

Background: HIV-2 Vpx modulates innate immunity by overcoming restrictions for infection of myeloid cells.
Results: Vpx binds IRF5 and inhibits its ability to bind to IL6, IL12, and TNFα promoters.
Conclusion: Effects of Vpx on the innate immune system are at least partially due to its inhibition of TLR activation of IRF5.
Significance: Understanding the mechanism of HIV immunomodulation is critical for vaccine development.
Keywords: Cytokine, HIV, Innate Immunity, Interferon, Myeloid Cell
Abstract
Interferon regulatory factor (IRF) family members have been implicated as critical transcription factors that function in immune responses, hematopoietic differentiation, and cell growth regulation. Activation of IRF5 results in the production of pro-inflammatory cytokines such as TNFα, IL6, and IL12, as well as type I interferons. In this study, we demonstrate that HIV-2 Vpx interacts with IRF5, and Vpx inhibits IRF5-mediated transactivation. Expression of Vpx in THP-1 cells reduced mRNA levels and protein production of Toll-like receptor-dependent IL6, IL12p40, and TNFα induced by lipopolysaccharide, R848, and ODN2216. Chromatin immunoprecipitation assays show that Vpx expression results in decreased promoter binding activity of IRF5. This study provides new insights into mechanisms employed by HIV-2 to counteract innate immune defenses against viral infection.
Introduction
The interferon (IFN) regulatory factor (IRF)2 family consists of nine cellular IRFs, each with distinct pleiotropic biological functions. As demonstrated by biochemical, molecular biological, and gene knock-out studies, IRFs are important in pathogen response, cytokine signaling, hematopoietic differentiation, regulation of the cell cycle, and apoptosis (1). IRF5 is involved in the induction of type I interferon and pro-inflammatory cytokines, and thus, it is a critical mediator of innate and adaptive immunity (2–4). IRF5 is a latent transcription factor that is constitutively expressed in B cells, dendritic cells, and macrophages, and its activity can be further enhanced by type I IFN or DNA damage (2, 5–7). IRF5 possesses two nuclear localization signals and one nuclear export signal that control dynamic shuttling between the cytoplasm and the nucleus. IRF5 resides in the cytoplasm of most unstimulated cells and becomes activated by post-translational modifications that include phosphorylation, acetylation, and/or ubiquitination, resulting in translocation to the nucleus (8–13). IRFs share significant homology in their N-terminal DNA binding domain, whereas the C terminus of each IRF is not well conserved and is thought to dictate specific interactions with other proteins and IRF family members that mediate their distinct functions (1, 13). Among the IRF family members, IRF5 and IRF7 share a signaling pathway that is initiated through TLR7/8 and TLR9 (3, 14). Upon stimulation with TLR7/8 and TLR9 ligands, IRF5 and IRF7 are recruited to the myeloid differentiation primary response protein (MyD) 88. The MyD88-bound IRF5 is activated by phosphorylation due to interaction with TNF receptor-associated factor 6 and a kinase that remains to be defined (11). IRF7 can be activated by the TRIF and MyD88 pathways, whereas IRF5 is activated only by TLR7/8 and TLR9 (11, 14). TLR activation results in the nuclear translocation of IRF5 to activate cytokine gene transcription (3, 14). In IRF5 knock-out mice, gene induction of pro-inflammatory cytokines, such as IL6, IL12p40, and TNFα, is severely impaired, indicating IRF5 is a key player in the TLR-MyD88 signaling pathway (3).
Vpx is a virion-associated viral accessory protein packaged through a specific interaction with the Gag protein of HIV-2 and selected simian immunodeficiency virus lineages (15–19). It is essential for efficient viral replication in macrophages (20, 21) and dendritic cells (22, 23), promoting the accumulation of viral DNA during reverse transcription (24, 25). Interestingly, the delivery of Vpx in trans through virus-like particles also enables HIV-1 to infect otherwise resistant primary human cells such as monocytes, macrophages, or dendritic cells (24, 26). SAMHD1 was recently identified as a potent restriction factor of HIV-1 in myeloid cells and resting CD4+ T cells (27–29). SAMHD1 is a deoxynucleotide triphosphohydrolase that blocks HIV-1 reverse transcription by depleting the intracellular pool of deoxynucleoside triphosphates (30–32). Vpx neutralizes the antiviral activity of SAMHD1 by promoting its proteasome-dependent degradation. Vpx binds SAMHD1 and recruits it to the CRL4DCAF1 E3 ubiquitin ligase via its interaction with DCAF1 to facilitate SAMHD1 ubiquitination and subsequent degradation (27, 28, 33, 34).
Vpx is not essential for virus replication in tissue culture, but it is important for viral replication and disease progression in animal models (35–37). The effect of Vpx in vivo is possibly linked to its ability to enhance virus replication in dendritic cells and macrophages in tissue culture (21, 23, 24, 26, 38). Myeloid cells are believed to be critical targets for HIV in vivo, because they are capable of productive infection, and they facilitate virus transmission to CD4+ T cells (39–41).
To gain an understanding of the diverse mechanisms of how Vpx counteracts restriction to HIV replication in nonpermissive cells, such as macrophages and dendritic cells, we performed a yeast two-hybrid genetic screen for DCAF1 substrates, and we then examined the potential candidates that bind to DCAF1 for their interaction with Vpx. By co-immunoprecipitation assay, IRF5 was found to interact with HIV-2 Vpx, but IRF5 did not restrict HIV-2 replication. Interestingly, Vpx inhibits the function of IRF5 as a transcription activator, and overexpression of Vpx reduces the production of IL6, IL12p40, and TNFα. ChIP assay results indicate that the interaction of Vpx with IRF5 interferes with the binding of IRF5 to its targeted promoters.
EXPERIMENTAL PROCEDURES
Plasmids, Viruses, and Cells
HIV-2 Vpx (GH-1) was expressed in a pcDNA3.1 vector with a FLAG-Myc-HA tag at its N terminus (pFMH-Vpx). IRF3, IRF5, and IRF7 expression plasmid and IFNβ-luciferase reporter constructs were kindly provided by Dr. Pitha. IRF3, IRF5, and IRF7 were re-cloned into pCNF (pcDNA3.1 with an N-terminal FLAG tag), and FLAG-IRF3, FLAG-IRF5, and FLAG-IRF7 were used in all experiments. pGFP-IRF5 was generated by inserting the IRF5 gene at the 3′ end of the GFP gene in pEGFP-C1(Clontech). The IL12p40 promoter luciferase reporter plasmid was a kind gift from Dr. Murphy. The lentiviral vector (pFLRu-MCS-YFP) was a gift from Dr. Thomson. HIV-2 vpx (GH-1) with an N-terminal FLAG-Myc-HA tag was cloned into EcoRI and AgeI sites to yield pLenti-Vpx, and verified by DNA sequencing. pCMV-HA-MyD88 was a gift of Dr. Beutler (Addgene plasmid 12287). pcDNA3-IKKϵ was from Dr. Maniatis (Addgene plasmid 26201). pEF-Bos huTBK1 FLAG-His was a gift from Dr. Fitzgerald (Addgene plasmid 27241). The pLKO.Puro plasmid expressing shRNA 5′ CCTTAACAAGAGCCGGGACTT or 5′ TGATAGTATCCGGCTACAGAT was used to deplete IRF5.
Infectious lentiviral particles encoding HIV-2 Vpx were generated by transfecting 293T cells using TransIT (Mirus) with 4 parts transfer vector (pLenti-vpx), 3 parts packaging plasmid (pGag-Pol), and 1 part vesicular stomatitis virus (VSV) glycoprotein (VSVg) expression plasmid. Seventy two hours after transfection, the virus was collected and concentrated by ultracentrifugation through a 20% sucrose cushion. Human macrophage cell line, THP-1, was transduced with the lentivirus expressing HIV-2 Vpx or an empty vector for 2 days and selected for puromycin resistance for 2–3 more days.
Transfection, Immunoprecipitation, and Western Blot Analysis
For all transient assays, plasmids were transfected into either 293T or HT1080 cells using TransIT (Mirus) according to the manufacturer's instructions. For immunoprecipitation, 293T cells were transfected with IRF5 and Vpx expression plasmids, and cell lysates were collected 48–72 h post-transfection in PBS, 0.5% Nonidet P-40, and protease inhibitors, and cell debris was removed by centrifugation in a microcentrifuge at 14,000 rpm for 5 min. Cell lysates were diluted with PBS so that the final concentration of Nonidet P-40 was 0.2% and incubated with a specific antibody at 4 °C overnight. Then 30 μl of protein A/G-agarose beads were mixed with the lysate for 1 h at 4 °C and washed with PBS, 0.1% Nonidet P-40 three times, 10 min each time. Finally the beads were resuspended in sample buffer and boiled, before subjecting to SDS-PAGE. Co-precipitated proteins were identified by Western blotting using a femto Supersignal detection kit (Thermo Scientific).
