

Background: Asymmetric and symmetric dimethylarginine (ADMA and SDMA) residues are biologically distinct products of protein arginine methyltransferase (PRMT) isoforms.
Results: Met-48 in PRMT1 regulates the regiochemistry of dimethylation, and SDMA formation is energetically costly.
Conclusion: Steric changes in the PRMT1 active site can reprogram product formation.
Significance: SDMA-forming PRMTs may require additional factors to overcome the energetic cost of SDMA.
Keywords: Computer Modeling, Enzyme Mechanisms, Histone Methylation, Protein Methylation, S-adenosylmethionine (SAM), Site-directed Mutagenesis, Dimethylarginine, Product Specificity, Protein Arginine Methyltransferase
Abstract
Protein arginine methylation is emerging as a significant post-translational modification involved in various cell processes and human diseases. As the major arginine methylation enzyme, protein arginine methyltransferase 1 (PRMT1) strictly generates monomethylarginine and asymmetric dimethylarginine (ADMA), but not symmetric dimethylarginine (SDMA). The two types of dimethylarginines can lead to distinct biological outputs, as highlighted in the PRMT-dependent epigenetic control of transcription. However, it remains unclear how PRMT1 product specificity is regulated. We discovered that a single amino acid mutation (Met-48 to Phe) in the PRMT1 active site enables PRMT1 to generate both ADMA and SDMA. Due to the limited amount of SDMA formed, we carried out quantum mechanical calculations to determine the free energies of activation of ADMA and SDMA synthesis. Our results indicate that the higher energy barrier of SDMA formation (ΔΔG‡ = 3.2 kcal/mol as compared with ADMA) may explain the small amount of SDMA generated by M48F-PRMT1. Our study reveals unique energetic challenges for SDMA-forming methyltransferases and highlights the exquisite control of product formation by active site residues in the PRMTs.
Introduction
Protein arginine methylation is a major mechanism for regulating protein function in eukaryotic cells. This post-translational modification is catalyzed by a family of enzymes called protein arginine methyltransferases (PRMTs)2, which are widely involved in a variety of fundamental biological processes, including transcription, RNA splicing, signal transduction, DNA repair (reviewed in Refs. 1 and 2), and chromatin remodeling (3). In recent years, PRMTs have been increasingly studied in human diseases, especially in cardiovascular disease (4, 5) and cancer (6, 7).
Nine human PRMT isoforms transfer the methyl group from S-adenosylmethionine (AdoMet) and form monomethylarginine (MMA), asymmetric dimethylarginine (ADMA), and symmetric dimethylarginine (SDMA) on targeted proteins (Fig. 1). Type I PRMTs (such as PRMT1) form MMA and/or ADMA and represent the majority of identified PRMTs. Type III enzymes produce only MMA. Like the Type I and III enzymes, Type II PRMTs catalyze monomethylation; however, a second round of turnover results in the synthesis of SDMA.
FIGURE 1.
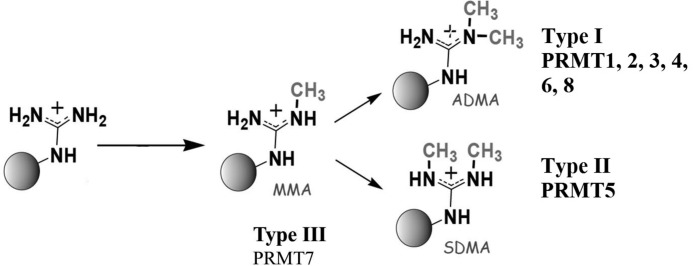
Methylation reactions catalyzed by PRMTs. Type I and Type II PRMTs make MMA. Type I PRMTs may then go on to make ADMA, whereas Type II PRMTs produce SDMA.
Each of the methylated arginines (MMA, ADMA, and SDMA) can induce different biological responses in the cell. Remarkably, histone arginines, such as Arg-3 of histone H4 (H4R3), can be either asymmetrically dimethylated by PRMT1 or symmetrically dimethylated by PRMT5, resulting in antagonistic effects on gene regulation (reviewed in Ref. 8). Moreover, after arginine-methylated proteins are hydrolyzed in vivo, free ADMA is an endogenous competitive inhibitor of nitric oxide synthase, which is intimately involved in cardiovascular health, whereas SDMA has no effect on nitric oxide synthase (reviewed in Refs. 9 and 10). Thus, free ADMA and free SDMA are two biologically distinct products generated by PRMTs. Although the biological significance of different methylarginines has become well accepted, it remains unclear how PRMTs govern the product specificity yielding asymmetric and symmetric dimethylation.
Guided by the crystal structure of PRMT1 (12), the active site of PRMT1 has been dissected to understand the kinetic mechanism (13–15) and product specificity (16). In our recent work, we identified that two conserved active site residues, Met-48 and Met-155, play a significant role in enzymatic activity and regulation of mono- versus asymmetric dimethylation (16). Although Met-155 was suggested to govern formation of ADMA over SDMA (12, 17), mutating either Met-155 or Met-48 to small amino acids, such as Ala or Leu, did not change the dimethylation product (14, 16). When the structure of PRMT5 was solved, a conserved phenylalanine, Phe-379, was surprisingly found at the corresponding location in the active site as Met-48 in rat PRMT1 (18). The phenylalanine is critical for PRMT5 to specify symmetric addition of the second methyl group as changing it to a methionine converts PRMT5 to an enzyme that catalyzes both symmetric and asymmetric dimethylation of arginine (18). Thus, we revisited our constructs bearing M48 mutations. Herein we report that mutation of Met-48 to phenylalanine in rat PRMT1 enables the remodeled PRMT1 to generate both ADMA and SDMA, although the amount of SDMA is relatively limited. The low amount of SDMA formed prompted us to also evaluate the free energies of activation, ΔG‡, of each methyl group transfer step in MMA, ADMA, and SDMA formation using gas-phase quantum mechanical (QM) calculations. Our results indicate that ADMA formation is energetically less costly compared with SDMA. The difference in the computed free energies of activation for ADMA and SDMA formation would represent several orders of magnitude difference in the enzymatic rate constant. The higher energy barrier for forming SDMA over ADMA may explain the limited amount of SDMA formed by the M48F-PRMT1 mutant. These data reveal a previously unknown obstacle that Type II PRMTs must overcome to synthesize SDMA.
