

Background: Genetic interaction between Runx2 and Pin1 is critical for embryonic bone formation.
Results: Pin1 is a critical modifying enzyme promoting both subnuclear accumulation and protein acetylation of Runx2.
Conclusion: Pin1 determines the fate of Runx2 protein in osteoblast differentiation.
Significance: The modulation of Pin1 activity may be a clinical target for the regulation of bone formation.
Keywords: Cell Differentiation, Osteoblasts, Protein Conformation, Protein Phosphorylation, Transcription Factors, FGF Signaling, Pin1, Runx2, Acetylation, Subnuclear Accumulation
Abstract
Fibroblast growth factor 2 (FGF2) signaling plays a pivotal role in bone growth/differentiation through the activation of osteogenic master transcription factor Runx2, which is mediated by the ERK/MAPK-dependent phosphorylation and the p300-dependent acetylation of Runx2. In this study, we found that Pin1-dependent isomerization of Runx2 is the critical step for FGF2-induced Runx2 transactivation function. We identified four serine or threonine residues in the C-terminal domain of Runx2 that are responsible for Pin1 binding and structural modification. Confocal imaging studies indicated that FGF2 treatment strongly stimulated the focal accumulation of Pin1 in the subnuclear area, which recruited Runx2. In addition, active forms of RNA polymerase-II also colocalized in the same subnuclear compartment. Dipentamethylene thiuram monosulfide, a Pin1 inhibitor, strongly attenuated their focal accumulation as well as Runx2 transactivation activity. The Pin1-mediated structural modification of Runx2 is an indispensable step connecting phosphorylation and acetylation and, consequently, transcriptional activation of Runx2 by FGF signaling. Thus, the modulation of Pin1 activity may be a target for the regulation of bone formation.
Introduction
Fibroblast growth factor (FGF) signaling represents a fundamental mechanism in modulating numerous cellular physiologies, including osteogenesis (1–3), neurogenesis (4–6), and homeostasis of vascular endothelium (7). Essential roles for FGF signaling have been defined in both endochondral (8, 9) and intramembranous bone formation (10, 11) by the identification of mutations in the receptors for FGFs, and it has been revealed that these signaling pathways are critical for skeletal development. FGF2 is one of the most powerful extracellular stimuli to regulate multiple regions of bone growth and osteoblast differentiation. FGF2 activates ERK and p38 MAPK propagation into the nucleus, which triggers the activation of nuclear transcription factors, thereby leading to downstream gene expression (12–15).
Runx2, a master transcription factor of bone formation, is a critical target of the FGF2 signaling pathway. Runx2, in response to FGF2 signaling, directly binds to the promoter region of the osteocalcin (Oc) gene, a hallmark of bone mineralization, and subsequently activates the expression of Oc and other bone marker genes (16). We have reported previously that the functional activity of Runx2 requires the MAPK signaling pathway, which is driven by both FGF2 binding to the receptor or the receptor activation by gain-of-function mutation (14, 15, 17, 18). These studies have consistently illuminated phosphorylational control of Runx2 in osteoblast differentiation. FGF2 signaling not only stimulates Runx2 phosphorylation and its transactivation activity (15, 19), it also regulates Runx2 acetylation and ubiquitination (17). In the course of the MAPK signaling pathway, Runx2 actively interplays with various modifying enzymes such as Ser/Thr kinases and histone acetyltransferase (HAT)2 (13, 15, 20, 21).
MAPK is integral in determining the functional activity of Runx2, especially in terms of Runx2 dosage at the physiological level. Specifically, cleidocranial dysplasia (CCD), caused by genetic insufficiency of Runx2, can be overcome by osteoblast-specific overexpression of constitutively active MEK1 (MEK1-Ca) or exaggerated by the expression of dominant-negative MEK (22). These results suggest that ERK/MAPK-induced phosphorylation of Runx2 may stimulate its transcriptional activity, thereby masking the effects of the genetic insufficiency of the Runx2 heterozygote. Recently, we found that the CCD phenotype develops in Pin1-deficient mice and identified that Pin1 plays a crucial role in osteogenesis and specifically targets Runx2 for further protein stabilization (23). Pin1 is a peptidyl-prolyl cis-trans isomerase (PPIase) that catalyzes the conformation of rigid peptide bonds in the phosphorylated serine-proline or phosphorylated threonine-proline backbone, thereby dramatically altering the structural conformation of the protein (24–26). Therefore, the molecular interaction between Runx2 and Pin1 implies a molecular mechanism beyond Runx2 phosphorylation.
Pin1 is a unique enzyme among PPIases. Its association with substrates involves a WW domain that preferentially recognizes sequence motifs containing a phosphoserine or a phosphothreonine followed by a proline ((pS/pT)P motif, proline-directed phosphorylation) in target substrates. For this reason, Pin1 frequently functions as a binary switch to lay target substrates under two states of conformations (cis or trans) in the protein structure, thereby leading to distinct fates of the phosphorylated proteins by Pin1 activity. Therefore, Pin1 is associated tightly with cell signaling and Ser/Thr kinase activity as an important structural modifier. Pin1, as a MAPK responder, has a crucial role in the oogenesis of a fruit fly (27). In addition, Pin1 target sequences are shared by several protein kinases, such as ERK (27–29), cyclin-dependent kinase (30, 31), and GSK3a (32), indicating that ERK-phosphorylated Runx2 may also be the target of Pin1-mediated conformational and functional alterations.
Here, we show that prolyl isomerization of Runx2 mediated by Pin1 is a crucial post-phosphorylation event that constitutes a fate-determining switch to link phosphorylation with further molecular modification events of Runx2 during FGF/FGFR2 signaling. Given the critical role of Runx2 fate, our data may represent a profound understanding beyond the previous knowledge for skeletal development and a therapeutic approach for the aberrant control of Runx2 in various pathologies, including osteoporosis, cleidocranial dysplasia, craniosynostosis, and cancers.
EXPERIMENTAL PROCEDURES
Cell Culture and Reagents
C2C12, HEK-293, and mouse embryonic fibroblast (MEF) cells were maintained in Dulbecco's modified Eagle's medium (DMEM) with 10% heat-inactivated fetal bovine serum (DMEM, 10% FBS) supplemented with antibiotics. MC3T3-E1, mouse calvarial osteoblast, and mouse bone marrow stromal cells were maintained in α-minimum essential medium. MEFs were isolated from E13.5 embryos with Pin1+/+, Pin1+/−, and Pin1−/− genotypes, and cells from passages 3 to 5 were used. For the cultivation of primary osteoblasts, calvarial cells were collected from E18.5 embryos using sequential collagenase digestion. For bone marrow stromal cells culture, bone marrow in culture medium was plated into a 100-mm tissue culture dish. The cells were incubated until ∼80% confluent and then washed with PBS to remove nonadherent cells as follows: U0126 (ERK/MAPK inhibitor), anacardic acid (histone acetyl transferase (HAT) inhibitor), trichostatin A (HDAC inhibitor), juglone (33) or dipentamethylene thiuram monosulfide (DTM) (34). Pin1 inhibitors were purchased from Thermo Scientific (Rockford, IL). Noncytotoxic effects of juglone (∼20 μm) and DTM (∼5 μm) were proved in the C2C12 and MC3T3-E1 cells by CCK-8 assay (data not shown).
