

Background: There are conflicting reports on the ability of granzyme A (GzmA) to kill cells.
Results: Substrate specificity mapping of human and mouse GzmA was used to identify Serpinb6b as a mouse-specific GzmA inhibitor.
Conclusion: Mouse but not human GzmA is controlled by an intracellular inhibitor.
Significance: The GzmA debate is partly explained by species-specific divergence in cytotoxicity.
Keywords: Enzyme Inhibitors, Immunology, Proteomics, Serine Protease, Serpin, COFRADIC, Granzyme A
Abstract
The granzyme family serine proteases are key effector molecules expressed by cytotoxic lymphocytes. The physiological role of granzyme (Gzm) A is controversial, with significant debate over its ability to induce death in target cells. Here, we investigate the natural inhibitors of GzmA. We employed substrate phage display and positional proteomics to compare substrate specificities of mouse (m) and human (h) GzmA at the peptide and proteome-wide levels and we used the resulting substrate specificity profiles to search for potential inhibitors from the intracellular serpin family. We identified Serpinb6b as a potent inhibitor of mGzmA. Serpinb6b interacts with mGzmA, but not hGzmA, with an association constant of 1.9 ± 0.8 × 105 m−1 s−1 and a stoichiometry of inhibition of 1.8. Mouse GzmA is over five times more cytotoxic than hGzmA when delivered into P815 target cells with streptolysin O, whereas transfection of target cells with a Serpinb6b cDNA increases the EC50 value of mGzmA 13-fold, without affecting hGzmA cytotoxicity. Unexpectedly, we also found that Serpinb6b employs an exosite to specifically inhibit dimeric but not monomeric mGzmA. The identification of an intracellular inhibitor specific for mGzmA only indicates that a lineage-specific increase in GzmA cytotoxic potential has driven cognate inhibitor evolution.
Introduction
The killing of target cells by cytotoxic T lymphocytes and natural killer cells proceeds via two main routes as follows: the ligation of cell surface death receptors and exocytosis of granular contents. The granule exocytosis pathway involves delivery of granzymes (Gzm),2 a family of serine proteases, into the cytoplasm of the target cell, a process mediated by the pore-forming protein perforin (1).
Of the five human granzymes, the cytotoxin GzmB is best characterized. Its Aspase activity leads primarily to cleavage of Bid and activation of the mitochondrial death pathway, although caspases and other death substrates are also directly activated. GzmB is regulated by the intracellular inhibitor SERPINB9, which serves to protect cytotoxic lymphocytes from GzmB-induced suicide (2). Although cytotoxic roles have been ascribed to all human granzymes, considerable controversy exists over their exact roles during the immune response in vivo, and physiologically relevant inhibitors analogous to SERPINB9 have not yet been convincingly demonstrated (3). Analyses are often complicated by the use of model systems, particularly mice, and the fact that orthologous granzymes from different species have distinct substrate preferences and can activate different pathways to death (4–7). For instance, although both human and mouse GzmB are cytotoxic, the mouse protease (in sharp contrast to human GzmB) cannot activate Bid (5, 8) and is primarily dependent on caspase activation to exert its function (8). This difference is reflected in the divergent substrate specificities of human and mouse granzyme B both N- and C-terminal to the cleavage point (5, 6).
The potential roles of GzmA have been particularly highlighted in recent years. Early evidence suggested a cytotoxic role as an auxiliary to GzmB (9, 10). Indeed, many reports have shown that cytotoxic cells purified from GzmA null mice are less effective killers than wild-type cells (10). Two cytotoxic pathways have been mapped for GzmA. The first is centered on the cleavage of SET complex proteins and characterized by production of reactive oxygen species and single-stranded DNA damage (11), whereas the second affects the actin cytoskeleton, without generation of reactive oxygen species or caspase activation (12). Others report that GzmA is not cytotoxic and instead is involved in the processing of cytokines, with the experimentally observed cytotoxicity suggested to arise from bombarding cells with nonphysiological levels of protease (13, 14). Some of these findings can be reconciled by observations suggesting a difference in cytotoxic potential between hGzmA and mGzmA (5, 12, 13).