Immunofluorescence Microscopy
Raw 267.4 cells grown on coverslips were transfected with pFMH-Vpx and pGFP-IRF5. THP-1 cells were transduced with the lentiviral vector encoding vpx for 72 h and then treated with 100 nm PMA. After 24 h, cells were treated with 10 μm R848 for 6 h and fixed with 2% paraformaldehyde, permeabilized with 0.2% Triton X-100 in PBS for 5 min, and stained with monoclonal anti-FLAG antibody and then with Alexa Fluor 954-conjugated goat anti-mouse IgG (Molecular Probes). Nuclei were visualized using 0.5 μg/ml Hoechst 33258 pentahydrate (Molecular Probes). Cells were visualized with a Tis epifluorescence microscope (Nikon) equipped with Metamorph software (objective ×40).
Luciferase Assays
293T or HT1080 cells in 12-well plates were transiently transfected with an IL12p40 promoter-firefly luciferase reporter plasmid (500 ng) and IRF5 and/or a Vpx expression plasmid. A pRL-TK (Renilla luciferase) reporter plasmid was used as an internal control for transfection efficiency using TransIT (Mirus) according to the instructions of the manufacturer. The total amount of DNA was kept constant by supplementation with an empty vector (pcDNA3.1). At 24 h post-transfection, the luciferase activity was measured by Dual-Luciferase assay.
RNA Isolation and qRT-PCR
Total RNA was extracted from THP-1 cells transduced with either an empty lentiviral vector or the vector encoding HIV-2 Vpx or was infected with HIV-2 viruses using the RNeasy mini kit (Qiagen). Real time qRT-PCR was performed using the one-step RT-PCR kit (Bio-Rad) to measure the mRNA level according to the manufacturer's instructions. Gene expression was determined by the comparative Ct method (2−ΔΔCt) (42) based on real time qRT-PCR with primer sets for IL-6, IL-12p40, TNFα, IκBα, and FLIP. The following primers were used: IL-6 primers, 5′-AAATTCGGTACATCCTCGACGGCA and 5′-AGTGCCTCTTTGCTGCTTTCACAC; IL-12p40 primers, 5′-TCATCAAACCTGACCCACCCAAGA and 5′-TTTCTCTCTTGCTCTTGCCCTGGA; TNFα primers, 5′-AAGCCCTGGTATGAGCCCATCTAT and 5′-ATGATCCCAAAGTAGACCTGCCCA; IκBα primers, 5′-GAGTTACCTACCAGGGCTATTC and 5′-CTCTCCTCATCCTCACTCTCT; and FLIP primers, 5′-GAGTGTGTATGGTGTGGATCA and 5′-CTTTGGCTTCCCTGCTAGATAA.
Enzyme-linked Immunosorbent Assay (ELISA)
THP-1 cells, transduced with a lentiviral vector encoding HIV-2 Vpx or empty vector, were treated with 100 nm PMA for 24 h and mock-stimulated (control) or stimulated with TLR4 ligand LPS (100 ng/ml) (Sigma), TLR7 ligand R848 (10 μm), or TLR9 ligand CpG OND2216 (5 μm) (InvivoGen) for 24 h. Secreted IL6, IL12, or TNFα was measured with an ELISA kit (eBiosciences) according to the manufacturer's instructions.
Chromatin Immunoprecipitation (ChIP) Assay
ChIP assay was performed as described (43) with some modifications. THP-1 cells transduced with an empty lentiviral vector or the vector encoding HIV-2 Vpx were treated with 100 nm PMA for 24 h and stimulated with TLR7 ligand R848 (10 μm) for 6–12 h. Cells were cross-linked with 1% formaldehyde for 10 min at room temperature and incubated with 125 mm glycine to stop the cross-linking. After a wash with PBS, cells (1 × 107) were collected and resuspended in 200 μl of lysis buffer (HEPES, pH 7.5, 0.5% Nonidet P-40, protease inhibitors) for 10 min on ice, and nuclei were pelleted by microcentrifugation at 6000 rpm for 5 min. The nuclei were resuspended in nuclear lysis buffer (20 mm Tris-HCl, pH 7.5, 1 mm EDTA, 1% SDS, protease inhibitors) on ice for 10 min. Chromatin was then sonicated for a total of 5 min (20-s burst each time followed by 3 min cooling on ice), which resulted in DNA fragment sizes of 100–600 bp. After debris was removed by centrifugation, the supernatant was diluted 5-fold in RIPA buffer (10 mm Tris-HCl, pH 7.5, 1 mm EDTA, 1% Triton X-100, 0.1% sodium deoxycholate, 0.1% SDS, 100 mm NaCl, and protease inhibitors), and 2 μg of antibody was added to each 250-μl aliquot of the sheared chromatin. After 12 h of incubation at 4 °C, immune complexes were collected with protein A/G-agarose beads for 1 h at 4 °C, washed with 3× RIPA buffer, 2× TE buffer, and eluted with 1% SDS, 0.1 m NaHCO3 by vortex. The eluate was incubated with 0.3 m NaCl/RNase A for 4 h at 65 °C to reverse formaldehyde cross-links. DNA was precipitated with ethanol and purified with a DNA gel extraction column (Qiagen). Real time qPCR was conducted to quantify the DNA containing interferon-stimulated response element (ISRE) sequence using the following primers. Each DNA sample was run as triplicates: IL6-ISRE (−277 to −200) primers, 5′-CGTGCATGACTTCAGCTTTAC and 5′-AGCAGAACCACTCTTCCTTTAC; IL6-UTR (+4728 to +4868) primers, 5′-GGAAAGTGTAGGCTTACCTCAA and 5′-GACACACTCAAAGTTGCTGAAT; IL12p40-ISRE (−132 to −32) primers, 5′-CACACACAGAGAGAGACAAACA and 5′-ACTCTACTCCTTTCTGATGGAAAC; IL12p40-UTR (+14,206 to +14,306) primers, 5′-CTGGCATGAAATCCCTGAAAC and 5′-GGATCAGAACCTGGAAGAGAAT.
RESULTS
HIV-2 Vpx Interacts with IRF5
To identify cellular proteins associated with Vpx that may play a role in HIV replication, we examined by co-immunoprecipitation (Co-IP) assay the candidates that interacted with DCAF1 from a yeast two-hybrid screen. One of the proteins that interacted with Vpx by Co-IP was IRF5. When we co-expressed Vpx with IRF5 in 293T cells and performed Co-IP, Vpx and IRF5 were co-precipitated when using anti-IRF5 antibody (Fig. 1A) or anti-Vpx antibody (Fig. 1B), whereas in control samples lacking either IRF5 (Fig. 1A) or Vpx (Fig. 1B), no Vpx or IRF5 was co-precipitated by anti-IRF5 or anti-Vpx antibody. These results indicate that HIV-2 Vpx specifically interacts with IRF5. To determine which part of IRF5 is responsible for the interaction, we co-expressed Vpx with IRF5 (1–160 amino acids, DNA binding domain) or IRF5 (161–489 amino acids, IRF interaction and autoinhibitory domains) in 293T cells and performed Co-IP. Fig. 1D shows that Vpx still binds to IRF5 (161–489 amino acids) but not IRF5 (1–160 amino acids).
FIGURE 1.