RNA Extraction and Quantitative Real Time PCR Expression Analyses
RNAs were isolated from cultured cells using the Qiazol Lysis Reagent from Qiagen (Mannheim, Germany), according to the manufacturer's protocol. cDNAs were synthesized from 1 μg of total RNA using the SuperScript II first-strand synthesis system reverse transcriptase kit (Invitrogen) and subsequently used for SYBR Green-based real-time PCRs using a standard protocol (Takara). PCR primers for mouse Oc and glyceraldehyde-3-phosphate dehydrogenase (Gapdh) genes were used as described previously (35)
Immunofluorescent Detection of Runx2 and Pin1
Immunofluorescent detection of Pin1 and Runx2 required antigen retrieval. Briefly, the specimens were fixed in 4% formaldehyde and boiled in Tris/EDTA buffer (pH 9.2) with 5% urea for 10 min. The cells were then stained with the appropriate primary antibody and fluorescent-conjugated secondary antibody, visualized with a Carl Zeiss LSM700 microscope, and analyzed with ZEN2011 software (Carl Zeiss, Oberkochen, Germany). For live cell measurements, MC3T3-E1 cells were transiently transfected with EGFP-Runx2 and DsRed-Pin1 plasmids using the neon transfection system (Invitrogen), and cells were monitored by time-lapse confocal microscopy (LSM700, Carl Zeiss) after 3 h of transfection. The supplemental movie covers 669 min and is composed of pictures taken every 15 min.
GST Pulldown Assay, Immunoprecipitation, and Immunoblot Analyses
Cellular proteins were prepared in a lysis buffer of 50 mm HEPES (pH 7.5), 150 mm NaCl, 100 mm NaF, 1 mm DTT, 1 mm EDTA, 0.25% sodium deoxycholate, 0.25% CHAPS, 1% Nonidet P-40, and 10% glycerol supplemented with protease and phosphatase inhibitors, including Na3VO4. For the acetylation, the buffer was supplemented with 1 mm NaB. For the GST pulldown assay, cell lysates (1 mg) were incubated with recombinant GST proteins and bead mixtures for 30 min at 4 °C.
Subtilisin Protection Assay
Subtilisin protection assay was performed as described previously (36) with slight modifications. Briefly, His-Runx2 protein was ectopically expressed together with MEK1-Ca in MEF-Pin1−/− cells and purified by nitrilotriacetic acid affinity purification. Purified Runx2 protein was directly dissolved in 50 mm HEPES (pH 7.4), 100 mm NaCl, and 1 mm MgCl2 supplemented with phosphatase inhibitors and incubated with Pin1-WT, Pin1-C113A, or Pin1-ΔPPIase proteins. After 30 min of incubation at room temperature, reaction mixtures were cooled on ice, followed by incubation with subtilisin for 5 s. The reaction was stopped by the addition of SDS sample buffer, and the proteolytic fragments of Runx2 were detected by immunoblot analysis with an anti-Runx2 monoclonal antibody. Recombinant Pin1 proteins for this assay were produced by an in vitro transcription and translation reaction.
Transactivation Activity of Runx2
The transcriptional activity of Runx2 was measured using the 6×OSE2-Luc or the rat Oc promoter-Luc reporter plasmid vectors, as described previously (15). MC3T3-E1 or C2C12 cells were transiently transfected with the indicated plasmids. Cells were treated with FGF2 (20 ng/ml) together with either juglone (10 μm) or U0126 (10 μm) 24 h after transfection, followed by the measurement of luciferase activity after an additional 18 h.
Cellular Transfections
Electroporation using the neon transfection system was performed for DNA transfection into the MC3T3-E1 or C2C12 cells. Transfection of the HEK-293 cells was performed using Polyget reagent (SignaGen Laboratory) according to the manufacturer's instructions.
Statistical Analyses
All quantitative data are presented as the mean ± S.D. Each experiment was performed at least three times, and the results from one representative experiment are shown. Statistical differences were analyzed by Student's t test. A p value of statistical significance was mentioned in the individual figure legend.
RESULTS
Pin1 Enhances the Transcriptional Activity of Runx2
We investigated whether phosphorylation was required for the transactivation function of Runx2 via FGF2 signaling. To test whether Pin1 has an important role in this process, we examined the transactivation activity of Runx2 on its specific reporter plasmids. C2C12 cells were transiently transfected with a promoter construct consisting of −208 to −23 bp of the rat Oc proximal promoter, which contains a Runx2-binding site (15), and exposed the cells to FGF2 in the presence or absence of juglone, a potent inhibitor of Pin1 (33), after which reporter activities were determined (Fig. 1A). FGF2 treatment increased the reporter activity 3-fold, whereas juglone treatment almost completely suppressed the reporter activity in the presence and absence of FGF2 treatment. In contrast, Runx2-mediated transactivation activity of the Oc promoter was synergistically enhanced by FGF2 treatment and Pin1 overexpression (Fig. 1B). Notably, only Pin1 overexpression could strongly promote Oc promoter activation in the absence of FGF2 treatment. Similar results were obtained when the cells were transfected with the osteoblast-specific element 2 (OSE2) reporter construct, which is derived from a genomic sequence within the promoter region of mouse Oc (16) (Fig. 1, C and D). The overexpression of MEK1-Ca or Runx2 independently enhanced 6×OSE2-Luc reporter activity in C2C12 cells, and the coexpression of these two components synergistically stimulated Luc reporter activity (Fig. 1C). Juglone-induced inhibition of Pin1 markedly suppressed the trans-activation activity of Runx2 that resulted from MEK1-Ca overexpression (Fig. 1C). In addition, FGF2 treatment stimulated the 6×OSE2-Luc reporter activity by 5–10-fold, and the overexpression of wild-type Pin1 synergistically enhanced the transcriptional activity of the FGF2-stimulated Runx2, as observed in the Oc promoter (Fig. 1D). However, the overexpression of dominant-negative Pin1 mutants suppressed FGF2-stimulated Runx2 transcriptional activity (Fig. 1D). These results indicate that Pin1 is indispensable for FGF2-stimulated Runx2 transcriptional activity.
FIGURE 1.

Pin1 is essential for FGF2-induced Runx2 transactivation activity. A, FGF2-induced Oc promoter activation was fully abrogated by the inhibition of Pin1 activity. C2C12 cells were transiently transfected with the Oc-Luc reporter vector. After 18 h of transfection, cells were treated with 20 ng/ml FGF2 in the presence or absence of juglone (10 μm) for 24 h, and the luciferase activity was measured from the harvested cells. Data represent the means ± S.D. (n = 3). *, p < 0.05 (compared with DMSO-treated group); **, p < 0.05 (compared with DMSO/FGF2-treated group). B, Oc-Luc reporter activity was strongly enhanced by Pin1 overexpression. C2C12 cells were transiently transfected with the Oc-Luc reporter vector either with the empty or the Pin1-expression plasmid. After 24 h, cells were treated with 20 ng/ml FGF2 for an additional 24 h, and luciferase activity was measured from the harvested cells. Data represent the means ± S.D. (n = 3). *, p < 0.05 (compared with EV/DMSO group); **, p < 0.05 (compared with Pin1/FGF2 group); #, p < 0.05 (compared with EV/FGF group). C, down-regulation of transactivation activity of Runx2 by the inhibition of Pin1 activity. C2C12 cells stably transfected with the p6×OSE2-Luc reporter vector (OSE2-C2C12) were transiently transfected with Runx2 along with either an empty vector or a MEK1-Ca plasmid. After 18 h of transfection, the cells were incubated with juglone (10 μm) for an additional 24 h, and luciferase activity was measured from the harvested cells. Data represent the means ± S.D. (n = 3). *, p < 0.0005 (compared with DMSO group expressing only Runx2 without MEK1-Ca expression); **, p < 0.0005 (compared with DMSO group expressing both Runx2 and MEK1-Ca). D, FGF2-induced Runx2 transactivation activity was dependent on Pin1 activity. MC3T3-E1 cells were transiently transfected with p6×OSE-Luc and either wild-type Pin1 or mutant Pin1 plasmids (C113A or Y23A) for 24 h. Cells were cultured in the presence or absence of FGF2 for an additional 18 h. Data represent the means ± S.D. (n = 3). *, p < 0.005 (compared with EV/FGF2 group); **, p < 0.005 (compared with Pin1-WT/FGF2 group). E and F, induction of Oc mRNA expression by FGF2 was abrogated by the treatment of the Pin1 inhibitor or the absence of Pin1. C2C12 cells were stimulated with 20 ng/ml FGF2 in the presence or absence of juglone (10 μm) for 24 h. E, equal numbers of primary mOBs from Pin1+/+ and Pin1−/− mice were treated with FGF2 (20 ng/ml) for 24 h after confluence. Data represent the means ± S.D. (n = 3). **, p < 0.001 (compared with DMSO/veh. group); *, p < 0.05 (compared with DMSO/FGF2 group). F, total RNAs were isolated, and the Oc mRNA levels were analyzed by quantitative real time PCR. Data represent the means ± S.D. (n = 3). **, p < 0.001 (compared with DMSO treated mOB-Pin1+/+); *, p < 0.005 (compared with FGF2 treated mOB-Pin1+/+). Veh, vehicle; EV, empty vector; RLU, relative luciferase units.