EXPERIMENTAL PROCEDURES
Expression and Purification of PRMT1 Mutant Proteins
PRMT1 mutant proteins were generated using the QuikChange® site-directed mutagenesis kit (Stratagene) with sets of complementary oligonucleotide primers spanning the desired site of mutation. For each PCR, the pET28b vector (Novagen) containing the gene that codes for N-terminal histidine-tagged rat WT-PRMT1 plasmid (pET28b-PRMT1) (17) was used as a template. Desired mutations were confirmed through DNA sequencing. Mutated proteins were purified using the same methods used to express and purify wild-type His-PRMT1 (described in Ref. 17). Purified proteins were more than 95% pure by SDS-PAGE. Mutant protein sequences were verified using mass spectrometry.
Reverse Phase-HPLC Analysis of Methylated Arginines
Methylation assays containing 4 μm WT or M48F-PRMT1 proteins, 800 μm AdoMet, 10 nm methylthioadenosine nucleosidase (MTAN, purified as in Ref. 19), 1 mm dithiothreitol (DTT), and 50 mm sodium phosphate buffer (pH 7.8) were equilibrated at 37 °C for 3 min. Reactions were initiated with 200 μm R3 peptide and were terminated after 3 h with 10% (v/v, final concentration) trichloroacetic acid (TCA). Reactions were spiked with 2.6 μm [3H]AdoMet (specific activity of 2.02 mCi/μmol). Peptide products were hydrolyzed and separated as reported previously (16). Hydrolyzed amino acids from WT- and M48F-catalyzed reactions were derivatized using o-phthaldialdehyde (20) and analyzed with a Gemini® 3-μm C18 110 Å LC column 75 × 4.6 mm (Phenomenex) as stated previously (16). MMA, ADMA, or SDMA standard amino acids were used to verify the identity of the methylated products generated. The detection limit for this method is ∼10 pmol of methylated arginine in a 20-μl sample.
Mass Spectrometry Analysis of Methylated Arginines
A Nano-RPLC system consisting of a Spark Endurance autosampler (Emmen, Holland) and an ultrahigh pressure Eksigent (Dublin, CA) Nano-2D Ultra capillary/Nano-LC system, which features low void volume and high chromatographic reproducibility (21–23), was employed for peptide separation. Mobile phases A and B were 0.1% formic acid in 2% acetonitrile and 0.1% formic acid in 88% acetonitrile, respectively. Samples were loaded onto a trap (300-μm inner diameter × 5 mm, packed with Zorbax 5-μm C18 material) with 1% phase B at 10 μl/min, and the trap was washed for 2 min. A series of nanoflow gradients (flow rate was 250 nl/min) was used to back-flush the trapped samples onto the Nano-LC column (75-μm inner diameter, 75-cm length, packed with Pepmap 3-μm C18 material) for separation. The column was heated at 52 °C to improve both chromatographic resolution and reproducibility. Gradient profile was as follows: 1) a linear increase from 3 to 8% B over 5 min; 2) an increase from 8 to 27% B over 65 min; 3) an increase from 27 to 45% B over 30 min; 4) an increase from 45 to 98% B over 20 min; and 5) isocratic at 98% B for 20 min.
An LTQ/Orbitrap/Electron transferring dissociation (ETD) hybrid mass spectrometer (Thermo Fisher Scientific) was used for the identification of methyl site(s) and types, under data-dependent MS/MS mode. One cycle included an MS1 scan (m/z 310–2000) at a resolution of 60,000 followed by alternating collision-induced dissociation (CID) and ETD to fragment the three most abundant precursors found in the MS1 spectrum. The CID activation time was 30 ms with isolation width of 1.5 atomic mass units, the normalized activation energy at 35%, and the activation q at 0.25. As for ETD, a mixture of ultrapure helium and nitrogen (25% helium and 75% nitrogen, purity >99.995%) was used as the reaction gas. The ETD reaction time was set at 110 ms, and the isolation width was 2 atomic mass units for the precursor and 10 atomic mass units for the fluoranthene anions. Singly charged precursors were rejected for ETD.
CID and ETD activation spectra were processed using BioWorks (3.3.1, Thermo Scientific) incorporating the SEQUEST algorithm. Briefly, the potential charge states of ETD precursors were assigned by the Charger program (Thermo Scientific) and then searched against a database containing the sequence of the R3 (GGRGGFGGRGGFGGRGGFG) peptide. Differential modifications of MMA (+14.0156 Da) and DMA (+28.0313 Da) on Arg residues, and a static modification of N terminus acetylation (+42.01057 Da,) were employed. Mass tolerances were 10 ppm and 1.0 Da, respectively, for the precursor and fragments. A stringent set of criteria was applied for result filtering including high Xcorr and Δ-CN cut-off values (Xcorr > 1.8 for 1+ charge (CID), > 2.1 for 2+ charge, > 3 for 3+ charge, and >4 for 4+ charge, and Δ-CN >0.1), probability = 0.05 for CID, and then using the Sf scores (final score, > 0.85) for ETD. Where multiple methylation patterns were speculated for one ETD spectrum, confirmation of the most probable assignment was obtained by manual inspection of c and z ions. For putatively identified methylated peptides, the charge states and accurate m/z of precursors were obtained by the Orbitrap, and any identification with incorrect charge state assignment or precursor mass error larger than 10 ppm was eliminated. Based on the data search, the methylation types were identified via characteristic neutral losses under CID activation, combined with high-resolution product ion scan (23, 24). For each methylated peptide identified by ETD, the corresponding CID spectrum was manually inspected to determine the symmetry of the methylation.