We hypothesize that cytotoxicity of GzmA, if physiologically important, would drive the co-evolution of a specific intracellular inhibitor, as it has for GzmB. The GzmB-specific inhibitor SERPINB9 belongs to the intracellular clade B of the serpin superfamily, and limited in vitro evidence suggests that GzmH and -M may also have cognate inhibitors from the same clade (15, 16). The serpin inhibitory mechanism uses an exposed region termed the reactive center loop (RCL) that mimics the substrate specificity of the target protease. Cleavage of the RCL initiates a conformational change in the serpin fold that traps the protease in a 1:1 covalent complex (17). Serpin RCL sequences are thus critical determinants of protease inhibition and closely resemble the substrate cleavage site of the protease. This is best illustrated by alterations in the RCL sequence of orthologous human (SERPINB9) (2) and mouse (Serpinb9a) GzmB inhibitors (18) to match alterations in the protease specificity (5, 6). However, RCLs often include suboptimal residues that would lead to inefficient cleavage by the target protease. This can be overcome by the presence of exosites and cofactors that increase association rates by several orders of magnitude (19). The combination of the RCL sequence and exosites or cofactors thus imparts specificity to the serpin-protease interaction.
Two extracellular human GzmA inhibitors have previously been reported, the Kazal-type pancreatic secretory trypsin inhibitor (20) and the serpin antithrombin III (SERPINC1) (21). The identification of a specific and efficient intracellular inhibitor would provide strong evidence for cytotoxicity of GzmA in vivo. Here, we probe the substrate specificity of mouse and human GzmA using phage display and positional proteomics. The deduced specificity profiles are then used to identify the intracellular serpin Serpinb6b as a candidate species-specific inhibitor of mGzmA but not hGzmA. We confirm a strong inhibitory interaction and show that Serpinb6b utilizes both the substrate specificity and an exosite on the mGzmA dimer to efficiently inhibit mGzmA. Our findings suggest evolutionary divergence in which mGzmA has increased cytotoxic potential.
EXPERIMENTAL PROCEDURES
Phage Display
Phage display using a random 9-mer library was performed as described (5).
Positional Proteomics
Culturing and SILAC labeling conditions of Jurkat T cells were performed as described (22). Jurkat lysates were either incubated with 200 nm hGrA ([12C6]Arg), mGrA ([13C6]Arg), or left untreated ([13C6,15N4]Arg) for 1 h at 37 °C. Samples were processed and analyzed according to the N-terminal COFRADIC protocol described previously (23).
Cloning Serpinb6b
A Serpinb6b EST was obtained from the IMAGE consortium (clone 2331842) and sequenced on both strands. The open reading frame was amplified with Pfu polymerase using the antisense primer 5′-GAATTCTCATGGGGAGGAGAACCG and the sense primers 5′-GGGAATTCATGGACTACAAAGACGATGACGATAAAATGGATCCACTGCTGG (introducing an N-terminal FLAG tag for expression in cells) or 5′-GAATTCATGCATCACCATCACCATCACAGTGGTAGTGGTATGGATCCACTGCTGGAAGCA (introducing an N-terminal hexahistidine tag for protein purification). Products were ligated to pCR-Blunt, sequence-verified, and subcloned using EcoRI into pEF-IRES-GFP (for mammalian expression) or pHIL-D2 (for purification from yeast).
Recombinant Proteins
Recombinant human and mouse granzyme A were produced in Pichia pastoris as described previously (12, 24) and assessed by native PAGE as fully dimeric. Recombinant serpins were also produced in P. pastoris according to Ref. 25. Protein concentrations were determined by absorbance at 280 nm using a NanoDrop 1000 spectrophotometer and extinction coefficients predicted from primary amino acid sequences. All batches of granzymes and serpins were assessed as >95% active by gel shift with an appropriate serpin or protease target. Serpinb6a and Serpinb6b were assayed against mGzmA, SERPINB6 against hGzmA, mGzmA against Serpinb6b, and hGzmA against α1-antitrypsin with the RCL mutated to include a Ser-Val-Ala-Arg motif at P4–P1.
Kinetics
Quenched fluorescence peptides Abz-VANRSAS(K-dnp)D and Abz-GLFRSLSS(K-dnp)D at >95% purity were purchased from Mimotopes. Inhibition was measured according to Ref. 26. Where indicated, GzmA was reduced to a monomer by the addition of 20 mm β-mercaptoethanol to all buffers.