Next, we determined whether Pin1 influences the downstream gene expression of Runx2, and we consistently observed that Oc mRNA induced by FGF2 stimuli was strongly down-regulated by juglone (Fig. 1E). The Oc mRNA level in C2C12 premyoblasts was increased ∼8-fold by FGF2 treatment, but the increase was completely abrogated by juglone (Fig. 1E). A similar result was obtained in primary osteoblast cultures from Pin1+/+ and Pin1−/− mice (Fig. 1F).
Pin1-mediated Prolyl Isomerization Is Required for Runx2 Stabilization in FGF2/MAPK Signaling
Previously, we demonstrated that the Runx2 protein level is strongly down-regulated in Pin1-null mice, which is not due to decreased Runx2 mRNA expression but is due to enhanced ubiquitination of Runx2, suggesting that Pin1 can mediate post-phosphorylational control of Runx2 (37). In addition, FGF2 signaling is required for Runx2 stabilization and its transactivation activity (17). These data indicate that Pin1 plays a critical role in the phosphorylation and further stabilization of Runx2 during FGF2 signaling. For this reason, we confirmed that ERK/MAPK phosphorylation is not affected by the absence of Pin1. FGF2 treatment still stimulated immediate early phosphorylation of ERK in both mouse calvarial osteoblast (mOB) from the Pin1+/+ or Pin1−/− mice (Fig. 2A). However, we observed that endogenous Runx2 protein levels were strongly down-regulated by Pin1 deficiency (Fig. 2A). As observed in previous literature (37), the Runx2 protein level was increased by FGF2 stimulation in Runx2+/+ cells but was still decreased in Pin1−/− cells (Fig. 2A). We also found that the Runx2 protein level is increased in MC3T3-E1 cells transfected with gain-of-function mutant FGFR2s, but the increase was attenuated in cells exposed to DTM, a Pin1 inhibitor (Fig. 2B). A protein life span analysis using tetracycline-inducible Runx2 expression system (38) demonstrated that Runx2 stability is strongly decreased by Pin1 deficiency (Fig. 2C), indicating that Pin1 is still functioning in FGFR2-enhanced Runx2 protein stabilization. These data suggest that the FGF2-stimulated ERK pathway and Runx2 phosphorylation are intact in the Pin1-null mice, indicating that phosphorylation occurs prior to prolyl isomerization and that the MAPK-dependent phosphorylation of the (S/T)P dipeptide is a target of Pin1 (27–29). Therefore, we examined the ability of MEK1 to increase the level of Runx2 protein, as MEK1 is known to act downstream of FGF2 and to stimulate ERK/MAPK. Coexpression of MEK1-Ca increased both ERK phosphorylation and Runx2 expression (Fig. 2D, 3rd lane) but still attenuated ERK phosphorylation and Runx2 expression in the juglone-treated cells (Fig. 2D, 4th lane), which demonstrates that Pin1 activity is important for the post-phosphorylational stabilization of Runx2. Similarly, blocking MAPK activity with U0126 treatment strongly down-regulated Runx2 stability even when Pin1 and MEK-Ca were coexpressed (Fig. 2E). Interestingly, endogenous Pin1 was sufficient to support Runx2 stabilization after its phosphorylation (Fig. 2E, 1st and 2nd lanes). The expression of MEK1-Ca with Pin1-C113A, but not with wild-type Pin1, was found to abolish the MEK-dependent increase in Runx2 protein levels (Fig. 2F). The Pin1-C113A mutant reduced the Runx2 levels below those that were observed in the control cells, indicating that this mutant interferes (dominant-negative action) with the stabilizing activity of endogenous Pin1 (Fig. 2F). This evidence demonstrates that phosphorylated Runx2 by ERK may be a critical target for Pin1 in FGF2 signaling.
FIGURE 2.

FGF2-induced increase of Runx2 protein level requires phosphorylation and subsequent structural modification by Pin1. A, determination of Runx2 protein levels depending on the Pin1 activity in mOBs. mOBs isolated from Pin1+/+ or Pin1−/− mice were cultured in the presence or absence of FGF2 for 24 h. B, gain-of-function mutations of FGFR2 increased Runx2 protein levels, which was attenuated by Pin1 inhibitor treatment. C342S and S354C are constitutively active FGFR2 mutants and have been shown to cause the craniosynostosis syndrome in humans. MC3T3-E1 cells were transiently transfected with the indicated constructs and were further incubated for 48 h under physiological culture conditions with or without the Pin1 inhibitor, DTM (1 μm). C, determination of Runx2 protein half-life in the presence or absence of Pin1 activity. Tet-On-inducible Runx2 expression vectors (38) were transiently expressed in MEF-Pin1+/+ and MEF-Pin1−/− cells. Cells were cultured for the induction of Runx2 expression in the presence of doxycycline (1 μg/ml). After 24 h, cells were washed and cultured in the fresh media without doxycycline to determine the remaining Runx2 protein level. The remaining Runx2 protein levels at the indicated time were determined by immunoblot assay. D, Runx2 stabilization requires both ERK/MAPK and Pin1 activation. Primary MEF cells were transiently transfected with Runx2. E, Runx2 stabilization requires ERK/MAPK activation. U0126 was used to inhibit the MEK/ERK pathway. Recombinant and endogenous Pin1 proteins are indicated by an arrow and an arrowhead, respectively. F, absence of Pin1 could not support ERK/MAPK-induced Runx2 stabilization. C2C12 cells were transiently transfected with indicated plasmids for 24 h, and the cells were further exposed to juglone or U0126 for 18 h (D–F). Veh, vehicle; EV, empty vector.
Colocalization of Runx2 with Pin1 and Pin1-mediated Subnuclear Accumulation of Runx2 Foci
Immunofluorescence studies demonstrated that interplay between Runx2 and Pin1 are more specific during FGF2 signaling. Above all, we found that the colocalization of endogenous Pin1 and Runx2 is highly inducible in the nuclei of MC3T3-E1 cells following FGF2 administration (Fig. 3A). This colocalization is composed of highly condensed and accumulated Runx2 foci and Pin1 foci in a subnuclear region. Moreover, focally accumulated Runx2 is only found in Pin1 foci, indicating a strong interaction. The cells preserving large (>2 mm in diameter) Runx2 foci were counted (Fig. 3B). Although Runx2 foci are highly accumulated in response to FGF2, the foci are strongly suppressed by DTM, a Pin1 inhibitor (Fig. 3B). Interestingly, the subnuclear accumulation of Runx2 is clearly coupled with active RNA polymerase II (Fig. 3C), indicating that the Pin1-mediated accumulation of Runx2 is not only associated with the active transcription complex, but the accumulation of Runx2 occurs in the transcriptionally active subnuclear regions. Next, we addressed whether the accumulated Runx2 proteins require Pin1 activity. Wild-type Pin1 clearly induced the subnuclear accumulation of Runx2 in transiently transfected MC3T3-E1 cells expressing fluorescently tagged Runx2 and Pin1 (Fig. 3D). In contrast, Pin1 was unable to induce the subnuclear accumulation of Runx2 through the overexpression of either a catalytically inactive form of Pin1 (Pin1-C113A, in Fig. 3D) or a form of Pin1 with a binding motif mutation (Pin1-Y23A, in Fig. 3D). Time-lapse observation of the distribution of both proteins in living cells also confirmed that Pin1 speckle formation is required for Runx2 foci formation (Fig. 3E). These data suggest that Pin1 has a critical role in structural alteration, thereby inducing the active complex of Runx2 in the specific subnuclear region.