The relative quantification of different methylated peptides was calculated by extracting ion currents of the precursors obtained by Orbitrap. For each identified methylated peptide, the extracting ion currents were extracted in a narrow m/z window (± 0.01 units) around the monoisotopic m/z for each available charge state. The area under the curve for each precursor at each charge state was calculated using Qualbrowser (Thermo Scientific) and then the percentage of each product was calculated. The calculation of different methylation type of the same molecular weight was based on the spectra count information generated by BioWorks software.
Gas-phase Calculation of the Activation Energy for Methylarginine Formation
All geometries and energies in the present study were computed using density functional theory (25) and the high-level composite method CBS-QB3 (26) as implemented in the Gaussian 09 program (27). For the density functional theory calculations, the B3LYP (28, 29) and M06-2X (30) methods with the 6-311++G(2d,p) basis set have been employed. Frequency calculations were used to characterize all structures as minima or first-order saddle points and to provide thermodynamic corrections.
RESULTS AND DISCUSSION
M48F-PRMT1 Generates ADMA and SDMA
Before the crystal structure of PRMT5 was solved, sequence alignment of Type I and II PRMTs indicated that the equivalent residue of Met-155 of PRMT1 in the Type II enzymes was a serine, a smaller amino acid. Therefore, Met-155 was previously hypothesized to specify ADMA and SDMA formation between Type I and II PRMTs by providing steric hindrance in the active site (12, 17, 31). However, M48L-, M48A-, M155L-, and M155A-PRMT1 exclusively generate MMA and ADMA (14, 16). When the crystal structure of human PRMT5 was solved (18), Phe-397 was found at the same location in the active site of PRMT5 as Met-48 in rat PRMT1. By Western blotting, a F397M-PRMT5 mutant was shown to generate both ADMA and SDMA (18).
We generated the M48F-PRMT1 mutant and analyzed the product formation by reverse phase HPLC with trace amounts of tritium-labeled AdoMet (Fig. 2). Separation of standard MMA, ADMA, and SDMA was clearly observed. Due to the low activity of M48F-PRMT1 (16), the radioactivity detected through the HPLC analysis was very low (Fig. 2, dotted line). A small amount of radioactivity eluted at the same retention time as MMA, and, notably, SDMA. Although the HPLC analysis method has been shown to be a reliable tool to identify methylarginine products (16), the low level of methylation activity exhibited by M48F mutant prompted us to confirm the presence of SDMA by another method.
FIGURE 2.
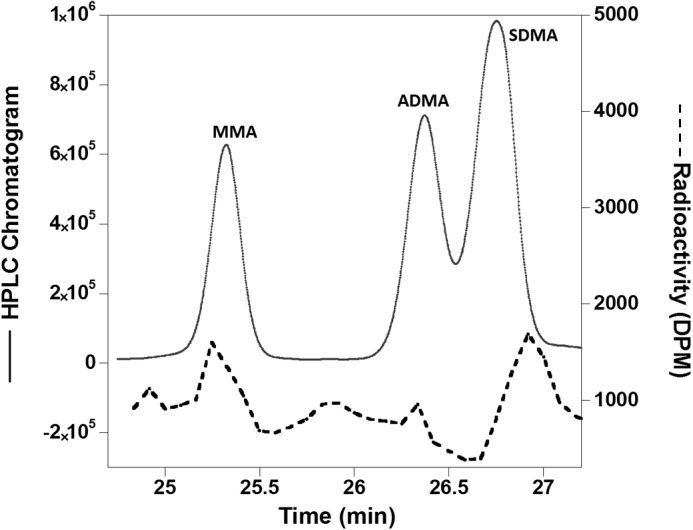
Methylation products of M48F-PRMT1 and the R3 peptide analyzed by reverse phase HPLC. The solid line shows the HPLC chromatogram of derivatized amino acid standards of MMA, ADMA, and SDMA. The dotted line shows peaks in the chromatogram where the radioactivity from tritiated AdoMet was incorporated into the methylated products.
We further employed high-resolution and accurate nano-flow liquid chromatography mass spectrometry (LC/MS) to analyze the methylation products of wild-type (WT) and M48F-PRMT1 (Fig. 3). Two orthogonal sequencing strategies, CID and ETD, were used with an Orbitrap MS detector. Although ETD is capable of accurately locating the methylation site, the alternating CID, with the help of the high resolution MS detector, can distinguish between asymmetric and symmetric dimethylation (23, 24). The comprehensive LC/MS analyses proved that WT-PRMT1 only forms MMA and ADMA, whereas M48F-PRMT1 generates MMA, ADMA, and a very small amount of SDMA with a relative abundance of 0.10% (Table 1). Thus, our results indicate that Met-48 in the active site of PRMT1 functions to guide ADMA synthesis over SDMA. Mutating Met-48 to phenylalanine in PRMT1 enables the mutated enzyme to generate SDMA as a new product, representing the first time that a Type I PRMT can be tuned to generate SDMA.
FIGURE 3.

Identification of ADMA and SDMA formation via neutral loss in wild type (top panel) and M48F-PRMT1 (bottom panel) reactions for triply charged dimethylated peptides. Peaks of m/z [M+3H-DMG]3+ and [M+2H-DMA]2+ indicate that there are ADMA sites, whereas the [M+3H-DMC]3+ peak indicates that there are SDMA sites.
TABLE 1.