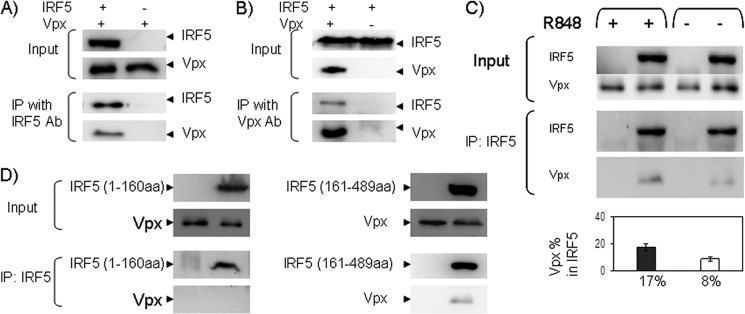
IRF5 interacts with Vpx. A, B, and D, 293T cells, co-transfected with full-length or truncated IRF5 and HIV-2 Vpx expression plasmids, were collected and lysed at 48 h post-transfection. Cell lysates were immunoprecipitated (IP) with anti-IRF5 (A and D) or anti-Vpx antibody (Ab) (B), and immunoblot assays were performed with anti-Vpx (A and D) or anti-FLAG (for IRF5) antibody (B). C, THP-1 cells transduced with the lentivector-encoding vpx gene were lysed, and cell lysates were immunoprecipitated with anti-IRF5 antibody, and immunoblot assays were performed with anti-Vpx and anti-IRF5 antibody. Immunoblot data are representative of three independent experiments. Vpx bound to IRF5 was quantified by densitometry using Image lab software 3.0 (Bio-Rad). Data are presented as the percentage of immunoprecipitated Vpx in IRF5 immune complexes. The input lanes represent IRF5 and Vpx proteins in 10% of the lysate, and half of the immunoprecipitation complexes were loaded onto the gel. Proteins in input and immunoprecipitation lanes were detected by the same concentration of antibody, and the blots were exposed for the same amount of time.
We also co-expressed IRF5 and Vpx in 293T cells in which DCAF1 was depleted by a shRNA against DCAF1. The Co-IP assay was used to investigate whether IRF5 binding to Vpx is DCAF1-dependent. Interestingly, IRF5 was co-precipitated with Vpx independent of the level of DCAF1 (data not shown), suggesting that the interaction between IRF5 and Vpx is not DCAF1-dependent.
To examine whether IRF5 and Vpx are co-localized in cells, we transfected Raw 264.7 cells with GFP-IRF5 and a Vpx expression plasmid. Immunofluorescence microscopy showed that GFP-IRF5 was predominantly localized in the cytoplasm prior to its activation, whereas Vpx was more concentrated in the nucleus, although Vpx could be found to a lesser extent in the cytoplasm (Fig. 2). The overlapping staining of the proteins was mainly in the cytoplasm. After IRF5 activation with the TLR7/8 ligand R848, more IRF5 accumulated in the nucleus, and more overlapped staining of IRF5 and Vpx was observed in the nucleus, as compared with unactivated cells. To confirm the co-localization result, we expressed Vpx in THP-1 cells and then conducted immunofluorescence microscopy. A similar co-localization of IRF5 and Vpx was observed before and after IRF5 activation (Fig. 2B). We also noticed that the level of nuclear IRF5 is slightly higher than the nuclear GFP-IRF5, suggesting that the fusion of GFP to IRF5 may affect the localization of IRF5. This variation may be also due to the cell line difference. The co-localization of the two proteins suggests a possible functional link in the cell.
FIGURE 2.
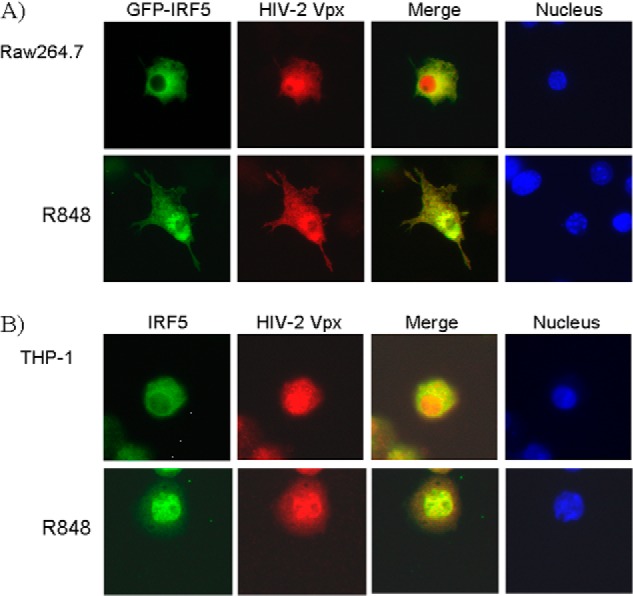
IRF5 and Vpx are co-localized in Raw 267.4 and THP-1 cells. A, Raw 267.4 cells transfected with pEGFP-IRF5 and pFMH-Vpx plasmids were stained with anti-FLAG antibody to detect Vpx, followed by Alexa Fluor 954-conjugated goat anti-mouse IgG. B, THP-1 cells transduced with the lentiviral vector encoding vpx gene were stained with anti-IRF5 and anti-Vpx antibody, followed by Alexa Fluor 488-conjugated goat anti-rabbit IgG (IRF5) and Alexa Fluor 954-conjugated goat anti-mouse IgG (Vpx). Nuclei were stained with Hoechst 33258 pentahydrate.
Because the amount of co-localized IRF5 and Vpx increased after IRF5 activation as shown above, we were interested in whether more IRF5 binds to Vpx after IRF5 was activated. To address this question, THP-1 cells transduced with the lentivector encoding the vpx gene were either stimulated with R848 or not stimulated. The interaction between IRF5 and Vpx was examined and quantified by Co-IP. As shown in Fig. 1C, Vpx bound to the activated IRF5 (17%) is 2-fold higher than Vpx bound to nonactivated IRF5 (8%).
Because IRF5 is constitutively expressed in myeloid cells such as primary monocytes, DCs, and macrophages, which are generally restrictive to HIV-2Δvpx (HIV-2 virus unable to express Vpx) replication, we were interested to determine whether IRF5 is a restrictive factor for HIV-2 infection. To test this idea, 293T cells were transfected with an IRF5 expression plasmid and then infected with VSVg-pseudotyped HIV-2 or HIV-2-luc viruses. Viral replication was examined by measuring luciferase activity (Fig. 3, A and B) or the level of capsid protein (p27) in the medium (D and E) of infected cells. Fig. 3, A and D, shows that overexpression of IRF5 did not affect HIV-2 replication as compared with the control sample. To examine whether HIV-2 infection is affected by depleting IRF5, THP-1 cells stably expressing shRNA against IRF5 (Fig. 3, B and E) and primary monocyte-derived macrophages transfected with IRF5 siRNA (Fig. 3C) were challenged with HIV-2 or HIV-2-luc viruses. Down-regulation of IRF5 did not affect HIV-2 replication, indicating that IRF5 is not a restrictive factor for HIV-2.
FIGURE 3.

Overexpression or down-regulation of IRF5 does not affect HIV-2 replication. A, 293T cells were transfected with either an IRF5 expression plasmid (pFLAG-IRF5) or empty vector (pCNF), and 48 h post-transfection the cells were infected with HIV-2 luciferase reporter viruses pseudotyped with the VSVg envelope. After 48 h, the cells were lysed, and luciferase activities were measured. Luciferase activities of the cells expressing IRF5 are normalized to that of cells expressing pCNF vector, which is set as 100%. B, THP-1 cells expressing either IRF5 or GFP shRNA were treated with PMA for 24 h and infected with HIV-2 luciferase reporter viruses pseudotyped with VSVg. After 48 h, cells were lysed, and luciferase activity was measured. Luciferase activities of cells expressing IRF5 shRNA are normalized to that of GFP shRNA-expressing cells, which is set as 100%. C, monocyte-derived macrophages were transfected with a smart pool siRNA against IRF5, and 48 h post-transfection the cells were infected with HIV-2 viruses pseudotyped with VSVg envelope. After 24 h, DNA was isolated, and the late viral gene product (Gag DNA) was measured by real time qPCR. 293T cells transfected with either pFLAG-IRF5 or pCNF (D) or PMA-treated THP-1 cells expressing either GFP or IRF5 shRNA (E) were infected with HIV-2 viruses pseudotyped with the VSVg envelope. After 72 h, the concentration of capsid protein (p27) in the medium was measured by ELISA. F, PMA-treated THP-1 cells expressing either GFP or IRF5 shRNA were transduced with increasing amounts of VLPvpx (virus-like particle containing Vpx) and collected at 24 h after transduction. Cell lysates were analyzed by immunoblot assay.
To test if IRF5 modulates the Vpx function, we examined Vpx-induced SAMHD1 degradation in the presence and absence of IRF5 in THP-1 cells. As shown in Fig. 3F, the degradation of SAMHD1 by Vpx in IRF5 knock-out cells is very similar to that of control cells, suggesting that IRF5 does not affect the function of Vpx.
HIV-2 Vpx Inhibits IRF5-mediated Transactivation Activity
IRF5 is a transcriptional activator that plays a key role in regulation of pro-inflammatory cytokines, such as TNFα, IL6, and IL12. To investigate whether Vpx affects IRF5-mediated transactivation activity, we measured expression of a luciferase reporter gene driven by the IL12p40 promoter in the presence of IRF5 and Vpx in 293T and HT1080 cells. As shown in Fig. 4, A and B, in both cell types the expression of IRF5 induced IL12p40 promoter activity. Interestingly, the induction of IL12p40 promoter activity was significantly reduced with increasing amounts of Vpx expression.