Culture and Transfection of COS-1
COS-1 cells were maintained as subconfluent monolayers in DMEM supplemented with 10% (v/v) fetal calf serum, 2 mm l-glutamine, 50 units/ml penicillin, and 50 μg/ml streptomycin. Human serpin cDNAs cloned into the NheI site of pEGFP-c2 in-frame with GFP were purchased from Genscript. 4–5 μg of each plasmid was added to 5 × 105 COS-1 cells in DMEM supplemented with 2 mm l-glutamine, 50 units/ml penicillin, 50 μg/ml streptomycin, 400 μg/ml DEAE-dextran, and 25 μm chloroquine for 3 h at 37 °C. The medium was replaced with 10% dimethyl sulfoxide in DMEM for 2 min, and cells then returned to complete DMEM.
Human GzmA Interactions with Clade B Serpins
Two days after transfection, cells were lysed in 100 μl of lysis buffer (10 mm Tris-HCl, pH 7.4, 1% (v/v) IGEPAL® CA-630, 1 mm EDTA, 1 μg/ml leupeptin, 1 μm pepstatin) on ice for 5 min, clarified by centrifugation at 16,000 × g for 5 min at 4 °C, and protein concentrations determined using the Bio-Rad protein assay kit. 10 μg of protein was incubated with or without 1 μg of hGzmA in a total volume of 20 μl of Tris-buffered saline (150 mm NaCl, 20 mm Tris-HCl, pH 7.4) for 15 min at 37 °C after which the reaction was stopped by addition of 10 μl of Laemmli sample buffer (125 mm Tris-HCl, pH 6.8, 4% (w/v) SDS, 20% (v/v) glycerol, 0.1% (w/v) bromphenol blue, 0.1 m dithiothreitol).
Samples were resolved by 10% reducing SDS-PAGE and transferred to nitrocellulose membranes. Membranes were blocked with 1% (w/v) skim milk powder and probed with a 1:1000 dilution of a monoclonal anti-GFP antibody (Roche Applied Science).
Culture and Transfection of P815
Mouse P815 mastocytoma cells were maintained in RPMI 1640 medium supplemented with 10% (v/v) heat-inactivated fetal calf serum, 2 mm l-glutamine, 55 μm β-mercaptoethanol, 50 units/ml penicillin, and 50 μg/ml streptomycin. 1 × 108 cells were electroporated as described previously (5) with either a 10:1 mixture of pEF-IRES-GFP/Serpinb6b:pEF-puro or pEF-puro alone, and transfectants were selected using 1 mg/ml puromycin. Serpinb6b transfectants were sorted on the basis of GFP expression and maintained as an uncloned pool.
Cytotoxicity Assays
Cell death assays were performed with recombinant streptolysin O and granzyme A as described previously (5).
Structural Models
A model of the mGzmA-Serpinb6b Michaelis complex was constructed by overlaying the crystal structure of dimeric hGzmA (PDB accession 1ORF) with the Michaelis complex between S195A trypsin and M358R antitrypsin (PDB accession 1OPH). Potential exosites were defined as regions in which the serpin was in close proximity to the noninhibited GzmA monomer.
RESULTS
Substrate Specificity of Mouse and Human GzmA Is Different
Our aim was to search for intracellular serpins targeting either hGzmA or mGzmA, because their existence would provide evidence of GzmA cytotoxicity in vivo. The serpin inhibitory mechanism requires the RCL to match the substrate specificity of the target protease, and therefore our first step was to define the substrate specificity of both hGzmA and mGzmA using substrate phage display (5). Sequencing of 62 phage after four rounds of selection with mGzmA revealed strong selectivity at the P4 and P2′ positions around a P1 Arg, with further selection at P3, P2, P1′, and P4′. The data gave a mGzmA substrate specificity sequence of Gly-Val-Phe-Arg-↓-Met-Leu/Phe-Xaa-Val (Fig. 1A). By contrast, analysis of 79 phage following four rounds of selection with hGzmA showed strongest selection at P3, within the pattern Ile-Gly-Xaa-Arg-↓-Xaa-Gly. By including weakly selected residues, some overlap between species could be observed, with Ile and Gly weakly selected by mGzmA at P4 and P3, and hGzmA weakly selected bulky residues Phe, Trp, and Tyr at P2 (Fig. 1A). These findings agree with the P4 to P1 positional scanning substrate combinatorial library analysis (PS-SCL) performed by Bell et al. (27) and the proteomic analysis of van Damme et al. (28) but expand our knowledge to prime side interactions. No selection for Lys at the P1 position was observed; however, this is likely due to the very strong preference of both human and mouse GzmA for Arg (see below), rather than an inability to accommodate Lys at P1 or under-representation of P1 Lys in the library.