Product analysis of M48F-PRMT1 by electron transfer dissociation and orbitrap mass spectrometry (ETD-MS)
Only intact peptide has been counted; the Xcorr versus charge state is 1.8(1+), 2.3(2+), 3(3+), 4.0(4+) for both CID and ETD activation; Sf = 0.85 for ETD. Probability = 0.05 for CID. * indicates mono-methylation; #(a) indicates asymmetric dimethylation, and #(s) indicates symmetric dimethylation.
| Sequence | m/z (3+) | Relative abundance (mean ± S.D.%) n = 3 | Sf/Xcorr | Mass error (ppm) |
|---|---|---|---|---|
| Methylation product of M48F-PRMT1 | ||||
| GGR*GGFGGRGGFGGRGGFG | 575.949 | 8.19 ± 1.23 | 0.95/6.46 | 1.70 |
| GGRGGFGGR*GGFGGRGGFG | 575.949 | 2.55 ± 0.40 | 0.88/5.20 | 1.70 |
| GGR#(a)GGFGGRGGFGGRGGFG | 580.621 | 2.35 ± 0.30 | 0.93/6.00 | 3.60 |
| GGR#(s)GGFGGRGGFGGRGGFG | 580.621 | 0.10 ± 0.04 | 0.93/4.20 | 3.60 |
| GGR#(a)GGFGGR*GGFGGRGGFG | 585.293 | 0.10 ± 0.03 | 0.90/6.18 | 6.45 |
| GGR#(a)GGFGGRGGFGGR*GGFG | 585.293 | 0.02 ± 0.008 | 0.85/0.98 | 4.64 |
| Methylation product of WT-PRMT1 | ||||
| GGR*GGFGGRGGFGGRGGFG | 575.949 | 7.69 ± 0.93 | 0.94/6.42 | 1.80 |
| GGRGGFGGR*GGFGGRGGFG | 575.949 | 3.96 ± 0.63 | 0.89/5.34 | 1.84 |
| GGR*GGFGGR*GGFGGRGGFG | 580.621 | 0.12 ± 0.042 | 0.86/5.57 | 3.61 |
| GGR#(a)GGFGGRGGFGGRGGFG | 580.621 | 1.68 ± 0.32 | 0.93/6.05 | 4.10 |
| GGRGGFGGR#(a)GGFGGRGGFG | 580.621 | 0.06 ± 0.027 | 0.87/5.30 | 4.24 |
| GGR#(a)GGFGGR*GGFGGRGGFG | 585.293 | 0.11 ± 0.024 | 0.93/6.40 | 6.52 |
Computational Simulation to Understand the Energetics for Different Methylarginine Formation
Although M48F-PRMT1 generates both SDMA and ADMA, the amount of SDMA is much lower than ADMA, and the overall activity of the mutant is severely impaired (16). To better understand why the formation of SDMA is limited, we questioned whether the formation of SDMA in the absence of protein was energetically more costly as compared with MMA and ADMA. Calculations have been applied for studies with protein lysine methyltransferases (PKMTs) suggesting that the energetics of the methyl transfer reactions, at least in part, may determine the product specificity of protein lysine methyltransferases (32–35). Recent calculations on PRMT1 predicted a more facile ability to form ADMA as compared with MMA, but SDMA formation was not investigated (36).
New gas-phase calculations were performed here to establish the free energies of activation for MMA, ADMA, and SDMA production in PRMTs (supplemental Computational Data). Modeling entire proteins in enzymatic reactions typically requires extensive computational resources and may not always be necessary to predict the energetics or product formation. For example, truncated methyl transfer reaction models involving dimethylammonium, tetramethylammonium, and trimethylsulfonium to dimethylamine have been successful in furthering the understanding of catechol O-methyl-transferase (37). As such, in our PRMT1 reaction model (Fig. 4), AdoMet was truncated at the Cα of the methionine moiety and at the oxolane ring of the adenosyl moiety. This is believed to appropriately represent the electronic structure of the cofactor because the main role of the positively charged sulfur atom of AdoMet is to attract electron density from the methyl group as the arginine abstracts the methyl group.
FIGURE 4.
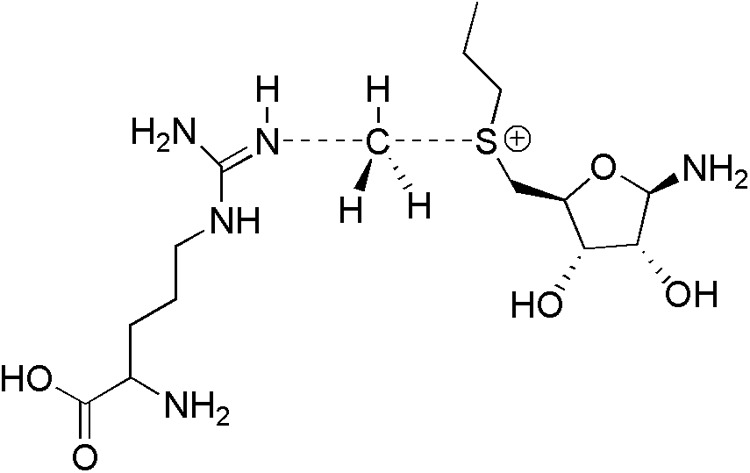
MMA-forming transition state model featuring a truncated AdoMet and a nucleophilic arginine.
The calculations were attempted with both protonated and deprotonated arginine in place of the nucleophilic arginine in Fig. 4 as experimental solvent isotope effects have indicated that prior deprotonation of the substrate guanidinium may not be required for PRMT1 methyl transfer (14). However, the energetics of the methyl transfer to the protonated arginine were predicted to be prohibitively large, i.e. ΔG‡ of 110.5, 98.0, and 107.2 for MMA, ADMA, and SDMA production, respectively (Table 2). The extreme activation barriers reflect the repulsive nature of the positively charged residue coming in close contact with the positively charged sulfur atom on AdoMet. The difference is also reflected in the perturbed transition structure distances (Table 3). It is clear that the protein environment is crucial for modulating the charges during the reaction and could be indicative of a concomitant proton transfer during reaction. Thus, we further focused on arginines in a nucleophilic state where the proton had been previously abstracted to have the electronics necessary to perform the nucleophilic attack.
TABLE 2.