FIGURE 4.

HIV-2 Vpx inhibits IRF5 transactivation activity. A, 293T cells in a 12-well plate were transfected with pRL-TK control plasmid (100 ng), IL12p40-luc reporter plasmid (300 ng), and IRF5 expression plasmid (1 μg) together with increasing amounts of Vpx expression plasmid (0, 1, and 3 μg) as indicated. Immunoblot analysis of whole cell lysates prepared for luciferase assay is shown below. B, HT1080 cells in a 12-well plate were transfected with pRL-TK control plasmid (200 ng), IL12p40-luc reporter plasmid (600 ng), and IRF5-expressing plasmid (1.5 μg) together with increasing amounts of Vpx expression plasmid (0, 1.5, and 4.5 μg) as indicated. Immunoblot analysis of whole cell lysates prepared for luciferase assay is shown below. C, 293T cells in a 12-well plate were transfected with pRL-TK control plasmid (100 ng), IL12p40-luc reporter plasmid (300 ng), IRF5 (1 μg), and MyD88 (0.5 μg) expression plasmids together with increasing amounts of Vpx expression plasmid (0, 2, and 6 μg) as indicated. Immunoblot analysis of whole cell lysates prepared for luciferase assay is shown below. In all transfections, the pCNF vector was added to bring the total plasmid to the same amount. Luciferase activity was measured at 24 h post-transfection by Dual-Luciferase reporter assay. Relative luciferase activity was determined as fold induction (relative to the basal level of reporter genes in the presence of pCNF vector after normalization with co-transfected relative luciferase units). Values are mean ± S.D. for three experiments.
Because IRF5 is activated through an MyD88-dependent signaling pathway, we next examined whether Vpx interferes with MyD88-mediated IRF5 activation. As shown in Fig. 4C, the IL12p40 promoter activity induced by MyD88 was decreased by Vpx expression. Western blot analysis showed that all proteins were expressed as expected, and IRF5 was present at similar levels in all tested samples. To investigate whether Vpx also affects the transcriptional activity of IRF3 and IRF7, we co-expressed IRF3 or IRF7 with Vpx in 293T cells and measured expression of a luciferase reporter gene driven by IFNβ promoter. Fig. 5 shows that the transcriptional activity of IRF3 and IRF7 was not significantly affected by Vpx. These results indicate that HIV-2 Vpx specifically inhibits the transcriptional function of IRF5.
FIGURE 5.
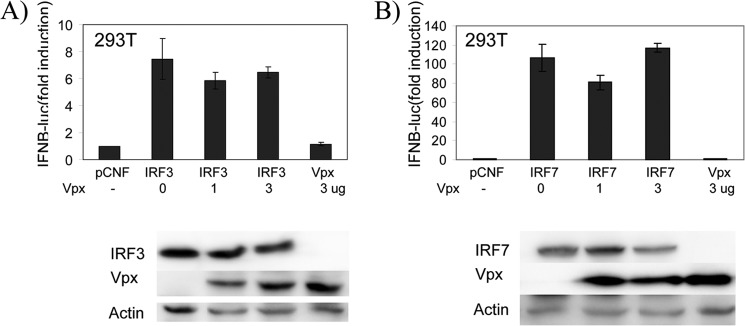
HIV-2 Vpx does not inhibit IRF3 and IRF7 transactivation activity. 293T cells in a 12-well plate were transfected with pRL-TK control plasmid (100 ng), IFNβ-luc reporter plasmid (300 ng), and IRF3 or IRF7 expression plasmid (1 μg) together with an increasing amounts of Vpx expression plasmid (0, 1, and 3 μg) as indicated. Immunoblot analysis of whole cell lysates prepared for luciferase assay is shown below. In all transfections, the pCNF vector was added to bring the total plasmid to the same amount. Luciferase activity was measured at 24 h post-transfection by Dual-Luciferase reporter assay. Relative luciferase activity was determined as fold induction (relative to the basal level of reporter genes in the presence of pCNF vector after normalization with co-transfected relative luciferase units). Values are mean ± S.D. for three experiments.
HIV-2 Vpx Inhibits IRF5-mediated Cytokine Production in THP-1 Cells
IRF5 is expressed constitutively in dendritic cells and macrophages, and it is activated through TLR7/8 and TLR9 (14). To further examine whether Vpx expression affects IRF5 signaling, we transduced the macrophage cell line THP-1 with a lentivector coding Vpx and then measured the expression of endogenous IL6, IL12p40, and TNFα genes in the presence or absence of TLR4 ligand LPS, TLR7/8 ligand R848, and TLR9 ligand CpG ODN2216 (ODN2216 is the TLR9 ligand CpG ODN synthetic oligonucleotide). Using qRT-PCR analyses, we found that Vpx significantly reduced TLR ligand-induced cytokine gene expression (Fig. 6A). To investigate whether TLR ligand-induced cytokine expression is affected by Vpx during virus infection, we infected THP-1 cells with wild type HIV-2 or HIV-2Δvpx virus pseudotyped with VSV glycoprotein and then examined the gene expression of the cytokines as described above. Notably, wild type HIV-2 infection significantly reduced the TLR ligand-induced cytokine gene expression, as compared with infection with HIV-2Δvpx (Fig. 7A). To determine whether the cells were equally infected by both viruses, we measured the levels of viral DNA in the infected cells. As shown in Fig. 7C, both wild type and Vpx deletion viruses replicated to similar levels in the target cells. In contrast, mRNA levels of IκBα and FLIP, which are regulated by NFκB, were not affected by Vpx (Figs. 6B and 7B) (44). These findings suggest that Vpx does not have global effects on transcription, and its effects are specific for IRF5-induced genes.
FIGURE 6.

HIV-2 Vpx represses TLR-mediated IL6, IL12p40, and TNFα induction. THP-1 cells transduced with an empty lentiviral vector or the vector encoding HIV-2 vpx (A and B) were treated with TLR ligands for various times as indicated, and then total RNA was isolated, and mRNA levels were measured by qRT-PCR of IL6, IL12p40, and TNFα (A) or FLIP or IκBα and β-actin (as an internal control) (B). Each mRNA induction was calculated based on untreated THP-1 cells. Each RT-PCR was run in triplicate. Data are shown as one representative example of three independent experiments, and values are mean ± S.D.
FIGURE 7.

HIV-2 Vpx represses TLR-mediated IL6, IL12p40, and TNFα induction. THP-1 cells infected for 48 h with equal amounts of HIV-2 viruses with or without vpx and pseudotyped with the VSVg envelope (A and B) were treated with TLR ligands for various times as indicated, and then total RNA was isolated, and mRNA levels were measured by qRT-PCR of IL6, IL12p40, and TNFα (A) or FLIP or IκBα, and β-actin (as an internal control) (B). Each mRNA induction was calculated based on untreated THP-1 cells. Each RT-PCR was run in triplicate. Data are shown as one representative example of three independent experiments, and values are mean ± S.D. C, total DNA was isolated from HIV-2-infected cells, and the level of viral DNA was determined by measuring Gag gene using real time qPCR.
We next measured TLR ligand-induced production of IL6, IL12, and TNFα by ELISA. As shown in Fig. 8, THP-1 cells expressing Vpx produced significantly less IL6, IL12, and TNFα in response to TLR4, TLR7/8, and TLR9 ligand induction than did control cells. These results demonstrate that HIV-2 Vpx specifically targets and inhibits IRF5 function.
FIGURE 8.