FIGURE 1.

Peptide substrate specificity profile of mouse and human granzyme A. A, following four rounds of selection, 62 (mGzmA) or 79 (hGzmA) phage-displayed nonameric peptides were sequenced, aligned, and analyzed to identify selected residues at each subsite. Shading of bars represents strength of selection as follows: white, <2σ; gray, 2σ to 4σ; black, >4σ. B, reaction rates were determined for mGzmA and hGzmA cleaving both of the optimized peptide substrates Abz-GLFRSLSS(K-dnp)D (white bars) and Abz-VANRSAS(K-dnp)D (gray bars). Data shown are the means ± S.D. of three independent replicates. *, p < 0.05; **, p < 0.01; n.s., p > 0.05 in Student's t test.
To confirm these differences in specificity, we produced an internally quenched fluorescence peptide substrate for mGzmA based on a phage sequence that most closely matched the overall pattern (GLFR↓SLSS), as well as a hGzmA substrate based on a combination of the phage display pattern and the PS-SCL pattern derived by Bell et al. (27) (VANR↓SAS). We then determined the activity of human and mouse GzmA against each peptide to determine whether the predicted substrate specificity could be recapitulated. The reaction rate for mGzmA cleaving VANRSAS was 3.70 ± 0.85 × 105 m−1 s−1 compared with a rate of 2.95 ± 0.48 × 105 m−1 s−1 when cleaving GLFRSLSS, and hGzmA rates were 1.27 ± 0.23 × 105 m−1 s−1 for VANRSAS and 4.69 ± 0.12 × 102 m−1 s−1 for the GLFRSLSS substrate (Fig. 1B and Table 1). This shows that mGzmA selects Phe residues strongly at P2, but this is not an absolute requirement as the difference in rate constants between peptides is not significant. Conversely, hGzmA shows a strong 280-fold preference for the VANR substrate, due to a dramatic decrease in turnover efficiency (Fig. 1B and Table 1).
TABLE 1.
Kinetic parameters for granzyme A cleavage of peptide substrates
Data shown are mean ± S.D. of three independent experiments. Substrates included an N-terminal aminobenzoyl fluorophore and a C-terminal dinitrophenyl lysine quencher and aspartic acid to enhance solubility.
| Mouse Granzyme A |
Human Granzyme A |
|||
|---|---|---|---|---|
| GLFRSLSS | VANRSAS | GLFRSLSS | VANRSAS | |
| kcat (s−1) | 33.5 ± 2.94 | 14.2 ± 2.13 | 2.51 ± 0.0642 × 10−2 | 4.88 ± 0.233 |
| Km (μm) | 114 ± 15.4 | 38.3 ± 6.62 | 53.5 ± 0.382 | 35.2 ± 3.46 |
| kcat/Km (m−1 s−1) | 2.95 ± 0.476 × 105 | 3.70 ± 0.848 × 105 | 4.69 ± 0.124 × 102 | 1.39 ± 0.152 × 105 |
Comparative Proteomic Analysis Suggests Exosite-driven Substrate Cleavage by GzmA
We further addressed macromolecular differences between human and mouse GzmA substrate specificities by performing a differential N-terminal combined fractional diagonal chromatography (COFRADIC) analysis. As such, the proteomes of Jurkat cells were SILAC-labeled with [13C6]Arg or [13C6,15N4]Arg or left unlabeled. Freeze-thaw lysates were then treated with 200 nm hGzmA (unlabeled), mGzmA ([13C6]Arg-labeled), or no protease ([13C6,15N4]Arg-labeled), and equal proteome quantities were mixed and processed accordingly, allowing for the isolation and quantification of neo-N-terminal peptides.