FIGURE 3.

Pin1 recruits Runx2 protein to subnuclear domains that are transcriptionally active. A, FGF2 enhances the colocalization of Runx2 and Pin1 protein in the same subnuclear foci. MC3T3-E1 cells were cultured in serum-free media and treated with vehicle (Veh) (0.2% BSA/PBS) or with 20 ng/ml FGF2. DTM (1 μm) was added to inhibit Pin1 activity, and the cells were cultured for 12 h. Runx2-Pin1 colocalization was detected by indirect immunofluorescence labeling in the cells. B, increase of Runx2-containing foci number in response to FGF2 stimulation was dependent on Pin1 activity. Cells containing at least three enlarged Runx2 foci (>2 μm diameter) were counted by confocal microscopy. Data represent means ± S.D. The number of cells (n) are 131, 145, and 150 in the groups for vehicle, FGF2, and FGF2 + DTM, respectively. **, p < 0.00001, compared with control group (vehicle); *, p < 0.0001, compared with FGF2-treated group. C, FGF2-induced focal accumulation of Runx2 was colocalized with active RNA polymerase II, and the accumulation was attenuated by the Pin1 inhibitor, DTM. D, Pin1-dependent subnuclear accumulation of Runx2. EGFP-Runx2 (green) was transiently expressed in MC3T3-E1 cells with DsRed-Pin1-WT, C113A, or Y23A (red). E, time-lapse analysis of Runx2 foci (green) accumulation in response to Pin1 (red). Dynamic subnuclear accumulation of Runx2 (green) was monitored by time-lapse confocal microscopy (LSM700, Carl Zeiss). The supplemental movie covers 669 min (m) and is composed of pictures taken every 15 min. Subnuclear accumulation of Runx2 foci began ∼213 min after complete formation of Pin1 speckles.
In a previous study, a C-terminal truncation mutation of Runx2 displayed bone phenotypes similar to those of the Runx2 knock-out mice (39). Moreover, FGF2, or the activated FGF receptor stimulation of ERK/MAPK, is critical for the pathogenesis of craniosynostosis (40) and cleidocranial dysplasia (22). Therefore, the subnuclear complex of Runx2 with Pin1, which was accentuated by FGF2 in our data, might indicate a relationship between Pin1 and Runx2 colocalization and FGF-associated bone phenotypes.
Identification of Pin1-binding Sites of Runx2 and Increased Binding of Pin1 by Runx2 Phosphorylation
Pin1 is known to directly interact with proteins containing phosphoserine-proline motifs (41–43). Because Runx2 is phosphorylated at such sites (13, 19, 20, 44–46), it seemed plausible that Runx2 and Pin1 interact. As anticipated, the administration of FGF2 stimulated the binding of Pin1 to Runx2 (Fig. 4A). Similarly, the overexpression of constitutively activated MEK1, a component of the FGFR signaling pathway, also enhanced the binding of Pin1 to Runx2 with an increase in p-ERK levels (Fig. 4B).
FIGURE 4.

FGF2-sitmulated ERK/MAPK signaling strongly enhanced Pin1 binding to the Runx2 C-terminal domain. A, elevated binding of Pin1 to FGF2-stimulated Runx2. HEK-293 cells were transiently transfected with Runx2 and cultured in the presence or absence of 10 ng/ml FGF2. After 60 min of FGF2 administration, lysates were prepared and used for GST-pulldown assays. B, ERK/MAPK-activated Runx2 strongly enhanced GST-Pin1 binding. For ERK activation, MEK1-Ca was transfected into HEK-293 cells with Runx2. After 24 h of transfection, cells were lysed, and the GST-pulldown assay was performed. C and D, identification of the Pin1 binding domain in Runx2. The ΔC-Runx2 construct was generated by excising amino acids 377–528, as described previously (39). FL-Runx2 indicates full-length Runx2. GST and GST-Pin1 are indicated as G and P, respectively. E, summary of the GST-pulldown assay for Pin1 binding to Runx2 deletion mutants. The functional domains of Runx2 conserved among Runx proteins are abbreviated as follows: RHD, runt-homology domain; NLS, nuclear localization sequence; PST, Pro/Ser/Thr-rich transactivation domain; NMTS, nuclear matrix target sequence. F, GST-pulldown assay of mutant Runx2 (Rx2–4AP) proteins in which the C-terminal Thr-408, Thr-449, Ser-472, and Ser-510 residues were substituted with Ala. A nonspecific band is indicated by n.s. G, determination of the structural alteration of Runx2 by subtilisin assay. HIS-Runx2 and MEK1-Ca were overexpressed in MEF-Pin1−/− cells and isolated by nitrilotriacetic acid affinity purification. For in vitro isomerization reaction, recombinant Pin1 proteins were synthesized in vitro and incubated with affinity-purified Runx2 protein-bead complex.
To identify Pin1-binding residue(s) of Runx2, in silico analysis was performed to identify conserved (pS/pT)P motifs in the Runx proteins. Twelve putative WW domain target motifs were identified in Runx2, and seven of these motifs were found to be conserved well across all Runx proteins (Table 1). These seven conserved target sites were then examined to identify the common motif(s) required for the interaction of Runx proteins with Pin1. A C-terminal deletion that removed four of the conserved (pS/pT)P motifs while conserving the three other conserved motifs exhibited drastically reduced Pin1 binding (Fig. 4C). To illuminate more specific sites, we analyzed and compared the binding strength of Runx2 fragments with two sets of WW motifs, Δ(397–434) and Δ(432–466). These deletion fragments are strongly inhibited in Pin1 binding but still maintained a weak interaction with recombinant GST-Pin1 protein, implying that all four WW motifs may be involved in Pin1 binding (Fig. 4D). Fig. 4E represents the summary of the GST-pulldown assay using the deletion constructs. Finally, the substitution of all four serine or threonine residues with alanine residues (Runx2–4AP) in these four motifs completely abrogated the interaction between Runx2 and Pin1 (Fig. 4F), indicating that the four C-terminal WW motifs are involved prominently in Pin1 binding.
TABLE 1.
Possible Pin1-binding sites in Runx2
The following abbreviations are used: AD, activating domain; NMTS, nuclear matrix target sequence; ID, inhibitory domain; R1, Runx1; R2, Runx2; R3, Runx3; R2-II, type II Runx2.
| WW motif (S/T)P | Functional domain | Putative kinasesa | Conservation among Runx |
|---|---|---|---|
| 11TP | AD | R2-II | |
| 28SP | AD | ERK1, PRKACG, AKT1, GSK3A, CDK5, CDC2 | R1/R2/R3 |
| 43SP | AD | R2 | |
| 125SP | Runt | ERK1, PKCμ, AKT1, AMPK | R1/R2/R3 |
| 282SP | AD | ERK1 | R2 |
| 301SP | AD | ERK1, GSK3 | R1/R2 |
| 319SP | AD | ERK1, CDK5, CDC2 | R1/R2 |
| 326TP | AD | CDK5, CDK5, CDC2, p38 | R1/R2/R3 |
| 408TP | NMTS | ERK1, CSNK1G2 | R1/R2/R3 |
| 449TP | NMTS | ERK1, GSK3A, CDK5, CDC2 | R1/R2/R3 |
| 472SP | ID | ERK1, GSK3A, CDK5 | R1/R2/R3 |
| 510SP | ID | ERK1, GSK3A, CDK5, CDC2 | R1/R2/R3 |
a Scansite Prediction was used.