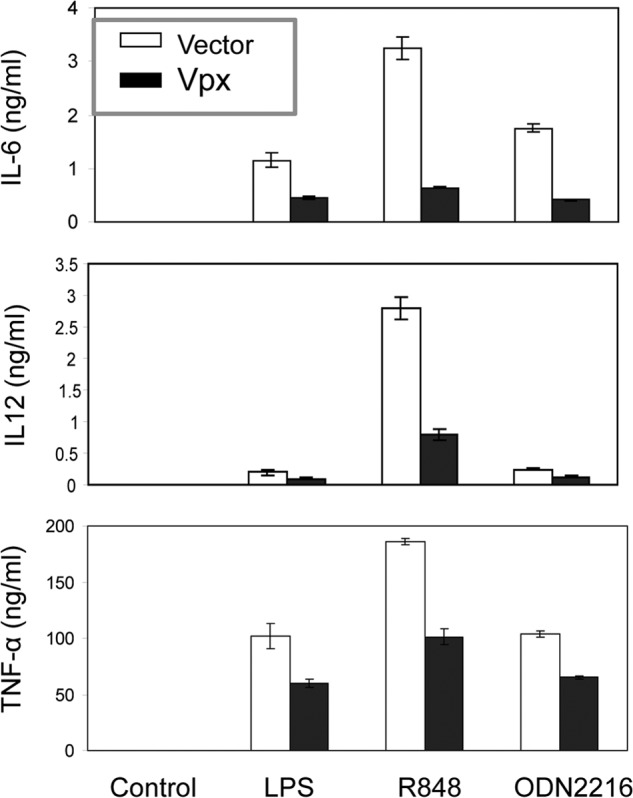
Expression of Vpx reduces production of IL6, IL12, and TNFα induced by LPS, R848, or ODN2216 in THP-1 cells. THP-1 cells transduced with either an empty lentiviral vector or the vector encoding HIV-2 Vpx were mock-stimulated (control) or stimulated with TLR ligands for 24 h, and IL6, IL12, or TNFα in the medium were measured by ELISA.
Vpx Interacts with IRF5 and Inhibits Its DNA Binding Activity
IRF5 is activated from its latent state by post-translational modifications that include phosphorylation and/or ubiquitination (11). The activated form of IRF5 is then translocated into the nucleus and binds to ISRE sequences in the promoters of target genes to activate their transcription. Although it is not clear which cellular kinases are responsible for the phosphorylation of IRF5 during viral infection and TLR signaling, TBK1 and IKKϵ have been shown to phosphorylate IRF5 (13, 45). To investigate whether the binding of Vpx to IRF5 interferes with IRF5 phosphorylation, we co-expressed IRF5 with TBK1 or IKKϵ in the presence or absence of Vpx in 293T cells. Expression of either TBK1 or IKKϵ generated a slower migrating IRF5 band, which was no longer evident if the sample was treated with calf intestinal phosphatase, suggesting that the retarded IRF5 band corresponded to phosphorylated IRF5. As shown in Fig. 9A, introduction of Vpx did not change the ratio of phosphorylated to nonphosphorylated IRF5, indicating that suppression of IRF5 activity by Vpx is not due to interference with phosphorylation.
FIGURE 9.

Vpx interferes with IRF5 DNA binding activity but does not affect IRF5 phosphorylation or nuclear translocation. A, 293T cells were transfected with plasmids expressing IRF5, IKKϵ, TBK1, and Vpx as indicated. After 48 h, cells were lysed, and aliquots of each cell lysate were either untreated or treated with calf intestinal phosphatase (CIP) for 30 min at 37 °C before subjected to SDS-PAGE. IRF5 and Vpx were immunoblotted separately with anti-FLAG or anti-Vpx antibody. Each phosphorylated and nonphosphorylated IRF5 band was quantified by densitometry using Image lab software 3.0 (Bio-Rad). Data are presented as the ratio of phosphorylated to nonphosphorylated IRF5. Values are mean ± S.D. of three experiments. B, THP-1 cells transduced with either an empty lentiviral vector or the vector encoding HIV-2 Vpx were treated with 100 nm PMA and then mock-stimulated (control) or stimulated with 10 μm R848 for 12 h. The cells were lysed and separated into cytoplasmic and nuclear fractions using a nuclear (N) and cytoplasmic (C) kit (Thermo Scientific). Equal amounts of protein from each fraction were loaded onto the gel and blotted with antibodies as indicated. Immunoblot data are representative of three independent experiments. Each IRF5 band was quantified by densitometry using Image lab software 3.0 (Bio-Rad) and presented as the ratio of nuclear to cytoplasmic IRF5. Values are mean ± S.D. of three experiments. C–E, THP-1 cells transduced with an empty lentiviral vector or the vector encoding HIV-2 Vpx were differentiated with 100 nm PMA for 24 h and then either stimulated with 10 μm R848 or not (E). After 6–12 h, cells were collected for ChIP assay as described under “Experimental Procedures.” Each PCR was run in triplicate, and data are representative of three independent experiments. Values are mean ± S.D. of the triplicate experiments.
To examine whether the nuclear translocation of IRF5 is affected by Vpx, THP-1 cells stably expressing either lentivector or Vpx were stimulated with TLR7 ligand R848 and separated into cytoplasmic and nuclear fractions. Endogenous cytoplasmic and nuclear IRF5 was immunoblotted and quantified. As shown in Fig. 9B, without stimulation, IRF5 is detected mostly in the cytoplasmic fraction in control and Vpx-expressing cells. Upon activation with R848, IRF5 accumulates in the nucleus. However, expression of Vpx did not change the level of IRF5 in the nuclear fraction as measured by the nuclear to cytoplasmic IRF5 ratio. The presence of histone H1 in the nuclear fraction and β-actin in the cytoplasmic fraction confirms a clean separation of nuclear and cytoplasmic proteins. These results suggest that Vpx expression does not affect the nuclear translocation of IRF5 upon its activation.
To determine whether the interaction of Vpx and IRF5 interferes with the binding of IRF5 to its target DNA sequence, we performed ChIP assays to specifically measure whether the binding activity of IRF5 to the ISRE within the promoter of the IL12p40 gene is affected by Vpx. THP-1 cells stably expressing Vpx or an empty lentivector were stimulated with R848 to induce cytokines. As shown in Fig. 9C, the PCR-amplified DNA corresponding to the IL12p40-ISRE was selectively detected in the anti-IRF5 antibody immunoprecipitate. Vpx expression reduced the amount of amplified DNA of IL12p40-ISRE bound in the anti-IRF5 immunoprecipitate (p < 0.05, 2-tailed t test). As a negative control, the untranslated region (UTR) at 3′ end of the gene was used and was not amplified in the anti-IRF5 antibody immunoprecipitate, suggesting that IRF5 specifically bound the ISRE sequence (Fig. 9D). When chromatin was immunoprecipitated with the anti-histone H3 antibody, the DNA corresponding to the IL12p40 ISRE or UTR from Vpx- expressing and control samples was equally amplified, indicating that expression of Vpx did not generate global toxicity causing structural damage of chromatin. IRF5 binding to IL12p40-ISRE-associated chromatin was not detected in cells not stimulated with R848, whether or not Vpx was present (Fig. 9E). Similarly, we carried out a ChIP assay to examine the binding activity of IRF5 to the promoter of the IL6 gene in the presence of Vpx. As expected, Vpx also decreased the DNA binding of IRF5 to the IL6 promoter (data not shown). These results suggest that Vpx interacts with IRF5, and the interaction hinders the DNA binding activity of IRF5, thus leading to the reduced transactivation activity of IRF5.
DISCUSSION
HIV-1 has evolved numerous mechanisms to evade various aspects of innate and adaptive immune responses and/or to hijack cellular factors to its own advantage (46). Among these host factors are members of the IRF family. These factors, primarily identified as activators of IFN genes (47, 48), are also key regulators of immune cell development, immune responses, and pattern recognition receptors (1, 49). A key component of innate immunity is the IFN system that is specialized in coordinating host responses against virus infections in a cell type-specific but virus-nonspecific manner. IFN production is controlled at the transcriptional level by IRFs (50). Downstream of TLRs, IRFs are activated by MyD88-dependent and -independent pathways, with different TLRs activating distinct or shared IRFs in a cell type- and stimulus-specific manner (51).
In T cells, HIV-1 disrupts IFN signaling induced by IRF3. HIV-1 infection of Jurkat T cells does not lead to IRF3 activation, but instead it causes a decrease in the level of IRF3 due to viral infectivity factor and viral protein R that target IRF3 for ubiquitin-mediated proteasome degradation (52). Depletion of IRF3 by viral protein U has also been reported during acute infection of primary CD4 T cells (53). In macrophages, the HIV-1 protease inhibits the initiation of the RIG-I (retinoic acid-inducible gene 1) signaling cascade and IRF3 activation, thus blocking type I interferon responses (54). In monocyte-derived dendritic cells (MDDCs), early after infection, HIV-1 similarly exploits IRF1 to subvert IFN induction and increase its own replication (55).
With the exception of plasmacytoid dendritic cells (pDCs) that produce robust type I IFN responses to HIV-1 infection, IFN and antiviral gene expression is not detected in other immune cells, despite sensing HIV-1 (56, 57). MDDCs are insensitive to HIV infection and produce very low levels of IFN when encountering HIV-1, and they also fail to mature or produce proinflammatory cytokines. In contrast, MDDCs induce T cell-dependent immunosuppression after exposure to HIV-1 (58).