Computed gas-phase free energies of activation, ΔG‡, (kcal/mol) for the MMA, ADMA, and SDMA methyl-transfer reactions using a protonated arginine
| MMA | ADMA | SDMA | |
|---|---|---|---|
| CBS-QB3 | 110.5 | 98.0 | 107.2 |
TABLE 3.
Computed transition structure distances (Å) between (AdoMet)S–C(methyl) and (methyl)C–N(protonated arginine) for the MMA, ADMA, and SDMA methyl-transfer reactions
| MMA |
ADMA |
SDMA |
||||
|---|---|---|---|---|---|---|
| S–C | C–N | S–C | C–N | S–C | C–N | |
| CBS-QB3 | 2.89 | 1.72 | 2.72 | 1.84 | 2.85 | 1.72 |
The computed transition structure distances between the SN2 reacting atoms (AdoMet)S–C(methyl) and (methyl)C–N(arginine) given in Table 4 are generally in close agreement between the different methods. CBS-QB3 has been shown to be particularly accurate in the prediction of methyl transfer reactions (37) and is assumed to produce the best gas-phase results for these reactions among the methods applied. Illustrations of the optimized transition structures for ADMA and SDMA formation are given in Fig. 5. Comparison of the computed ΔG‡ between MMA and ADMA formation finds that the preferred product differs based on the method used, e.g. CBS-QB3 and B3LYP favor MMA over ADMA in contrast to M06-2X (Table 5). Comparison with recent simulations by Zhang et al. (36) that carried out mixed quantum and molecular mechanical (QM/MM) ONIOM calculations on the entire protein utilizing B3LYP/6-31G(d) on a protonated arginine, part of AdoMet, and residues Arg-54, Glu-144, and Glu-153 found nearly identical energies and geometries as the current gas-phase work. For example, the QM/MM B3LYP/6-31G(d) method and the current gas-phase B3LYP/6-311++G(2d,p) method yielded energy barriers of 11.76 and 11.6 kcal/mol, respectively. The QM/MM calculation of ADMA production gave a nearly identical activation energy (11.63 kcal/mol) to MMA production; however, an MP2/6-31G(d) single point energy calculation significantly increased the difference in the energy barriers of MMA and ADMA, i.e. 19.08 and 14.94 kcal/mol, respectively. This strongly suggests a more facile formation of ADMA. The large energetic preference for MMA of ∼4 kcal/mol is unrealistic as this value would suggest an increase of several orders of magnitude in the rate constant for ADMA, whereas experimental kcat measurements yielded more muted values of 0.39 min−1 as compared with 0.79 min−1 for the unmodified and monomethylated substrate, respectively (38). The current M06-2X and B3LYP methods also overestimate the energy differences. This highlights that higher theory levels such as CBS-QB3, despite their large computational costs, may be required to properly reproduce experimental rates.
TABLE 4.
Computed transition structure distances (Å) between (AdoMet)S–C(methyl) and (methyl)C–N(arginine) for the MMA, ADMA, and SDMA methyl-transfer reactions
| MMA |
ADMA |
SDMA |
||||
|---|---|---|---|---|---|---|
| S–C | C–N | S–C | C–N | S–C | C–N | |
| CBS-QB3 | 2.29 | 2.14 | 2.26 | 2.17 | 2.27 | 2.15 |
| M06-2Xa | 2.24 | 2.07 | 2.22 | 2.08 | 2.23 | 2.08 |
| B3LYPa | 2.30 | 2.13 | 2.27 | 2.16 | 2.29 | 2.14 |
a 6-311++G(2d,p) basis set.
FIGURE 5.
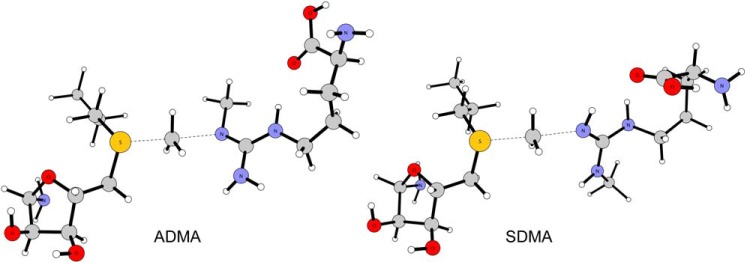
Transition structures for the ADMA and SDMA reactions computed using CBS-QB3.
TABLE 5.
Computed gas-phase free energies of activation, ΔG‡, (kcal/mol) for the MMA, ADMA, and SDMA methyl-transfer reactions
| MMA | ADMA | SDMA | |
|---|---|---|---|
| CBS-QB3 | 9.5 | 10.2 | 13.4 |
| M06-2Xa | 13.3 | 10.9 | 17.0 |
| B3LYPa | 11.6 | 13.5 | 17.2 |
a 6-311++G(2d,p) basis set.
The computed free energies of activation, ΔG‡, predicted a lower barrier for the ADMA reaction relative to SDMA at all theory levels (Table 4). The CBS-QB3 ΔΔG‡ of 3.2 kcal/mol favoring ADMA formation is a significant value and would represent several orders of magnitude difference in the enzymatic rate constant. The energy difference is in line with the exclusive formation of ADMA observed experimentally for WT-PRMT1. The higher ΔΔG‡ for SDMA can be rationalized when considering that methyl groups are electron-donating, making the unmethylated guanidino nitrogen on the SDMA-forming arginine a weaker nucleophile relative to the adjacent ADMA-forming methylated nitrogen atom.
Understanding Product Specificity in the PRMTs
Methyl transfer reactions catalyzed by AdoMet-dependent PRMTs proceed via an SN2 mechanism where the reacting arginine substrate acts as a nucleophile attacking the methyl group present on AdoMet, resulting in MMA or ADMA/SDMA (Figs. 1 and 4). The reaction has been proposed to involve 3 key conserved residues in the active site of PRMT1: Arg-54, Glu-144, and Glu-153 (Fig. 6A) (14). Arg-54 and Glu-144 help to properly position the substrates for the nucleophilic attack, whereas Glu-153 is hypothesized to play a role in increasing the nucleophilicity of the guanidinium moiety of the substrate via enhanced electronic effects. In addition, Glu-144 has also been postulated to act as the active site base, abstracting a proton from the reacting arginine either during or immediately after methyl transfer.