To confirm Pin1-mediated structural alteration of Runx2, we tried to achieve subtilisin proteolysis protection assay that is a functional assay of Pin1 isomerization activity. Binding of Pin1 to substrates results in structural changes that confer resistance to subtilisin proteolysis; thus, acquisition of subtilisin resistance indicates that the protein interacts with and is structurally modified by Pin1. Runx2 was resistant to subtilisin proteolysis in the presence of wild-type (WT) Pin1, whereas Pin1-C113A, a mutant deficient in prolyl isomerase activity, or Pin1-ΔPPIase, a prolyl isomerase domain deletion mutant, failed to render Runx2 resistant to proteolysis by subtilisin (Fig. 4G).
Runx2 Acetylation Is Mediated Critically by Pin1
The ubiquitination of Runx2 is inhibited by p300-mediated acetylation and is promoted by HDAC-dependent deacetylation (47). Because Pin1 prevented Runx2 ubiquitination (37), we next examined the importance of Runx2 acetylation via the prolyl isomerase activity of Pin1. Juglone-induced inhibition of Pin1 isomerization activity strongly reduced the level of acetylated Runx2 (Fig. 5A). Although the overexpression of wild-type FGFR2 did not affect Runx2 acetylation, the overexpression of a constitutively active FGFR2 mutant (S354C) strongly increased Runx2 acetylation. However, treatment with DTM, a Pin1 inhibitor, attenuated this FGFR2 mutant-mediated increase in Runx2 acetylation (Fig. 5B). The level of Runx2 acetylation was dramatically up-regulated by wild-type Pin1 but not by Pin1-C113A even in the overexpression of MEK-Ca and p300 (Fig. 5C), which are modifying enzymes that promote Runx2 acetylation, indicating that the Pin1-mediated conformational change of Runx2 is a very important process for further acetylation. Pin1-dependent acetylation was also exhibited by Runx1 and Runx3 (other Runx proteins), which both contain conserved lysines and (S/T)P dipeptides. The expression of wild-type Pin1, but not Pin1-C113A, was found to increase the acetylation of all three Runx proteins (Fig. 5D).
FIGURE 5.

FGF2-induced Runx2 acetylation and activation requires Pin1. A, increased acetylation of Runx2 protein by MEK-Ca depends on Pin1 activity. Levels of acetylated Runx2 were determined by immunoprecipitation (IP) assay with an anti-acetyl-lysine (AcK) antibody. An arrowhead indicates the heavy chain of the antibody. B, activated FGFR2-induced increase of Runx2 acetylation requires Pin1 activity. MC3T3-E1 cells were transiently transfected with FGFR2 mutant plasmids for 24 h and cultured an additional 24 h with or without the Pin1 inhibitor, DTM. The protein extract from the cultures was immunoprecipitated with an acetyl-lysine antibody and immunoblotted (IB) with anti-Runx2 antibody. Arrowhead, acetylated Runx2; arrow, antibody heavy chain. EV, empty vector. C, Pin1 (WT) overexpression enhances Runx2 acetylation, but catalytically inactive Pin1 mutant (C113A) overexpression could not support Runx2 acetylation. HEK-293 cells were transiently transfected with the indicated plasmids and culture for 24 h. The protein extract from the cultures was immunoblotted with anti-Myc antibody. D, Runx protein acetylation depends on Pin1 activity. HEK-293 cells were transfected with Myc-tagged plasmids for Runx1, Runx2, or Runx3 with either empty, Pin1-WT, or Pin1-C113A vector. Comparable amounts of Runx proteins were used for each immunoprecipitation. E, mutant Runx2 proteins exhibited decreased transcriptional activity due to defective Pin1 binding, subnuclear targeting, or acetylation. 2AP#1 and 2AP#2 indicate substitutions of serine or threonine residues to alanine at Thr-408/Thr-449 or Ser-472/Ser-510, respectively. The KR mutation of Runx2 was previously described (47). The substitution of these four lysine residues with arginine residues (KR) dramatically increased the stability of Runx2 due to the absence of its ubiquitination sites, although the mutants do not have transcriptional activity. Data represent the means ± S.D. (n = 3). **, p < 0.0001, compared with empty vector (EV) transfection; *, p < 0.001, compared with wild-type Runx2 overexpression group. F, MC3T3-E1 cells were transiently transfected with the 6×OSE2-Luc reporter vector. After 24 h, cells were treated with 20 ng/ml FGF2 for an additional 24 h in the presence of DMSO (vehicle), 50 nm trichostatin A (TSA) (HDAC inhibitor), 25 μm anacardic acid (AA) (HAT inhibitor), or 1 μm DTM (Pin1 inhibitor). **, p < 0.0001, compared with control group (DMSO).
Next, we addressed whether the deletion of the Pin1 target domain (ΔC) or mutations in the Pin1 target elements (2AP#1, 2AP#2 and 4AP; refer to Fig. 5E, legend) influence the transcriptional activity of Runx2. In addition, we also investigated whether the interrelationship between Runx2 acetylation and its trans-activation function attributed to Pin1 activity. The C-terminal deletion of Runx2 abrogated the FGF2-stimulated Runx2 transcriptional activity, indicating that Pin1 binding to the C-terminal region of Runx2 is required for its activation by FGF2 (Fig. 5E). In addition, the KR mutant of Runx2 (47) was not activated by FGF2, indicating that the acetylation of these four lysine residues is critical for the FGF2-mediated transcriptional activity of Runx2 (Fig. 5E). Finally, the inhibition of p300 by anacardic acid (an inhibitor of HAT) treatment dramatically suppressed FGF2-stimulated Runx2 activity, whereas the administration of trichostatin A (HDAC inhibitor) synergistically augmented the Runx2 activity (Fig. 5F). These findings demonstrate that the Runx2 activation by FGF2 is closely associated with the lysine acetylation by p300 or the HDACi and that the post-phosphorylation events, including prolyl isomerization and subsequent acetylation, are crucial for Runx2 activity. This finding might explain our previous observation of the phosphorylation-dependent transactivation of Runx2 target genes (17).
Taken together, our results indicate that the Pin1-mediated regulation of Runx2 acetylation and stabilization are controlled by multiple post-translational modification cascades that occur in the following sequence: phosphorylation, prolyl isomerization, and acetylation (Fig. 6).
FIGURE 6.
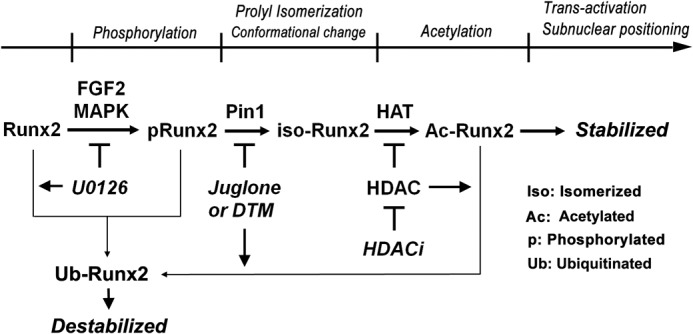
Positioning of Pin1-mediated prolyl isomerization in post-phosphorylational Runx2 activation cascade. Functional activation of Runx2 is tightly controlled by multiple post-translational modification cascades that occur in the following sequence: phosphorylation, prolyl isomerization, and acetylation. In these processes, Pin1 has a role as a molecular switch to determine the fate of Runx2 acetylation and further transactivation.