In total, 755 tryptic neo-N termini were identified from 570 unique substrates. Of these, 355 cleavage sites were specific to mGzmA, and six were uniquely cleaved by hGzmA, and 394 were cleaved by both proteases. IceLogos (29) were generated from the 749 mGzmA (Fig. 2A) and 400 hGzmA (Fig. 2B) cleavage sites to define their substrate specificity profiles. Weighting all cleavage events equally produces almost identical profiles. The tryptase activity of both is easily apparent, with only Arg or Lys occupying P1, although ∼72.5% of all human and mouse GzmA cleavage sites carry Arg at this position. As with many other proteases, the most common residues at P1′ (covering ∼30% of cleavage sites) are the small amino acids Ser and Ala; however, a significant proportion (16–18%) of GzmA substrates had a Lys at this position. Equal preferences for either small amino acids (Gly, Ala, or Ser) or Leu were found at P2′, each accounting for ∼30% occurrence in cleavage sites of both granzymes. Of note, proline is strongly under-represented at P2′ in GzmA cleavage sites. Although proline occurrence in the human subsection of the Swiss-Prot database is 6.3%, it is only present in 0.25% (1 cleavage site) and 0.40% (3 cleavage sites) of all human and mouse GzmA cleavage sites, respectively. No clear preferences could be identified beyond P3′ or on the nonprime side except for a general preference for charged residues. The similar specificity profiles between mouse and human GzmA obtained using this approach, in contrast to phage display, can be explained by exosite interactions between the proteases and protein substrates that override the primary cleavage specificity evident on peptide substrates.
FIGURE 2.
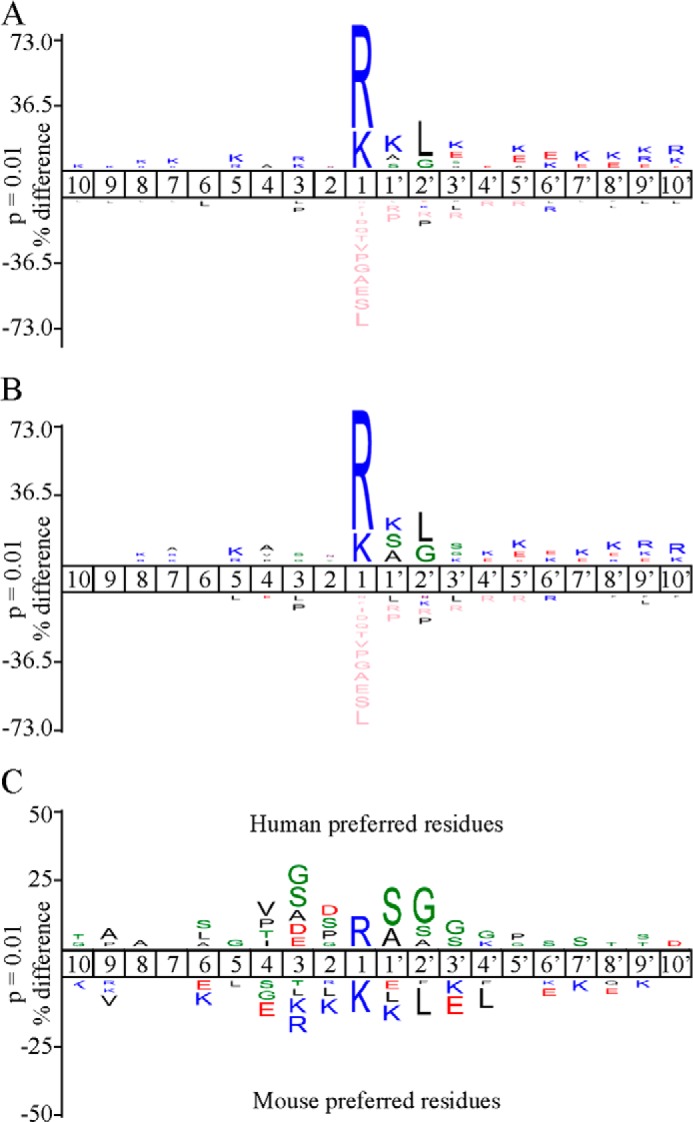
Proteomic analysis of mouse and human granzyme A cleavage specificity. IceLogo (29) visualization of 749 nonredundant mGzmA (A) or 400 nonredundant hGzmA (B) cleavage site motifs identified in the Jurkat proteome. Multiple sequence alignments (29) of peptide substrate motifs from P10 to P10′ were used to identify statistically significant over- or under-representation of residues, relative to the human Swiss-Prot database, with a p value threshold of 0.01. The amino acid heights are indicative of the degree of conservation at the indicated position (pink coloring represent the complete absence of the amino acid indicated). C, differential IceLogo indicating active site specificities based on hGzmA/mGzmA cleavage ratios. Residues cleaved more efficiently by hGzmA are shown above the subsite position, and residues cleaved more efficiently by mGzmA are shown below.