FIGURE 6.

A, the PRMT1 active site bound with S-adenosyl-l-homocysteine (SAH) and the substrate Arg (12) as observed in the crystal structure (Protein Data Bank (PDB) ID 1OR8). B, the PRMT5 active site bound with S-adenosyl-l-homocysteine (PDB ID 3UA3) (18) with the substrate Arg modeled into the active site. The position of the reacting Arg was approximated by overlaying with the 4GQB PRMT5 crystal structure that contained the Arg substrate and an analog of S-adenosyl-l-homocysteine (44). C, the PRMT1 active site with methyl substituents manually added to S-adenosyl-l-homocysteine to form AdoMet, and to Arg to make MMA, respectively. D, the PRMT5 active site with AdoMet and MMA modeled by manually adding a methyl group to each substrate.
Differences in the active sites of PRMT1 (12) (Type I) and PRMT5 (18) (Type II) structures can help elucidate the origin of ADMA versus SDMA specificity, respectively. The residues surrounding the substrate in PRMT1 are Arg 54, Met-48 (the residue mutated in the current study), Glu-144, Glu-153, Met-155, and His-293 with the corresponding residues in PRMT5 being Lys-385, Phe-379, Glu-499, Glu-508, Leu-509, and Ser-669, respectively (Fig. 6, A and B). The two Glu residues are conserved in all protein arginine methyltransferases (18), consistent with their proposed roles in methyl transfer in the PRMT enzymes.
Two potential avenues for controlling product specificity in the PRMTs are: 1) to use the protein active site to sterically control binding of MMA to preclude the formation of the undesired dimethylated product, and 2) to use active site residues to control the nucleophilicity of the desired guanidino nitrogen. Inspection of the crystal structures for both PRMT1 and PRMT5 with AdoMet and MMA modeled in the active sites (Fig. 6, C and D) suggests that the amino acid residues Met-48 in PRMT1 and Phe-379 in PRMT5 are positioned to sterically affect how MMA and the subsequent dimethylated products may bind. This suggests that different binding modes for MMA in PRMT1 and PRMT5 may function to control product specificity. These hypotheses, as well as more details on how the protein environment influences product specificity, are being investigated through molecular dynamic simulations and will be presented in a future publication.
The higher energy barrier for forming SDMA over ADMA may explain the limited amount of SDMA formed by the M48F-PRMT1 mutant. From an energetic point of view, it may be easier to convert a Type II PRMT (forms SDMA) into a Type I PRMT (forms ADMA) than the converse. Indeed, experimentally, the F379M-PRMT5 mutant, which generates SDMA and ADMA, showed significantly higher methylation activity than WT-PRMT5 (18).
Interestingly, Thompson and co-workers (39) recently reported that the symmetric dimethylation rate of PRMT5 is considerably lower than the monomethylation rate, as well as lower than the asymmetric dimethylation rate of PRMT1. These data may suggest that the meager amounts of SDMA observed in several studies with recombinant PRMT5 (39–41) are a reflection of the high energetic cost of synthesizing SDMA. This brings to light the exciting question of how this barrier is overcome in vivo.
Conclusion
Following our previous studies on the product specificity of PRMT1 (13, 16), we further investigated how the PRMT1 active site discriminates between ADMA and SDMA formation. We analyzed the product formation of M48F-PRMT1 with both reverse phase HPLC and ETD-MS. Under both methods, a small amount of SDMA can be detected in the methylation product of M48F, but not WT-PRMT1. Our present study revealed that a single amino acid mutation from Met-48 to Phe alters the product specificity of rat PRMT1 and allows both ADMA and SDMA generation. Gas-phase QM to compute the free energies of activation for MMA, ADMA, and SDMA formation revealed that SDMA formation is more energetically costly than ADMA, which would lead to a significant difference in methylation rate constants for asymmetric and symmetric dimethylation. An energetic argument is consistent with the activity displayed by the M48F-PRMT1 mutant as well as the activity displayed by recombinant PRMT5. Nevertheless, SDMA is formed in vivo at a ratio with ADMA of 1:3 (42, 43). This indicates that SDMA-forming methyltransferases overcome the energetic challenges in vivo, using mechanisms that may represent another exciting layer of enzymatic regulation.
Supplementary Material
This work was supported by National Science Foundation Award 0920776 (to J. M. H.); the American Recovery and Reinvestment Act, Auburn University, and the Alabama Supercomputer Center (to O. A.); National Institutes of Health Grants U54HD071594, DA027528, AI060260, HD075363, and HL103411 (to J. Q.); the Center for Protein Therapeutics (to J. Q.); and American Heart Association (AHA) Award 12SDG9450036 (to J. Q.).

This article contains supplemental Computational Data.
- PRMT
- protein arginine methyltransferase
- ADMA
- asymmetric dimethylarginine
- CID
- collision-induced dissociation
- ETD
- electron transferring dissociation
- AdoMet
- S-adenosylmethionine
- SDMA
- symmetric dimethylarginine
- MMA
- monomethylarginine
- QM
- quantum mechanical
- MM
- molecular mechanical.