DISCUSSION
Acetylation and ubiquitination are mutually exclusive reactions for each lysine residue of Runx2 (47). Previously, we reported that four lysine residues in Runx2 are targets for p300-mediated acetylation. The substitution of these four lysine residues with arginine residues (KR) dramatically increased the stability of Runx2 but almost completely abrogated its transactivation activity due to the absence of its ubiquitination and acetylation sites (47). We also reported that FGF2-induced phosphorylation (14, 15) and subsequent acetylation (17) can stimulate Runx2 transactivation activity. In this study, we demonstrate that Pin1-dependent isomerization is a critical intermediate step between the phosphorylation and the acetylation of Runx2, thereby enhancing the functional activity of Runx2. In the presence of normal Pin1 activity, FGF2 stimulation induced the phosphorylation, isomerization, and acetylation of Runx2; however, the absence of Pin1 or the inhibition of Pin1 activity abrogated Runx2 acetylation, which resulted in Runx2 destabilization. Therefore, Pin1 is a critical mediator for the determination of the functional level of Runx2 in the cell. Unfortunately, we could not determine how Runx2 acetylation could be enhanced by conformational changes, but we assume that prolyl isomerization alters the conformation of Runx2 to an acetylation-friendly structure or enhances the interaction with HAT, which consequently stabilizes and activates the protein. In fact, we identified that both Runx1 and Runx3 could also be important as Pin1 substrates, and we eventually found that the acetylation of all Runx proteins is strongly enhanced by Pin1 (Fig. 5). Therefore, our data suggest that Pin1-mediated modification is a common regulatory mechanism for the promotion of all Runx protein acetylations.
Recently, we reported findings that Pin1 deficiency developed CCD phenotypes exhibiting hypoplastic clavicle and open fontanelle in mouse embryonic stages (37). CCD is characterized by delayed cranial suture closure secondary to RUNX2 deficiency. The CCD phenotypes of Pin1-deficient mice and the necessity for Pin1 in FGF2-mediated Runx2 activation should help further the understanding of bone development. Gain-of-function mutations in FGFR2 have consequences for other diseases of bone development, referred to as craniosynostosis, including Apert syndrome and Crouzen syndrome. Based on our findings, the cis-trans isomerization of Runx2 is a critical fate determinant of MAPK signaling in mouse calvarial cells. The inhibition of Pin1 activity accelerated Runx2 protein destabilization, suggesting that Pin1 could be targeted to correct for abnormal Runx2 activity and to ensure the optimal fate determination of osteogenic cell differentiation in premature bone development.
FGFR2 activation, secondary to ligand binding, leads to MEK1/2 activation, which subsequently stimulates Runx2 transactivation activity (14, 15). Previous reports have indicated that the regulation of MEK activity might be a useful intervention in genetic diseases caused by a deficiency in Runx2 (22) or by activating mutations in FGFR2 (40). Based on our results, the modulation of Pin1 or HDAC activity might provide another therapeutic avenue for the treatment of these congenital disorders.
The genotype-phenotype correlation regarding various RUNX2 mutations revealed that the Runx2 protein was differentially distributed between the nucleus and the cytosol in different mutants, demonstrating the importance of nuclear localization in bone development (48). This result suggests that the nuclear positioning of Runx2 in the nucleus promotes its transactivation activity, which provides an alternative molecular etiology for CCD development. However, the link between Runx2 localization and its transactivation activity is not completely understood. Additionally, the C-terminal deletion of Runx2 in mice resulted in the development of human CCD-like phenotypes (39). Later, it was shown that the ordered nuclear localization of Runx2, mediated by its C terminus, is necessary for the lineage commitment of osteoblastic cells (49–52). In this study, we identified four (pS/pT)P sites in the C terminus of Runx2 that are crucial target sites for Pin1 binding. The administration of FGF2 increased the focal accumulation of Pin1 and the recruitment of Runx2 to these foci in subnuclear regions. Because FGF2-induced focal accumulation of Runx2 was disrupted by the inhibition of Pin1 activity, the subnuclear targeting of Runx2 or the formation of Runx2 foci appear to be phosphorylation- and isomerization-dependent. These results suggest that the post-translational modifications that stabilize Runx2 also facilitate its focal accumulation. Therefore, it is likely that the insufficient subnuclear focal accumulation of Runx2 is directly responsible for the development of the CCD phenotype in the Pin1-deficient mice (37).
Until now, it has been widely accepted that Pin1 is a molecular switch to determine the fate of numerous phosphoproteins, especially regarding the regulation of protein stabilization (53). In addition, Pin1 has been shown to function in critical roles in the promotion of tumor suppressor activity through enhanced acetylation (54, 55). In this context, our evidence suggests that the acetylation of proteins could be a molecular target of post-isomerized Pin1.
In conclusion, our study demonstrates that Pin1 is a critical fate determinant for the post-phosphorylation modification of Runx2 during osteogenic cell differentiation. Thus, modifying enzymes, including Pin1, might represent valuable drug targets to correct abnormal Runx2 activity and to ensure the optimal fate determination of osteogenic cells.
Supplementary Material
Acknowledgment
Tet-inducible Runx2 plasmid was a kind gift from Dr. Baruch Frenkel (University of Southern California).
This work was supported by The Bio & Medical Technology Development Program Grant 20100030015, the General Researcher Program Grant 20100010590, and the Korean-Japanese International Research Program Grant K20802001314-09B1200-11110 from the National Research Foundation of Korea.

This article contains a supplemental movie.
- HAT
- histone acetyltransferase
- AA
- anacardic acid
- DTM
- dipentamethylene thiuram monosulfide
- HDAC
- histone deacetylase
- CCD
- cleidocranial dysplasia
- PPIase
- peptidyl-prolyl cis-trans isomerase
- mOB
- mouse osteoblast cell
- MEF
- mouse embryonic fibroblast
- MEK1-Ca
- constitutively active form of MEK1.