Recently, SAMHD1 was found to be a restriction factor responsible for the inhibition of HIV reverse transcription by depleting cellular dNTP pools (28, 31). Interestingly, the restriction of HIV-1 replication in DCs can be overcome by Vpx. Resistance to productive HIV-1 infection in DCs can be circumvented by providing Vpx. This results in HIV-1-induced DC maturation and type I IFN production (59).
IFN induction by DCs depends on the interaction of newly synthesized viral capsid with cellular cyclophilin and subsequent activation of IRF3. This suggests the existence of a cell-intrinsic sensor for HIV-1 in MDDCs that can engage an immune response but is subverted by infection (59). Recently, cyclic GMP-AMP synthase was identified as an innate immune sensor of HIV and other retroviruses. Reverse transcribed viral DNA binds and activates cyclic GAMP synthase to synthesize the cyclic dinucleotide, cyclic GMP-AMP. Cyclic GMP-AMP activates NFκB and IRF3 through STING and IKK/TBK1 signaling pathways to induce IFN and other cytokines. In monocyte-derived macrophages and MDDCs, HIV infection leads to the generation of cyclic GMP-AMP and IFN induction under conditions that are permissive to viral replication (60).
IRF5 was originally described as an inducer of pro-inflammatory cytokines (e.g. IL6, IL12, and TNFα) but subsequently suggested to contribute to the type I IFN antiviral response (3, 4, 61, 62). Irf5−/− mice have increased susceptibility to viral infections, slightly reduced levels of type I IFN in serum, and more significantly reduced levels of pro-inflammatory cytokines (3, 4). IRF5 expression and antiviral activity, however, appear to be restricted to a limited set of cell types, including monocytes and DCs. In this study we demonstrate that HIV-2 Vpx interacts with IRF5, and this interaction negatively affects the transcriptional activity of IRF5. We also show that expression of Vpx leads to the reduced DNA binding of IRF5 in the promoter region of its targeted genes, which likely contributes to the inhibition of IRF5 transcriptional function.
Association of Vpx with IRF5 may impede the direct contact of IRF5 to its targeted promoter sequence, leading to decreased transcriptional activity of IRF5. Alternatively, Vpx could hinder the binding of IRF5 to co-activator proteins important for its transcriptional activity or factors required for IRF5 stability, thus indirectly affecting IRF5 function. For instance, it has been demonstrated that IRF5 is recruited to the TNFα gene via interaction with the NFκB Rel A protein, and the interaction of Vpx with IRF5 could block the binding of IRF5 to Rel A and thus reduce the recruitment of IRF5 to the target sequence (63). Recently, the COP9 signalosome has been shown to interact with and stabilize IRF5. The complex formation with COP9 signalosome subunits prevents IRF5 from being degraded through a ubiquitin-proteasome pathway (64). The binding of Vpx to IRF5 may hinder or disrupt the association of IRF5 with COP9 signalosome subunits and therefore destabilize IRF5, which could result in decreased transcriptional activity of IRF5.
We also detected degradation of IRF5 in the presence of Vpx in 293T cells (data not shown). About one-third of IRF5 was degraded over 24 h in the presence of cycloheximide when IRF5 and Vpx were co-expressed at a ratio of 1:5. The degradation was less significant when Vpx was expressed at a lower level. Surprisingly, Vpx-induced IRF5 degradation was not fully rescued by the proteasome inhibitor MG132. Because both Vpx and IRF5 bind to DCAF1 (data not shown), the interaction domains of DCAF1 for both proteins are probably very close. By interacting with IRF5, Vpx may facilitate the binding of IRF5 to CRLDCAF1 E3 ligase and therefore promote degradation of IRF5 via the proteasome-dependent pathway. However, no significant protein degradation was observed in HIV-2-infected THP-1 cells over 48 h. It is possible that the binding affinity of Vpx to IRF5 is low; thus, a large amount of Vpx is required for sufficient binding. During early stages of infection, Vpx protein is carried into the infected cell by the virus in limited quantities, and thus IRF5 degradation is not noticeable. Additional studies are necessary to investigate whether HIV-2 infection in other cell types causes observable IRF5 degradation and whether this degradation contributes to the inhibition of IRF5 transcriptional function.
We examined the phosphorylation of IRF5 by IKKϵ and TBK1. Because other kinases may be responsible for phosphorylation of IRF5, we cannot completely exclude the possibility that Vpx inhibits IRF5 phosphorylation. We were not successful in determining whether Vpx affects the phosphorylation of endogenous IRF5 in THP-1 cells stimulated with R848, because we could not detect a distinct, slower migrating band representing phosphorylated IRF5, and an antibody specific for phosphorylated IRF5 was not available.
Unlike HIV-1, HIV-2 efficiently infects DCs because the virus particle carries Vpx. HIV-2 replication activates DCs presumably through IRF3 signaling. Although inhibitory effects of HIV-2 viral infectivity factor and viral protein R on IRF3 have not been reported, HIV-2 likely utilizes similar mechanisms as HIV-1 to antagonize IRF3 function. During the early stages of infection of myeloid cells, Vpx overcomes the SAMHD1-mediated block to viral reverse transcription. Our current data demonstrate that Vpx also acts to inhibit IRF5, suggesting that targeted viral antagonism of IRF5 by Vpx may provide an additional level of viral control of the innate immune system. Although the role of IRF5 in induction of innate immunity in HIV infection has not been defined, gene knock-out studies have demonstrated that IRF5 is essential for induction of pro-inflammatory cytokines and type I IFN production in Newcastle disease virus, VSV, and West Nile virus-infected mice (4, 61, 65). This suggests an important role of IRF5 in the innate immune response to viral infection. By interacting with IRF5, Vpx acts to prevent the expression of genes regulated by IRF5 and involved in innate immune defenses against HIV-2. Therefore, IRF5 regulation by HIV-2 is expected to contribute to immune dysfunction in HIV-2-infected patients, thus serving to establish a permissive environment for seeding the initial infection.
Our preliminary data showed that endogenous IRF5 in PMA-differentiated THP-1 cells did not translocate into the nucleus when the cells were infected with HIV-2, indicating that IRF5 was not activated by the viral infection. Additional experiments need to be conducted to confirm this observation in other types of cells, especially DCs and macrophages which constitutively express IRF5, to understand how activation of IRF5 is inhibited. It is also of great interest to know whether HIV-1 infection activates IRF5 and whether HIV-1 suppression of IRF5 enhances permissiveness for infection. More studies are necessary to clarify the link between Vpx inhibition of IRF5 function and clinical implications with regard to HIV pathogenesis and disease progression. These findings may shed light on the development of new strategies to effectively control HIV.
This work was supported, in whole or in part, by National Institutes of Health Grant R21 AI093175.
- IRF
- interferon-regulatory factor
- IKK
- IκB kinase
- ISRE
- interferon-sensitive response element
- MDDC
- monocyte-derived dendritic cell
- MyD88
- myeloid differentiation primary response 88
- SAMHD1
- SAM and HD domain-containing protein 1
- TBK
- TANK-binding kinase
- Vpx
- viral protein X
- VSV
- vesicular stomatitis virus
- VSVg
- vesicular stomatitis virus glycoprotein
- DC
- dendritic cell
- qRT
- quantitative RT
- Co-IP
- co-immunoprecipitation
- PMA
- phorbol 12-myristate 13-acetate.