REFERENCES
- 1. Bedford M. T., Clarke S. G. (2009) Protein arginine methylation in mammals: who, what, and why. Mol. Cell 33, 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bedford M. T. (2007) Arginine methylation at a glance. J. Cell Sci. 120, 4243–4246 [DOI] [PubMed] [Google Scholar]
- 3. Huang S., Litt M., Felsenfeld G. (2005) Methylation of histone H4 by arginine methyltransferase PRMT1 is essential in vivo for many subsequent histone modifications. Genes Dev. 19, 1885–1893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Levine T. B., Levine A. B. (2006) Metabolic Syndrome and Cardiovascular Disease, p. 421, Wiley-Blackwell, Chichester, UK [Google Scholar]
- 5. Vallance P., Leiper J. (2004) Cardiovascular biology of the asymmetric dimethylarginine:dimethylarginine dimethylaminohydrolase pathway. Arterioscler. Thromb. Vasc. Biol. 24, 1023–1030 [DOI] [PubMed] [Google Scholar]
- 6. Cloos P. A., Christensen J., Agger K., Helin K. (2008) Erasing the methyl mark: histone demethylases at the center of cellular differentiation and disease. Genes Dev. 22, 1115–1140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jansson M., Durant S. T., Cho E. C., Sheahan S., Edelmann M., Kessler B., La Thangue N. B. (2008) Arginine methylation regulates the p53 response. Nat. Cell Biol. 10, 1431–1439 [DOI] [PubMed] [Google Scholar]
- 8. Wysocka J., Allis C. D., Coonrod S. (2006) Histone arginine methylation and its dynamic regulation. Front. Biosci. 11, 344–355 [DOI] [PubMed] [Google Scholar]
- 9. De Gennaro Colonna V., Bianchi M., Pascale V., Ferrario P., Morelli F., Pascale W., Tomasoni L., Turiel M. (2009) Asymmetric dimethylarginine (ADMA): an endogenous inhibitor of nitric oxide synthase and a novel cardiovascular risk molecule. Med. Sci. Monit. 15, RA91–R101 [PubMed] [Google Scholar]
- 10. Böger R. H. (2003) The emerging role of asymmetric dimethylarginine as a novel cardiovascular risk factor. Cardiovasc Res. 59, 824–833 [DOI] [PubMed] [Google Scholar]
- 11. Deleted in proof.
- 12. Zhang X., Cheng X. (2003) Structure of the predominant protein arginine methyltransferase PRMT1 and analysis of its binding to substrate peptides. Structure 11, 509–520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gui S., Wooderchak-Donahue W. L., Zang T., Chen D., Daly M. P., Zhou Z. S., Hevel J. M. (2013) Substrate-induced control of product formation by protein arginine methyltransferase 1. Biochemistry 52, 199–209 [DOI] [PubMed] [Google Scholar]
- 14. Rust H. L., Zurita-Lopez C. I., Clarke S., Thompson P. R. (2011) Mechanistic studies on transcriptional coactivator protein arginine methyltransferase 1. Biochemistry 50, 3332–3345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Obianyo O., Osborne T. C., Thompson P. R. (2008) Kinetic mechanism of protein arginine methyltransferase 1. Biochemistry 47, 10420–10427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gui S., Wooderchak W. L., Daly M. P., Porter P. J., Johnson S. J., Hevel J. M. (2011) Investigation of the molecular origins of protein-arginine methyltransferase I (PRMT1) product specificity reveals a role for two conserved methionine residues. J. Biol. Chem. 286, 29118–29126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhang X., Zhou L., Cheng X. (2000) Crystal structure of the conserved core of protein arginine methyltransferase PRMT3. EMBO J. 19, 3509–3519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sun L., Wang M., Lv Z., Yang N., Liu Y., Bao S., Gong W., Xu R. M. (2011) Structural insights into protein arginine symmetric dimethylation by PRMT5. Proc. Natl. Acad. Sci. U.S.A. 108, 20538–20543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cornell K. A., Swarts W. E., Barry R. D., Riscoe M. K. (1996) Characterization of recombinant Escherichia coli 5′-methylthioadenosine/S-adenosylhomocysteine nucleosidase: analysis of enzymatic activity and substrate specificity. Biochem. Biophys. Res. Commun. 228, 724–732 [DOI] [PubMed] [Google Scholar]
- 20. Markowski P., Baranowska I., Baranowski J. (2007) Simultaneous determination of l-arginine and 12 molecules participating in its metabolic cycle by gradient RP-HPLC method: application to human urine samples. Anal. Chim. Acta 605, 205–217 [DOI] [PubMed] [Google Scholar]
- 21. Duan X., Young R., Straubinger R. M., Page B., Cao J., Wang H., Yu H., Canty J. M., Qu J. (2009) A straightforward and highly efficient precipitation/on-pellet digestion procedure coupled with a long gradient nano-LC separation and Orbitrap mass spectrometry for label-free expression profiling of the swine heart mitochondrial proteome. J. Proteome Res. 8, 2838–2850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Qu J., Qu Y., Straubinger R. M. (2007) Ultra-sensitive quantification of corticosteroids in plasma samples using selective solid-phase extraction and reversed-phase capillary high-performance liquid chromatography/tandem mass spectrometry. Anal Chem. 79, 3786–3793 [DOI] [PubMed] [Google Scholar]
- 23. Wang H., Straubinger R. M., Aletta J. M., Cao J., Duan X., Yu H., Qu J. (2009) Accurate localization and relative quantification of arginine methylation using nanoflow liquid chromatography coupled to electron transfer dissociation and orbitrap mass spectrometry. J. Am. Soc. Mass Spectrom 20, 507–519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Fisk J. C., Li J., Wang H., Aletta J. M., Qu J., Read L. K. (2013) Proteomic analysis reveals diverse classes of arginine methylproteins in mitochondria of trypanosomes. Mol. Cell. Proteomics 12, 302–311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Parr R. G., Yang W. (1989) Density-Functional Theory of Atoms and Molecules, Oxford University Press, New York [Google Scholar]