REFERENCES
- 1. Ornitz D. M., Marie P. J. (2002) FGF signaling pathways in endochondral and intramembranous bone development and human genetic disease. Genes Dev. 16, 1446–1465 [DOI] [PubMed] [Google Scholar]
- 2. Canalis E., Centrella M., McCarthy T. (1988) Effects of basic fibroblast growth factor on bone formation in vitro. J. Clin. Invest. 81, 1572–1577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mayahara H., Ito T., Nagai H., Miyajima H., Tsukuda R., Taketomi S., Mizoguchi J., Kato K. (1993) In vivo stimulation of endosteal bone formation by basic fibroblast growth factor in rats. Growth Factors 9, 73–80 [DOI] [PubMed] [Google Scholar]
- 4. Yoshimura S., Takagi Y., Harada J., Teramoto T., Thomas S. S., Waeber C., Bakowska J. C., Breakefield X. O., Moskowitz M. A. (2001) FGF-2 regulation of neurogenesis in adult hippocampus after brain injury. Proc. Natl. Acad. Sci. U.S.A. 98, 5874–5879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zhou D., DiFiglia M. (1993) Basic fibroblast growth factor enhances the growth of postnatal neostriatal GABAergic neurons in vitro. Exp. Neurol. 122, 171–188 [DOI] [PubMed] [Google Scholar]
- 6. Walshe J., Mason I. (2000) Expression of FGFR1, FGFR2, and FGFR3 during early neural development in the chick embryo. Mech. Dev. 90, 103–110 [DOI] [PubMed] [Google Scholar]
- 7. Murakami M., Nguyen L. T., Zhuang Z. W., Zhang Z. W., Moodie K. L., Carmeliet P., Stan R. V., Simons M. (2008) The FGF system has a key role in regulating vascular integrity. J. Clin. Invest. 118, 3355–3366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sullivan R., Klagsbrun M. (1985) Purification of cartilage-derived growth factor by heparin affinity chromatography. J. Biol. Chem. 260, 2399–2403 [PubMed] [Google Scholar]
- 9. Peters K. G., Werner S., Chen G., Williams L. T. (1992) Two FGF receptor genes are differentially expressed in epithelial and mesenchymal tissues during limb formation and organogenesis in the mouse. Development 114, 233–243 [DOI] [PubMed] [Google Scholar]
- 10. Mundy G., Garrett R., Harris S., Chan J., Chen D., Rossini G., Boyce B., Zhao M., Gutierrez G. (1999) Stimulation of bone formation in vitro and in rodents by statins. Science 286, 1946–1949 [DOI] [PubMed] [Google Scholar]
- 11. Montero A., Okada Y., Tomita M., Ito M., Tsurukami H., Nakamura T., Doetschman T., Coffin J. D., Hurley M. M. (2000) Disruption of the fibroblast growth factor-2 gene results in decreased bone mass and bone formation. J. Clin. Invest. 105, 1085–1093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hurley M. M., Marcello K., Abreu C., Kessler M. (1996) Signal transduction by basic fibroblast growth factor in rat osteoblastic Py1a cells. J. Bone Miner. Res. 11, 1256–1263 [DOI] [PubMed] [Google Scholar]
- 13. Ge C., Xiao G., Jiang D., Yang Q., Hatch N. E., Roca H., Franceschi R. T. (2009) Identification and functional characterization of ERK/MAPK phosphorylation sites in the Runx2 transcription factor. J. Biol. Chem. 284, 32533–32543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kim H. J., Park H. D., Kim J. H., Cho J. Y., Choi J. Y., Kim J. K., Kim H. J., Shin H. I., Ryoo H. M. (2004) Establishment and characterization of a stable cell line to evaluate cellular Runx2 activity. J. Cell. Biochem. 91, 1239–1247 [DOI] [PubMed] [Google Scholar]
- 15. Kim H. J., Kim J. H., Bae S. C., Choi J. Y., Kim H. J., Ryoo H. M. (2003) The protein kinase C pathway plays a central role in the fibroblast growth factor-stimulated expression and transactivation activity of Runx2. J. Biol. Chem. 278, 319–326 [DOI] [PubMed] [Google Scholar]
- 16. Geoffroy V., Ducy P., Karsenty G. (1995) A PEBP2 α/AML-1-related factor increases osteocalcin promoter activity through its binding to an osteoblast-specific cis-acting element. J. Biol. Chem. 270, 30973–30979 [DOI] [PubMed] [Google Scholar]
- 17. Park O. J., Kim H. J., Woo K. M., Baek J. H., Ryoo H. M. (2010) FGF2-activated ERK mitogen-activated protein kinase enhances Runx2 acetylation and stabilization. J. Biol. Chem. 285, 3568–3574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Park J., Park O. J., Yoon W. J., Kim H. J., Choi K. Y., Cho T. J., Ryoo H. M. (2012) Functional characterization of a novel FGFR2 mutation, E731K, in craniosynostosis. J. Cell. Biochem. 113, 457–464 [DOI] [PubMed] [Google Scholar]
- 19. Kim B. G., Kim H. J., Park H. J., Kim Y. J., Yoon W. J., Lee S. J., Ryoo H. M., Cho J. Y. (2006) Runx2 phosphorylation induced by fibroblast growth factor-2/protein kinase C pathways. Proteomics 6, 1166–1174 [DOI] [PubMed] [Google Scholar]
- 20. Ge C., Yang Q., Zhao G., Yu H., Kirkwood K. L., Franceschi R. T. (2011) Interactions between extracellular signal-regulated kinase 1/2 and p38 MAP kinase pathways in the control of RUNX2 phosphorylation and transcriptional activity. J. Bone Miner. Res., 27, 538–551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sierra J., Villagra A., Paredes R., Cruzat F., Gutierrez S., Javed A., Arriagada G., Olate J., Imschenetzky M., Van Wijnen A. J., Lian J. B., Stein G. S., Stein J. L., Montecino M. (2003) Regulation of the bone-specific osteocalcin gene by p300 requires Runx2/Cbfa1 and the vitamin D3 receptor but not p300 intrinsic histone acetyltransferase activity. Mol. Cell. Biol. 23, 3339–3351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ge C., Xiao G., Jiang D., Franceschi R. T. (2007) Critical role of the extracellular signal-regulated kinase-MAPK pathway in osteoblast differentiation and skeletal development. J. Cell Biol. 176, 709–718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yoon W. J., Islam R., Cho Y. D., Woo K. M., Baek J. H., Uchida T., Komori T., van Wijnen A., Stein J. L., Lian J. B., Stein G. S., Choi J. Y., Bae S. C., Ryoo H. M. (2013) Pin1-mediated Runx2 modification is critical for skeletal development. J. Cell. Physiol. 228, 2377–2385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lu K. P., Zhou X. Z. (2007) The prolyl isomerase PIN1: a pivotal new twist in phosphorylation signalling and disease. Nat. Rev. Mol. Cell Biol. 8, 904–916 [DOI] [PubMed] [Google Scholar]
- 25. Lu K. P., Finn G., Lee T. H., Nicholson L. K. (2007) Prolyl cis-trans isomerization as a molecular timer. Nat. Chem. Biol. 3, 619–629 [DOI] [PubMed] [Google Scholar]
- 26. Yeh E. S., Means A. R. (2007) PIN1, the cell cycle and cancer. Nat. Rev. Cancer 7, 381–388 [DOI] [PubMed] [Google Scholar]
- 27. Hsu T., McRackan D., Vincent T. S., Gert de Couet H. (2001) Drosophila Pin1 prolyl isomerase Dodo is a MAP kinase signal responder during oogenesis. Nat. Cell. Biol. 3, 538–543 [DOI] [PubMed] [Google Scholar]
- 28. Monje P., Hernández-Losa J., Lyons R. J., Castellone M. D., Gutkind J. S. (2005) Regulation of the transcriptional activity of c-Fos by ERK. A novel role for the prolyl isomerase PIN1. J. Biol. Chem. 280, 35081–35084 [DOI] [PubMed] [Google Scholar]
- 29. Yeh E., Cunningham M., Arnold H., Chasse D., Monteith T., Ivaldi G., Hahn W. C., Stukenberg P. T., Shenolikar S., Uchida T., Counter C. M., Nevins J. R., Means A. R., Sears R. (2004) A signalling pathway controlling c-Myc degradation that impacts oncogenic transformation of human cells. Nat. Cell. Biol. 6, 308–318 [DOI] [PubMed] [Google Scholar]