REFERENCES
- 1. Savitsky D., Tamura T., Yanai H., Taniguchi T. (2010) Regulation of immunity and oncogenesis by the IRF transcription factor family. Cancer Immunol. Immunother. 59, 489–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Barnes B. J., Moore P. A., Pitha P. M. (2001) Virus-specific activation of a novel interferon regulatory factor, IRF-5, results in the induction of distinct interferon alpha genes. J. Biol. Chem. 276, 23382–23390 [DOI] [PubMed] [Google Scholar]
- 3. Takaoka A., Yanai H., Kondo S., Duncan G., Negishi H., Mizutani T., Kano S., Honda K., Ohba Y., Mak T. W., Taniguchi T. (2005) Integral role of IRF-5 in the gene induction programme activated by Toll-like receptors. Nature 434, 243–249 [DOI] [PubMed] [Google Scholar]
- 4. Yanai H., Chen H. M., Inuzuka T., Kondo S., Mak T. W., Takaoka A., Honda K., Taniguchi T. (2007) Role of IFN regulatory factor 5 transcription factor in antiviral immunity and tumor suppression. Proc. Natl. Acad. Sci. U.S.A. 104, 3402–3407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mori T., Anazawa Y., Iiizumi M., Fukuda S., Nakamura Y., Arakawa H. (2002) Identification of the interferon regulatory factor 5 gene (IRF-5) as a direct target for p53. Oncogene 21, 2914–2918 [DOI] [PubMed] [Google Scholar]
- 6. Barnes B. J., Kellum M. J., Pinder K. E., Frisancho J. A., Pitha P. M. (2003) Interferon regulatory factor 5, a novel mediator of cell cycle arrest and cell death. Cancer Res. 63, 6424–6431 [PubMed] [Google Scholar]
- 7. Mancl M. E., Hu G., Sangster-Guity N., Olshalsky S. L., Hoops K., Fitzgerald-Bocarsly P., Pitha P. M., Pinder K., Barnes B. J. (2005) Two discrete promoters regulate the alternatively spliced human interferon regulatory factor-5 isoforms. Multiple isoforms with distinct cell type-specific expression, localization, regulation, and function. J. Biol. Chem. 280, 21078–21090 [DOI] [PubMed] [Google Scholar]
- 8. Lin R., Yang L., Arguello M., Penafuerte C., Hiscott J. (2005) A CRM1-dependent nuclear export pathway is involved in the regulation of IRF-5 subcellular localization. J. Biol. Chem. 280, 3088–3095 [DOI] [PubMed] [Google Scholar]
- 9. Hu G., Mancl M. E., Barnes B. J. (2005) Signaling through IFN regulatory factor-5 sensitizes p53-deficient tumors to DNA damage-induced apoptosis and cell death. Cancer Res. 65, 7403–7412 [DOI] [PubMed] [Google Scholar]
- 10. Barnes B. J., Kellum M. J., Field A. E., Pitha P. M. (2002) Multiple regulatory domains of IRF-5 control activation, cellular localization, and induction of chemokines that mediate recruitment of T lymphocytes. Mol. Cell. Biol. 22, 5721–5740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Balkhi M. Y., Fitzgerald K. A., Pitha P. M. (2008) Functional regulation of MyD88-activated interferon regulatory factor 5 by K63-linked polyubiquitination. Mol. Cell. Biol. 28, 7296–7308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Feng D., Sangster-Guity N., Stone R., Korczeniewska J., Mancl M. E., Fitzgerald-Bocarsly P., Barnes B. J. (2010) Differential requirement of histone acetylase and deacetylase activities for IRF5-mediated proinflammatory cytokine expression. J. Immunol. 185, 6003–6012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cheng T. F., Brzostek S., Ando O., Van Scoy S., Kumar K. P., Reich N. C. (2006) Differential activation of IFN regulatory factor (IRF)-3 and IRF-5 transcription factors during viral infection. J. Immunol. 176, 7462–7470 [DOI] [PubMed] [Google Scholar]
- 14. Schoenemeyer A., Barnes B. J., Mancl M. E., Latz E., Goutagny N., Pitha P. M., Fitzgerald K. A., Golenbock D. T. (2005) The interferon regulatory factor, IRF5, is a central mediator of toll-like receptor 7 signaling. J. Biol. Chem. 280, 17005–17012 [DOI] [PubMed] [Google Scholar]
- 15. Pancio H. A., Ratner L. (1998) Human immunodeficiency virus 2 Vpx-Gag interaction. J. Virol. 72, 5271–5275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yu X. F., Ito S., Essex M., Lee T. H. (1988) A naturally immunogenic virion-associated protein specific for HIV-2 and SIV. Nature 335, 262–265 [DOI] [PubMed] [Google Scholar]
- 17. Henderson L. E., Sowder R. C., Copeland T. D., Benveniste R. E., Oroszlan S. (1988) Isolation and characterization of a novel protein (X-ORF product) from SIV and HIV-2. Science 241, 199–201 [DOI] [PubMed] [Google Scholar]
- 18. Selig L., Pages J. C., Tanchou V., Prévéral S., Berlioz-Torrent C., Liu L. X., Erdtmann L., Darlix J., Benarous R., Benichou S. (1999) Interaction with the p6 domain of the gag precursor mediates incorporation into virions of Vpr and Vpx proteins from primate lentiviruses. J. Virol. 73, 592–600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Accola M. A., Bukovsky A. A., Jones M. S., Göttlinger H. G. (1999) A conserved dileucine-containing motif in p6(gag) governs the particle association of Vpx and Vpr of simian immunodeficiency viruses SIV(mac) and SIV(agm). J. Virol. 73, 9992–9999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Guyader M., Emerman M., Montagnier L., Peden K. (1989) Vpx mutants of HIV-2 are infectious in established cell lines but display a severe defect in peripheral blood lymphocytes. EMBO J. 8, 1169–1175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yu X. F., Yu Q. C., Essex M., Lee T. H. (1991) The vpx gene of simian immunodeficiency virus facilitates efficient viral replication in fresh lymphocytes and macrophage. J. Virol. 65, 5088–5091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mangeot P. E., Duperrier K., Nègre D., Boson B., Rigal D., Cosset F. L., Darlix J. L. (2002) High levels of transduction of human dendritic cells with optimized SIV vectors. Mol. Ther. 5, 283–290 [DOI] [PubMed] [Google Scholar]
- 23. Goujon C., Rivière L., Jarrosson-Wuilleme L., Bernaud J., Rigal D., Darlix J. L., Cimarelli A. (2007) SIVSM/HIV-2 Vpx proteins promote retroviral escape from a proteasome-dependent restriction pathway present in human dendritic cells. Retrovirology 4, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sharova N., Wu Y., Zhu X., Stranska R., Kaushik R., Sharkey M., Stevenson M. (2008) Primate lentiviral Vpx commandeers DDB1 to counteract a macrophage restriction. PLoS Pathog. 4, e1000057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fujita M., Otsuka M., Miyoshi M., Khamsri B., Nomaguchi M., Adachi A. (2008) Vpx is critical for reverse transcription of the human immunodeficiency virus type 2 genome in macrophages. J. Virol. 82, 7752–7756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Srivastava S., Swanson S. K., Manel N., Florens L., Washburn M. P., Skowronski J. (2008) Lentiviral Vpx accessory factor targets VprBP/DCAF1 substrate adaptor for cullin 4 E3 ubiquitin ligase to enable macrophage infection. PLoS Pathog. 4, e1000059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hrecka K., Hao C., Gierszewska M., Swanson S. K., Kesik-Brodacka M., Srivastava S., Florens L., Washburn M. P., Skowronski J. (2011) Vpx relieves inhibition of HIV-1 infection of macrophages mediated by the SAMHD1 protein. Nature 474, 658–661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Laguette N., Sobhian B., Casartelli N., Ringeard M., Chable-Bessia C., Ségéral E., Yatim A., Emiliani S., Schwartz O., Benkirane M. (2011) SAMHD1 is the dendritic- and myeloid-cell-specific HIV-1 restriction factor counteracted by Vpx. Nature 474, 654–657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Baldauf H. M., Pan X., Erikson E., Schmidt S., Daddacha W., Burggraf M., Schenkova K., Ambiel I., Wabnitz G., Gramberg T., Panitz S., Flory E., Landau N. R., Sertel S., Rutsch F., Lasitschka F., Kim B., König R., Fackler O. T., Keppler O. T. (2012) SAMHD1 restricts HIV-1 infection in resting CD4(+) T cells. Nat. Med. 18, 1682–1687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lahouassa H., Dragin L., Transy C., Margottin-Goguet F. (2012) SAMHD1 deprives HIV of nucleotides, essential for viral DNA synthesis. Med. Sci. 28, 909–910 [DOI] [PubMed] [Google Scholar]