- 26. Ochterski J. W., Petersson G. A., Montgomery J. A. (1996) A complete basis set model chemistry. V. Extensions to six or more heavy atoms. J. Chem. Phys. 104, 2598–2619 [Google Scholar]
- 27. Frisch M. J., Trucks G. W., Schlegel H. B., Scuseria G. E., Robb M. A., Cheeseman J. R., Scalmani G., Barone V., Mennucci B., Petersson G. A., Nakatsuji H., Caricato M., Li X., Hratchian H. P., Izmaylov A. F., Bloino J., Zheng G., Sonnenberg J. L., Hada M., Ehara M., Toyota K., Fukuda R., Hasegawa J., Ishida M., Nakajima T., Honda Y., Kitao O., Nakai H., Vreven T., Montgomery J. A., Jr., Peralta J. E., Ogliaro F., Bearpark M., Heyd J. J., Brothers E., Kudin K. N., Staroverov V. N., Kobayashi R., Normand J., Raghavachari K., Rendell A., Burant J. C., Iyengar S. S., Tomasi J., Cossi M., Rega N., Millam J. M., Klene M., Knox J. E., Cross J. B., Bakken V., Adamo C., Jaramillo J., Gomperts R., Stratmann R. E., Yazyev O., Austin A. J., Cammi R., Pomelli C., Ochterski J. W., Martin R. L., Morokuma K., Zakrzewski V. G., Voth G. A., Salvador P., Dannenberg J. J., Dapprich S., Daniels A. D., Farkas Ö., Foresman J. B., Ortiz J. V., Cioslowski J., Fox D. J. (2009) Gaussian 09, Revision B. 01, Gaussian, Inc., Wallingford, CT [Google Scholar]
- 28. Becke A. D. (1993) Density-Functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 98, 5648–5652 [Google Scholar]
- 29. Lee C., Yang W., Parr R. G. (1988) Development of the Colle-Salvetti correlation-energy formula into a functional of the electron-density. Phys. Rev. B Condens. Matter 37, 785–789 [DOI] [PubMed] [Google Scholar]
- 30. Zhao Y., Truhlar D. G. (2008) The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor Chem. Acc. 120, 215–241 [Google Scholar]
- 31. Branscombe T. L., Frankel A., Lee J. H., Cook J. R., Yang Z., Pestka S., Clarke S. (2001) PRMT5 (Janus kinase-binding protein 1) catalyzes the formation of symmetric dimethylarginine residues in proteins. J. Biol. Chem. 276, 32971–32976 [DOI] [PubMed] [Google Scholar]
- 32. Chu Y., Yao J., Guo H. (2012) QM/MM MD and free energy simulations of G9a-like protein (GLP) and its mutants: understanding the factors that determine the product specificity. PLoS One 7, e37674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hu P., Zhang Y. (2006) Catalytic mechanism and product specificity of the histone lysine methyltransferase SET7/9: an ab initio QM/MM-FE study with multiple initial structures. J Am. Chem. Soc. 128, 1272–1278 [DOI] [PubMed] [Google Scholar]
- 34. Yao J., Chu Y., An R., Guo H. (2012) Understanding product specificity of protein lysine methyltransferases from QM/MM molecular dynamics and free energy simulations: the effects of mutation on SET7/9 beyond the Tyr/Phe switch. J. Chem. Inf. Model 52, 449–456 [DOI] [PubMed] [Google Scholar]
- 35. Xu Q., Chu Y. Z., Guo H. B., Smith J. C., Guo H. (2009) Energy triplets for writing epigenetic marks: insights from QM/MM free-energy simulations of protein lysine methyltransferases. Chemistry 15, 12596–12599 [DOI] [PubMed] [Google Scholar]
- 36. Zhang R., Li X., Liang Z., Zhu K., Lu J., Kong X., Ouyang S., Li L., Zheng Y. G., Luo C. (2013) Theoretical insights into catalytic mechanism of protein arginine methyltransferase 1. PLoS One 8, e72424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gunaydin H., Acevedo O., Jorgensen W. L., Houk K. N. (2007) Computation of accurate activation barriers for methyl-transfer reactions of sulfonium and ammonium salts in aqueous solution. J. Chem. Theory Comput. 3, 1028–1035 [DOI] [PubMed] [Google Scholar]
- 38. Osborne T. C., Obianyo O., Zhang X., Cheng X., Thompson P. R. (2007) Protein arginine methyltransferase 1: positively charged residues in substrate peptides distal to the site of methylation are important for substrate binding and catalysis. Biochemistry 46, 13370–13381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wang M., Xu R. M., Thompson P. R. (2013) Substrate specificity, processivity, and kinetic mechanism of protein arginine methyltransferase 5. Biochemistry 52, 5430–5440 [DOI] [PubMed] [Google Scholar]
- 40. Zurita-Lopez C. I., Sandberg T., Kelly R., Clarke S. G. (2012) Human protein arginine methyltransferase 7 (PRMT7) is a Type III enzyme forming ω-NG-monomethylated arginine residues. J. Biol. Chem. 287, 7859–7870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Rho J., Choi S., Seong Y. R., Cho W. K., Kim S. H., Im D. S. (2001) Prmt5, which forms distinct homo-oligomers, is a member of the protein-arginine methyltransferase family. J. Biol. Chem. 276, 11393–11401 [DOI] [PubMed] [Google Scholar]
- 42. Paik W. K., Kim S. (1980) Natural occurrence of various methylated amino acid derivatives. In: Protein Methylation (Meister A., ed) pp. 8–25, John Wiley & Sons, New York [Google Scholar]
- 43. Matsuoka M. (1972) [Epsilon-N-methylated lysine and guanidine-N-methylated arginine of proteins. 3. Presence and distribution in nature and mammals]. Seikagaku 44, 364–370 [PubMed] [Google Scholar]
- 44. Antonysamy S., Bonday Z., Campbell R. M., Doyle B., Druzina Z., Gheyi T., Han B., Jungheim L. N., Qian Y., Rauch C., Russell M., Sauder J. M., Wasserman S. R., Weichert K., Willard F. S., Zhang A., Emtage S. (2012) Crystal structure of the human PRMT5:MEP50 complex. Proc. Natl. Acad. Sci. U.S.A. 109, 17960–17965 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.