Active site preferences can be deduced from the COFRADIC data using the cleavage ratios (supplemental Table S1) of shared substrates as a measure of relative cleavage efficiency between hGzmA and mGzmA (Fig. 2C). This analysis shows that mGzmA cleaves P1 Lys positions over 10-fold better than hGzmA. Similarly, substrates carrying a P1–P2′ Lys-Lys-Leu motif were cleaved far more efficiently by mGzmA (∼87% of these are unique mGzmA cleavage events). In contrast, cleavages holding an Arg-Ser-Gly P1–P2′ motif tend to be cleaved more efficiently by hGzmA.
Serpinb6b Inhibits Mouse, but Not Human, GzmA
Cytotoxicity requires the intracellular delivery and function of granzymes, and therefore any cognate regulator must have an intracellular distribution. The clade B serpins are the only mammalian serpins found in the cytosol (30, 31), and some have already been characterized as granzyme inhibitors (2, 15, 16, 32, 33). Therefore, to identify potential serpin regulators of mouse and human GzmA, we aligned the derived substrate specificity patterns with clade B serpin RCLs (Fig. 3, A and B). None of the human serpins appeared to be good hGzmA inhibitor candidates. However, Serpinb6b was identified as a potential interactor for mGzmA. As shown in Fig. 3C, the RCL sequence of Serpinb6b (AANIGFR↓CMVPY) matches a mGzmA strongly selected residue at P2 and weakly selected residues at P4, P3, P1′, and P2′ (hGzmA strongly selected residues reside at P4 and P3 with weakly selected residues at P2 and P1′).
FIGURE 3.

Identification of Serpinb6b as a potential mouse granzyme A inhibitor. Alignment of all human (A) or mouse (B) clade B serpins with a predicted P1 Arg or Lys. Basic residues are shown enlarged in bold. C, peptide substrate specificity derived from phage display for mGzmA and hGzmA aligned to the Serpinb6b reactive center loop sequence. Strongly selected (>4σ) residues are shown in uppercase, and weakly selected residues (2σ to 4σ) are in lowercase. D, interaction of hGzmA with human clade B serpins. Serpins were expressed in COS-1 cells as GFP fusions and cell lysates incubated with or without hGzmA. Reactions were resolved by 10% SDS-PAGE and immunoblotted with a monoclonal GFP antibody.
Furthermore, although the proteomic substrate specificity profiles of human and mouse GzmA are quite similar, differences in cleavage efficiencies can readily be observed in substrates holding a cleavage site similar to the Serpinb6b RCL. Similar efficiencies were observed for mGzmA or hGzmA cleavage of TPM4 at the sequence Ile-Glu-Asn-Arg (Fig. 4A). By contrast, cleavage of SPF45 occurs 20-fold more efficiently by mGzmA at the sequence Tyr-Gly-Phe-Arg, which is highly similar to the Serpinb6b RCL motif Ile-Gly-Phe-Arg (Fig. 4B). Furthermore, MTr1 (P4 to P1 cleavage motif Met-Gly-Phe-Arg) was cleaved 10 times more efficiently by mGzmA, and DDX39A (cleavage motif Ser-Gly-Phe-Arg) was a unique mGzmA substrate (supplemental Table S1). In this way, phage display data and N-terminal COFRADIC results are complementary in suggesting Serpinb6b as a physiological mGzmA inhibitor.
FIGURE 4.
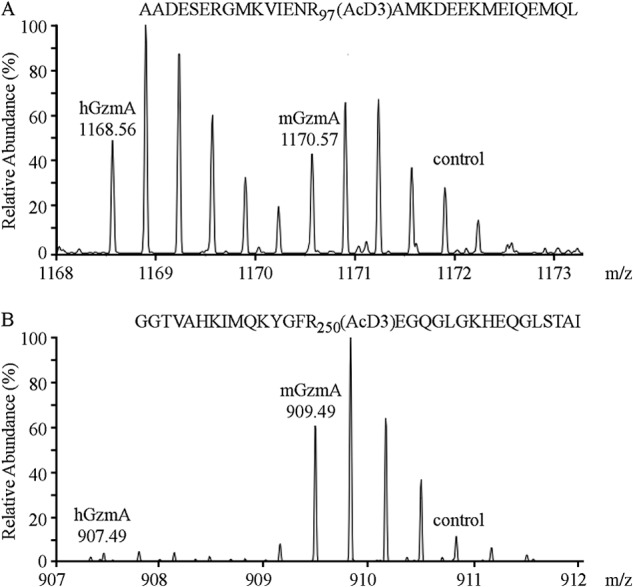
Proteome-wide identification of species-specific granzyme A substrates. A, mass spectra of a neo-N terminus derived from tropomyosin 4 (AcD3-98AMKDEEKMEIQEMQLKEAKHIAEEADR124), generated by both hGzmA and mGzmA. B, AcD3-250EGQGLGKHEQGLSTALSVEKTSKR273 from the splicing factor 45, preceded by a Serpinb6b-like tetrapeptide motif (YGFR), was cleaved almost exclusively by mGzmA. AcD3 denotes a trideutero-acetylated α-amino group.