- 31. Lahouassa H., Daddacha W., Hofmann H., Ayinde D., Logue E. C., Dragin L., Bloch N., Maudet C., Bertrand M., Gramberg T., Pancino G., Priet S., Canard B., Laguette N., Benkirane M., Transy C., Landau N. R., Kim B., Margottin-Goguet F. (2012) SAMHD1 restricts the replication of human immunodeficiency virus type 1 by depleting the intracellular pool of deoxynucleoside triphosphates. Nat. Immunol. 13, 223–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Goldstone D. C., Ennis-Adeniran V., Hedden J. J., Groom H. C., Rice G. I., Christodoulou E., Walker P. A., Kelly G., Haire L. F., Yap M. W., de Carvalho L. P., Stoye J. P., Crow Y. J., Taylor I. A., Webb M. (2011) HIV-1 restriction factor SAMHD1 is a deoxynucleoside triphosphate triphosphohydrolase. Nature 480, 379–382 [DOI] [PubMed] [Google Scholar]
- 33. Ahn J., Hao C., Yan J., DeLucia M., Mehrens J., Wang C., Gronenborn A. M., Skowronski J. (2012) HIV/simian immunodeficiency virus (SIV) accessory virulence factor Vpx loads the host cell restriction factor SAMHD1 onto the E3 ubiquitin ligase complex CRL4DCAF1. J. Biol. Chem. 287, 12550–12558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Brandariz-Nuñez A., Valle-Casuso J. C., White T. E., Laguette N., Benkirane M., Brojatsch J., Diaz-Griffero F. (2012) Role of SAMHD1 nuclear localization in restriction of HIV-1 and SIVmac. Retrovirology 9, 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gibbs J. S., Lackner A. A., Lang S. M., Simon M. A., Sehgal P. K., Daniel M. D., Desrosiers R. C. (1995) Progression to AIDS in the absence of a gene for vpr or vpx. J. Virol. 69, 2378–2383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hirsch V. M., Sharkey M. E., Brown C. R., Brichacek B., Goldstein S., Wakefield J., Byrum R., Elkins W. R., Hahn B. H., Lifson J. D., Stevenson M. (1998) Vpx is required for dissemination and pathogenesis of SIV(SM) PBj: evidence of macrophage-dependent viral amplification. Nat. Med. 4, 1401–1408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Belshan M., Kimata J. T., Brown C., Cheng X., McCulley A., Larsen A., Thippeshappa R., Hodara V., Giavedoni L., Hirsch V., Ratner L. (2012) Vpx is critical for SIVmne infection of pigtail macaques. Retrovirology 9, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kawamura M., Sakai H., Adachi A. (1994) Human immunodeficiency virus Vpx is required for the early phase of replication in peripheral blood mononuclear cells. Microbiol. Immunol. 38, 871–878 [DOI] [PubMed] [Google Scholar]
- 39. Hu J., Gardner M. B., Miller C. J. (2000) Simian immunodeficiency virus rapidly penetrates the cervicovaginal mucosa after intravaginal inoculation and infects intraepithelial dendritic cells. J. Virol. 74, 6087–6095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sewell A. K., Price D. A. (2001) Dendritic cells and transmission of HIV-1. Trends Immunol. 22, 173–175 [DOI] [PubMed] [Google Scholar]
- 41. Haase A. T. (2010) Targeting early infection to prevent HIV-1 mucosal transmission. Nature 464, 217–223 [DOI] [PubMed] [Google Scholar]
- 42. Schmittgen T. D., Livak K. J. (2008) Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 3, 1101–1108 [DOI] [PubMed] [Google Scholar]
- 43. O'Geen H., Echipare L., Farnham P. J. (2011) Using ChIP-seq technology to generate high-resolution profiles of histone modifications. Methods Mol. Biol. 791, 265–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yang L., Zhao T., Shi X., Nakhaei P., Wang Y., Sun Q., Hiscott J., Lin R. (2009) Functional analysis of a dominant negative mutation of interferon regulatory factor 5. PLoS One 4, e5500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chang Foreman H. C., Van Scoy S., Cheng T. F., Reich N. C. (2012) Activation of interferon regulatory factor 5 by site specific phosphorylation. PLOS One 7, e33098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kirchhoff F. (2010) Immune evasion and counteraction of restriction factors by HIV-1 and other primate lentiviruses. Cell Host Microbe 8, 55–67 [DOI] [PubMed] [Google Scholar]
- 47. Taniguchi T., Ogasawara K., Takaoka A., Tanaka N. (2001) IRF family of transcription factors as regulators of host defense. Annu. Rev. Immunol. 19, 623–655 [DOI] [PubMed] [Google Scholar]
- 48. Battistini A. (2009) Interferon regulatory factors in hematopoietic cell differentiation and immune regulation. J. Interferon Cytokine Res. 29, 765–780 [DOI] [PubMed] [Google Scholar]
- 49. Honda K., Taniguchi T. (2006) IRFs: master regulators of signaling by Toll-like receptors and cytosolic pattern-recognition receptors. Nat. Rev. Immunol. 6, 644–658 [DOI] [PubMed] [Google Scholar]
- 50. Honda K., Takaoka A., Taniguchi T. (2006) Type I interferon gene induction by the interferon regulatory factor family of transcription factors. Immunity 25, 349–360 [DOI] [PubMed] [Google Scholar]
- 51. Akira S. (2006) TLR signaling. Curr. Top. Microbiol. Immunol. 311, 1–16 [DOI] [PubMed] [Google Scholar]
- 52. Okumura A., Alce T., Lubyova B., Ezelle H., Strebel K., Pitha P. M. (2008) HIV-1 accessory proteins Vpr and Vif modulate antiviral response by targeting IRF-3 for degradation. Virology 373, 85–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Doehle B. P., Chang K., Rustagi A., McNevin J., McElrath M. J., Gale M. (2012) Vpu mediates depletion of interferon regulatory factor 3 during HIV infection by a lysosome-dependent mechanism. J. Virol. 86, 8367–8374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Solis M., Nakhaei P., Jalalirad M., Lacoste J., Douville R., Arguello M., Zhao T., Laughrea M., Wainberg M. A., Hiscott J. (2011) RIG-I-mediated antiviral signaling is inhibited by HIV-1 infection by a protease-mediated sequestration of RIG-I. J. Virol. 85, 1224–1236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Harman A. N., Lai J., Turville S., Samarajiwa S., Gray L., Marsden V., Mercier S. K., Mercier S., Jones K., Nasr N., Rustagi A., Cumming H., Donaghy H., Mak J., Gale M., Jr., Churchill M., Hertzog P., Cunningham A. L. (2011) HIV infection of dendritic cells subverts the IFN induction pathway via IRF-1 and inhibits type 1 IFN production. Blood 118, 298–308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Pitha P. M. (2011) Innate antiviral response: role in HIV-1 infection. Viruses 3, 1179–1203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Mogensen T. H., Melchjorsen J., Larsen C. S., Paludan S. R. (2010) Innate immune recognition and activation during HIV infection. Retrovirology 7, 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Granelli-Piperno A., Golebiowska A., Trumpfheller C., Siegal F. P., Steinman R. M. (2004) HIV-1-infected monocyte-derived dendritic cells do not undergo maturation but can elicit IL-10 production and T cell regulation. Proc. Natl. Acad. Sci. U.S.A. 101, 7669–7674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Manel N., Hogstad B., Wang Y., Levy D. E., Unutmaz D., Littman D. R. (2010) A cryptic sensor for HIV-1 activates antiviral innate immunity in dendritic cells. Nature 467, 214–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Gao D., Wu J., Wu Y. T., Du F., Aroh C., Yan N., Sun L., Chen Z. J. (2013) Cyclic GMP-AMP synthase is an innate immune sensor of HIV and other retroviruses. Science 341, 903–906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Paun A., Reinert J. T., Jiang Z., Medin C., Balkhi M. Y., Fitzgerald K. A., Pitha P. M. (2008) Functional characterization of murine interferon regulatory factor 5 (IRF-5) and its role in the innate antiviral response. J. Biol. Chem. 283, 14295–14308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Barnes B. J., Richards J., Mancl M., Hanash S., Beretta L., Pitha P. M. (2004) Global and distinct targets of IRF-5 and IRF-7 during innate response to viral infection. J. Biol. Chem. 279, 45194–45207 [DOI] [PubMed] [Google Scholar]
- 63. Krausgruber T., Saliba D., Ryzhakov G., Lanfrancotti A., Blazek K., Udalova I. A. (2010) IRF5 is required for late-phase TNF secretion by human dendritic cells. Blood 115, 4421–4430 [DOI] [PubMed] [Google Scholar]
- 64. Korczeniewska J., Barnes B. J. (2013) The COP9 signalosome interacts with and regulates interferon regulatory factor 5 protein stability. Mol. Cell. Biol. 33, 1124–1138 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 65. Lazear H. M., Lancaster A., Wilkins C., Suthar M. S., Huang A., Vick S. C., Clepper L., Thackray L., Brassil M. M., Virgin H. W., Nikolich-Zugich J., Moses A. V., Gale M., Jr., Früh K., Diamond M. S. (2013) IRF-3, IRF-5, and IRF-7 coordinately regulate the type I IFN response in myeloid dendritic cells downstream of MAVS signaling. PLoS Pathog. 9, e1003118. [DOI] [PMC free article] [PubMed] [Google Scholar]