova NA, Kapralov AA, Kini V, et al. Cytochrome c Acts as a Cardiolipin Oxygenase Required for Release of Proapoptotic Factors. Nat Chem Biol. 2005;1:223–232. doi: 10.1038/nchembio727. [DOI] [PubMed] [Google Scholar]
- 31.Kapralov AA, Kurnikov IV, Vlasova II, Belikova NA, Tyurin VA, Basova LV, Zhao Q, Tyurina YY, Jiang J, Bayir H, et al. The Hierarchy of Structural Transitions Induced in Cytochrome c by Anionic Phospholipids Determines Its Peroxidase Activation and Selective Peroxidation During Apoptosis in Cells. Biochemistry. 2007;46:14232–14244. doi: 10.1021/bi701237b. [DOI] [PubMed] [Google Scholar]
- 32.Kagan VE, Bayir HA, Belikova NA, Kapralov O, Tyurina YY, Tyurin VA, Jiang J, Stoyanovsky DA, Wipf P, Kochanek PM, et al. Cytochrome c/Cardiolipin Relations in Mitochondria: A Kiss of Death. Free Radic Biol Med. 2009;46:1439–1453. doi: 10.1016/j.freeradbiomed.2009.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bradley JM, Silkstone G, Wilson MT, Cheesman MR, Butt JN. Probing a Complex of Cytochrome c and Cardiolipin by Magnetic Circular Dichroism Spectroscopy: Implications for the Initial Events in Apoptosis. J Am Chem Soc. 2011;133:19676–19679. doi: 10.1021/ja209144h. [DOI] [PubMed] [Google Scholar]
- 34.Silkstone G, Kapetanaki SM, Husu I, Vos MH, Wilson MT. Nitric Oxide Binding to the Cardiolipin Complex of Ferric Cytochrome C. Biochemistry. 2012;51:6760–6766. doi: 10.1021/bi300582u. [DOI] [PubMed] [Google Scholar]
- 35.Hanske J, Toffey JR, Morenz AM, Bonilla AJ, Schiavoni KH, Pletneva EV. Conformational Properties of Cardiolipin-Bound Cytochrome c. Proc Natl Acad Sci USA. 2012;109:125–130. doi: 10.1073/pnas.1112312108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Balakrishnan G, Hu Y, Spiro TG. His26 Protonation in Cytochrome c Triggers Microsecond Β-Sheet Formation and Heme Exposure: Implications for Apoptosis. J Am Chem Soc. 2012;134:19061–19069. doi: 10.1021/ja307100a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Beales PA, Bergstrom CL, Geerts N, Groves JT, Vanderlick TK. Single Vesicle Observations of the Cardiolipin-Cytochrome c Interaction: Induction of Membrane Morphology Changes. Langmuir. 2011;27:6107–6115. doi: 10.1021/la104924c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Patriarca A, Polticelli F, Piro MC, Sinibaldi F, Mei G, Bari M, Santucci R, Fiorucci L. Conversion of Cytochrome c into a Peroxidase: Inhibitory Mechanisms and Implication for Neurodegenerative Diseases. Arch Biochem Biophys. 2012;522:62–69. doi: 10.1016/j.abb.2012.03.028. [DOI] [PubMed] [Google Scholar]
- 39.Snider EJ, Muenzner J, Toffey JR, Hong Y, Pletneva EV. Multifaceted Effects of Atp on Cardiolipin-Bound Cytochrome c. Biochemistry. 2013;52:993–995. doi: 10.1021/bi301682c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hong Y, Muenzner J, Grimm SK, Pletneva EV. Origin of the Conformational Heterogeneity of Cardiolipin-Bound Cytochrome c. J Am Chem Soc. 2012;134:18713–18723. doi: 10.1021/ja307426k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Margoliash E, Frohwirt N. Spectrum of Horse-Heart Cytochrome c. Biochem J. 1959;71:570–572. doi: 10.1042/bj0710570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. Ucsf Chimera--a Visualization System for Exploratory Research and Analysis. J Comp Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 43.Balakrishnan G, Hu Y, Oyerinde OF, Su J, Groves JT, Spiro TG. A Conformational Switch to Β-Sheet Structure in Cytochrome c Leads to Heme Exposure. Implications for Cardiolipin Peroxidation and Apoptosis. J Am Chem Soc. 2007;129:504–505. doi: 10.1021/ja0678727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sinibaldi F, Fiorucci L, Patriarca A, Lauceri R, Ferri T, Coletta M, Santucci R. Insights into Cytochrome c-Cardiolipin Interaction. Role Played by Ionic Strength. Biochemistry. 2008;47:6928–6935. doi: 10.1021/bi800048v. [DOI] [PubMed] [Google Scholar]