We then confirmed an interaction between mGzmA and Serpinb6b by producing recombinant serpin in yeast and showing it forms a SDS-stable complex with mGzmA in vitro (Fig. 5A). Complex formation was increased between 1:1 and 2:1 ratios but was unchanged between 2:1 and 4:1, indicating a stoichiometry of inhibition between 1 and 2. Assays of residual activity indicated that the stoichiometry of inhibition (SI) is 1.8 ± 0.4 based on protein concentration (Fig. 5B); however, mGzmA is a dimer with two independent active sites (34); therefore, the SI per active site is 0.9 ± 0.2. We next used continuous kinetics to determine the association constant (ka) under pseudo-first order conditions. The derived value of 1.9 ± 0.8 × 105 m−1 s−1, combined with the low SI, suggests that the interaction is physiologically relevant (35).
FIGURE 5.
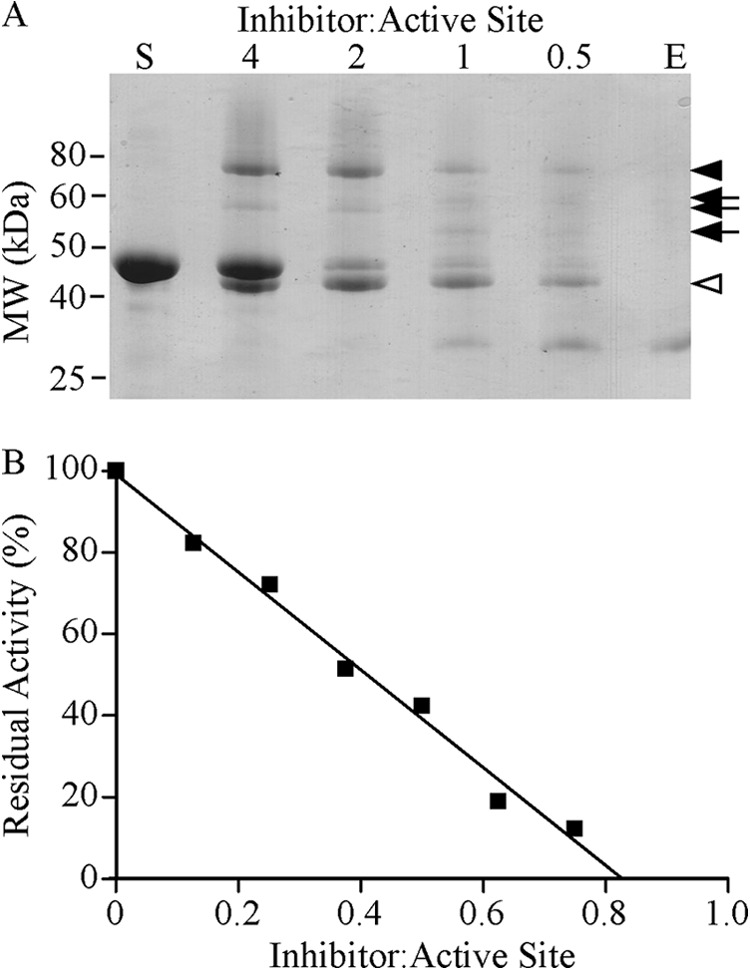
Kinetic analyses of the mouse granzyme A-Serpinb6b interaction. A, formation of a SDS-stable complex between mGzmA and Serpinb6b. 2 μg of mGzmA was incubated with the indicated molar excess of Serpinb6b at 37 °C for 20 min. The reaction was subsequently stopped by addition of Laemmli sample buffer with β-mercaptoethanol and boiling. Products were resolved by 12.5% reducing SDS-PAGE and visualized with Coomassie Brilliant Blue R-250. Arrowhead indicates complex; arrows indicate complex breakdown products, and open arrowhead indicates cleaved serpin. S, Serpinb6b alone; E, mGzmA alone. B, SI. Dimeric mGzmA was incubated with various concentrations of Serpinb6b at 37 °C for 30 min, and residual activity was assayed. Linear regression was used to extrapolate the concentration of Serpinb6b required to fully inhibit mGzmA.