- 30. Muñoz J. P., Huichalaf C. H., Orellana D., Maccioni R. B. (2007) cdk5 modulates β- and δ-catenin/Pin1 interactions in neuronal cells. J. Cell. Biochem. 100, 738–749 [DOI] [PubMed] [Google Scholar]
- 31. Patra D., Wang S. X., Kumagai A., Dunphy W. G. (1999) The Xenopus Suc1/Cks protein promotes the phosphorylation of G2/M regulators. J. Biol. Chem. 274, 36839–36842 [DOI] [PubMed] [Google Scholar]
- 32. Chen S. Y., Wulf G., Zhou X. Z., Rubin M. A., Lu K. P., Balk S. P. (2006) Activation of β-catenin signaling in prostate cancer by peptidyl-prolyl isomerase Pin1-mediated abrogation of the androgen receptor-β-catenin interaction. Mol. Cell. Biol. 26, 929–939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hennig L., Christner C., Kipping M., Schelbert B., Rücknagel K. P., Grabley S., Küllertz G., Fischer G. (1998) Selective inactivation of parvulin-like peptidyl-prolyl cis/trans isomerases by juglone. Biochemistry 37, 5953–5960 [DOI] [PubMed] [Google Scholar]
- 34. Tatara Y., Lin Y. C., Bamba Y., Mori T., Uchida T. (2009) Dipentamethylene thiuram monosulfide is a novel inhibitor of Pin1. Biochem. Biophys. Res. Commun. 384, 394–398 [DOI] [PubMed] [Google Scholar]
- 35. Cho Y. D., Yoon W. J., Woo K. M., Baek J. H., Lee G., Cho J. Y., Ryoo H. M. (2009) Molecular regulation of matrix extracellular phosphoglycoprotein expression by bone morphogenetic protein-2. J. Biol. Chem. 284, 25230–25240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zita M. M., Marchionni I., Bottos E., Righi M., Del Sal G., Cherubini E., Zacchi P. (2007) Post-phosphorylation prolyl isomerisation of gephyrin represents a mechanism to modulate glycine receptors' function. EMBO J. 26, 1761–1771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yoon W. J., Islam R., Cho Y. D., Woo K. M., Baek J. H., Uchida T., Komori T., van Wijnen A., Stein J. L., Lian J. B., Stein G. S., Choi J. Y., Bae S. C., Ryoo H. M. (2013) Pin1-mediated Runx2 modification is critical for skeletal development. J. Cell. Physiol., 228, 2377–2385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Baniwal S. K., Khalid O., Gabet Y., Shah R. R., Purcell D. J., Mav D., Kohn-Gabet A. E., Shi Y., Coetzee G. A., Frenkel B. (2010) Runx2 transcriptome of prostate cancer cells: insights into invasiveness and bone metastasis. Mol. Cancer 9, 258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Choi J. Y., Pratap J., Javed A., Zaidi S. K., Xing L., Balint E., Dalamangas S., Boyce B., van Wijnen A. J., Lian J. B., Stein J. L., Jones S. N., Stein G. S. (2001) Subnuclear targeting of Runx/Cbfa/AML factors is essential for tissue-specific differentiation during embryonic development. Proc. Natl. Acad. Sci. U.S.A. 98, 8650–8655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Shukla V., Coumoul X., Wang R. H., Kim H. S., Deng C. X. (2007) RNA interference and inhibition of MEK-ERK signaling prevent abnormal skeletal phenotypes in a mouse model of craniosynostosis. Nat. Genet. 39, 1145–1150 [DOI] [PubMed] [Google Scholar]
- 41. Lu P. J., Zhou X. Z., Shen M., Lu K. P. (1999) Function of WW domains as phosphoserine- or phosphothreonine-binding modules. Science 283, 1325–1328 [DOI] [PubMed] [Google Scholar]
- 42. Ranganathan R., Lu K. P., Hunter T., Noel J. P. (1997) Structural and functional analysis of the mitotic rotamase Pin1 suggests substrate recognition is phosphorylation-dependent. Cell 89, 875–886 [DOI] [PubMed] [Google Scholar]
- 43. Yaffe M. B., Schutkowski M., Shen M., Zhou X. Z., Stukenberg P. T., Rahfeld J. U., Xu J., Kuang J., Kirschner M. W., Fischer G., Cantley L. C., Lu K. P. (1997) Sequence-specific and phosphorylation-dependent proline isomerization: a potential mitotic regulatory mechanism. Science 278, 1957–1960 [DOI] [PubMed] [Google Scholar]
- 44. Rajgopal A., Young D. W., Mujeeb K. A., Stein J. L., Lian J. B., van Wijnen A. J., Stein G. S. (2007) Mitotic control of RUNX2 phosphorylation by both CDK1/cyclin B kinase and PP1/PP2A phosphatase in osteoblastic cells. J. Cell. Biochem. 100, 1509–1517 [DOI] [PubMed] [Google Scholar]
- 45. Wee H. J., Huang G., Shigesada K., Ito Y. (2002) Serine phosphorylation of RUNX2 with novel potential functions as negative regulatory mechanisms. EMBO Rep. 3, 967–974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Xiao G., Jiang D., Gopalakrishnan R., Franceschi R. T. (2002) Fibroblast growth factor 2 induction of the osteocalcin gene requires MAPK activity and phosphorylation of the osteoblast transcription factor, Cbfa1/Runx2. J. Biol. Chem. 277, 36181–36187 [DOI] [PubMed] [Google Scholar]
- 47. Jeon E. J., Lee K. Y., Choi N. S., Lee M. H., Kim H. N., Jin Y. H., Ryoo H. M., Choi J. Y., Yoshida M., Nishino N., Oh B. C., Lee K. S., Lee Y. H., Bae S. C. (2006) Bone morphogenetic protein-2 stimulates Runx2 acetylation. J. Biol. Chem. 281, 16502–16511 [DOI] [PubMed] [Google Scholar]
- 48. Yoshida T., Kanegane H., Osato M., Yanagida M., Miyawaki T., Ito Y., Shigesada K. (2002) Functional analysis of RUNX2 mutations in Japanese patients with cleidocranial dysplasia demonstrates novel genotype-phenotype correlations. Am. J. Hum. Genet. 71, 724–738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Harrington K. S., Javed A., Drissi H., McNeil S., Lian J. B., Stein J. L., Van Wijnen A. J., Wang Y. L., Stein G. S. (2002) Transcription factors RUNX1/AML1 and RUNX2/Cbfa1 dynamically associate with stationary subnuclear domains. J. Cell Sci. 115, 4167–4176 [DOI] [PubMed] [Google Scholar]
- 50. Lian J. B., Javed A., Zaidi S. K., Lengner C., Montecino M., van Wijnen A. J., Stein J. L., Stein G. S. (2004) Regulatory controls for osteoblast growth and differentiation: role of Runx/Cbfa/AML factors. Crit. Rev. Eukaryot. Gene Expr. 14, 1–41 [PubMed] [Google Scholar]
- 51. Stein G. S., Lian J. B., Stein J. L., van Wijnen A. J., Choi J. Y., Pratap J., Zaidi S. K. (2003) Temporal and spatial parameters of skeletal gene expression: targeting RUNX factors and their coregulatory proteins to subnuclear domains. Connect. Tissue Res. 44, 149–153 [PubMed] [Google Scholar]
- 52. Zaidi S. K., Javed A., Choi J. Y., van Wijnen A. J., Stein J. L., Lian J. B., Stein G. S. (2001) A specific targeting signal directs Runx2/Cbfa1 to subnuclear domains and contributes to transactivation of the osteocalcin gene. J. Cell Sci. 114, 3093–3102 [DOI] [PubMed] [Google Scholar]
- 53. Liou Y. C., Zhou X. Z., Lu K. P. (2011) Prolyl isomerase Pin1 as a molecular switch to determine the fate of phosphoproteins. Trends Biochem. Sci. 36, 501–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Mantovani F., Tocco F., Girardini J., Smith P., Gasco M., Lu X., Crook T., Del Sal G. (2007) The prolyl isomerase Pin1 orchestrates p53 acetylation and dissociation from the apoptosis inhibitor iASPP. Nat. Struct. Mol. Biol. 14, 912–920 [DOI] [PubMed] [Google Scholar]
- 55. Mantovani F., Piazza S., Gostissa M., Strano S., Zacchi P., Mantovani R., Blandino G., Del Sal G. (2004) Pin1 links the activities of c-Abl and p300 in regulating p73 function. Mol. Cell 14, 625–636 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.