- 45.Pletneva EV, Gray HB, Winkler JR. Snapshots of Cytochrome c Folding. Proc Natl Acad Sci USA. 2005;102:18397–18402. doi: 10.1073/pnas.0509076102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lorch M, Booth PJ. Insertion Kinetics of a Denatured Α Helical Membrane Protein into Phospholipid Bilayer Vesicles. J Mol Biol. 2004;344:1109–1121. doi: 10.1016/j.jmb.2004.09.090. [DOI] [PubMed] [Google Scholar]
- 47.Allen SJ, Curran AR, Templer RH, Meijberg W, Booth PJ. Folding Kinetics of an α-Helical Membrane Protein in Phospholipid Bilayer Vesicles. J Mol Biol. 2004;342:1279–1291. doi: 10.1016/j.jmb.2004.07.040. [DOI] [PubMed] [Google Scholar]
- 48.Deng Z, Thontasen N, Malinowski N, Rinke G, Harnau L, Rauschenbach S, Kern K. A Close Look at Proteins: Submolecular Resolution of Two- and Three-Dimensionally Folded Cytochrome c at Surfaces. Nano Lett. 2012;12:2452–2458. doi: 10.1021/nl3005385. [DOI] [PubMed] [Google Scholar]
- 49.Salamon Z, Tollin G. Surface Plasmon Resonance Studies of Complex Formation between Cytochrome c and Bovine Cytochrome c Oxidase Incorporated into a Supported Planar Lipid Bilayer. I. Binding of Cytochrome c to Cardiolipin/Phosphatidylcholine Membranes in the Absence of Oxidase. Biophys J. 1996;71:848–857. doi: 10.1016/S0006-3495(96)79286-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kostrzewa A, Pali T, Froncisz W, Marsh D. Membrane Location of Spin-Labeled Cytochrome c Determined by Paramagnetic Relaxation Agents. Biochemistry. 2000;39:6066–6074. doi: 10.1021/bi992559l. [DOI] [PubMed] [Google Scholar]
- 51.Rytömaa M, Kinnunen PK. Evidence for Two Distinct Acidic Phospholipid-Binding Sites in Cytochrome c. J Biol Chem. 1994;269:1770–1774. [PubMed] [Google Scholar]
- 52.Bandi S, Bowler BE. Probing the Dynamics of a His73-Heme Alkaline Transition in a Destabilized Variant of Yeast Iso-1-Cytochrome c with Conformationally Gated Electron Transfer Methods. Biochemistry. 2011;50:10027–10040. doi: 10.1021/bi201082h. [DOI] [PubMed] [Google Scholar]
- 53.Krishna MM, Maity H, Rumbley JN, Englander SW. Branching in the Sequential Folding Pathway of Cytochrome c. Prot Sci. 2007;16:1946–1956. doi: 10.1110/ps.072922307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sauder JM, MacKenzie NE, Roder H. Kinetic Mechanism of Folding and Unfolding of Rhodobacter Capsulatus Cytochrome c2. Biochemistry. 1996;35:16852–16862. doi: 10.1021/bi961976k. [DOI] [PubMed] [Google Scholar]
- 55.Oellerich S, Lecomte S, Paternostre M, Heimburg T, Hildebrandt P. Peripheral and Integral Binding of Cytochrome c to Phospholipids Vesicles. J Phys Chem B. 2004;108:3871–3878. [Google Scholar]
- 56.Jones S, Holm T, Mager I, Langel U, Howl J. Characteriation of Bioactive Cell Penetrating Peptides from Human Cytochrome c: Protein Mimicry and the Development of a Novel Apoptogenic Agent. Chem Biol. 2010;17:735–744. doi: 10.1016/j.chembiol.2010.05.018. [DOI] [PubMed] [Google Scholar]
- 57.Bergstrom CL, Beales PA, Lv Y, Vanderlick KT, Groves JT. Cytochrome c Causes Pore Formation in Cardiolipin-Containing Membranes. Proc Natl Acad of Sci USA. 2013;110:6269–6274. doi: 10.1073/pnas.1303819110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bushnell GW, Louie GV, Brayer GD. High-Resolution 3-Dimensional Structure of Horse Heart Cytochrome c. J Mol Biol. 1990;214:585–595. doi: 10.1016/0022-2836(90)90200-6. [DOI] [PubMed] [Google Scholar]
- 59.Pletneva EV, Gray HB, Winkler JR. Many Faces of the Unfolded State: Conformational Heterogeneity in Denatured Yeast Cytochrome c. J Mol Biol. 2005;345:855–867. doi: 10.1016/j.jmb.2004.10.085. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.