Inhibition of hGzmA by Serpinb6b was extremely poor, and because Serpinb6b is a mouse-specific serpin (36), we could not assess a human orthologue with hGzmA. Instead, we tested human SERPINB6 (RCL sequence AAIMMMR↓CARFVPR), the nearest homologue of Serpinb6b to determine whether it can interact with hGzmA; however, no inhibition was observed. Likewise, Serpinb6a (RCL sequence AGMMTVR↓CMRFTPR), the mouse orthologue of SERPINB6 from which Serpinb6b evolved, showed no inhibition of mGzmA.
Because we were unable to identify an inhibitor of hGzmA using the substrate specificity profiles, we instead tested the ability of human clade B serpins to form an inhibitory complex with hGzmA. Clade B serpins were overexpressed in COS-1 cells fused to GFP at the C terminus (Fig. 3D). Only SERPINB5 and SERPINB9 were excluded as SERPINB9 is a well characterized GzmB inhibitor (2), whereas SERPINB5 is noninhibitory (37). Western blot of lysates incubated with hGzmA showed no interaction with SERPINB1, -B3, -B4, or -B13. A small proportion of cleavage was seen with SERPINB2, -B7, -B8, and -B10 indicating that they are weak substrates of hGzmA, whereas SERPINB11 was almost totally degraded. The only complexes identified were with SERPINB6 or SERPINB12 (Fig. 3D). In the case of SERPINB12, almost all of the serpin has been cleaved, with only a very small fraction forming an inhibitory complex, suggesting that this is a very good substrate but a poor serpin-type inhibitor of hGzmA. SERPINB6 showed the greatest degree of complex formation with hGzmA. However, the proportion of serpin that is cleaved is much greater than that forming an inhibitory complex (Fig. 3D). This agrees with the in vitro inhibition data showing that SERPINB6 is not an efficient inhibitor of hGzmA, although it is most efficient among the human clade B serpins.
Serpinb6b Is an Inhibitor of Dimeric, but Not Monomeric, mGzmA
Previous studies (27, 28) have indicated the presence of an exosite on dimeric GzmA that drives selection of macromolecular substrates. To test whether an exosite drives the interaction between Serpinb6b and mGzmA, we chemically reduced the mGzmA dimer and tested inhibition of monomeric mGzmA by Serpinb6b. As expected, reduction had no effect on the efficiency of peptide substrate cleavage by mGzmA (data not shown) (27); however, the serpin was no longer able to inhibit, suggesting that an important interaction has been lost.
The loss of inhibition upon chemical reduction of the mGzmA dimer indicates that the exosite plays an important role in the inhibition reaction with Serpinb6b, as well as during standard substrate cleavage interactions. The use of exosites during serpin inhibition is well established (19); however, the particular mechanism employed between mGzmA and Serpinb6b is somewhat unusual. Most of the well studied exosites in serpins are glycosaminoglycan-binding domains. The association with glycosaminoglycans accelerates the serpin protease interaction by promoting a conformational change in the serpin or bringing the serpin and protease into close proximity (38). Some serpins use exosites to directly bind their target protease; however, these exosites are linear extensions of the RCL and form interactions only between the serpin and the protease domain being targeted for inhibition (39). By contrast, the mGzmA-Serpinb6b interaction appears to involve an exosite formed by the protease dimer to guide the serpin interaction, meaning that the nontargeted protease monomer contributes to inhibitor binding.
A model of the initial Michaelis complex between mGzmA and Serpinb6b (Fig. 6) shows that this exosite interaction likely occurs between a positive charge patch on GzmA encompassing lysine residues 125, 126, and 232 and a corresponding polyglutamate stretch on the serpin from residues 265 to 268. Encouragingly, the positive patch on GzmA corresponds to the previously identified substrate exosite (27, 40), suggesting a conservation of selection mechanism between the inhibitor and substrates. The region identified in the serpin spans the top of the H-helix and the linker into strand 2C, which corresponds to the exosite of SERPINA4 (41). Similar to SERPINA4, the acidic residues in Serpinb6b constitute a small insertion, relative to the superfamily archetype, SERPINA1.
FIGURE 6.
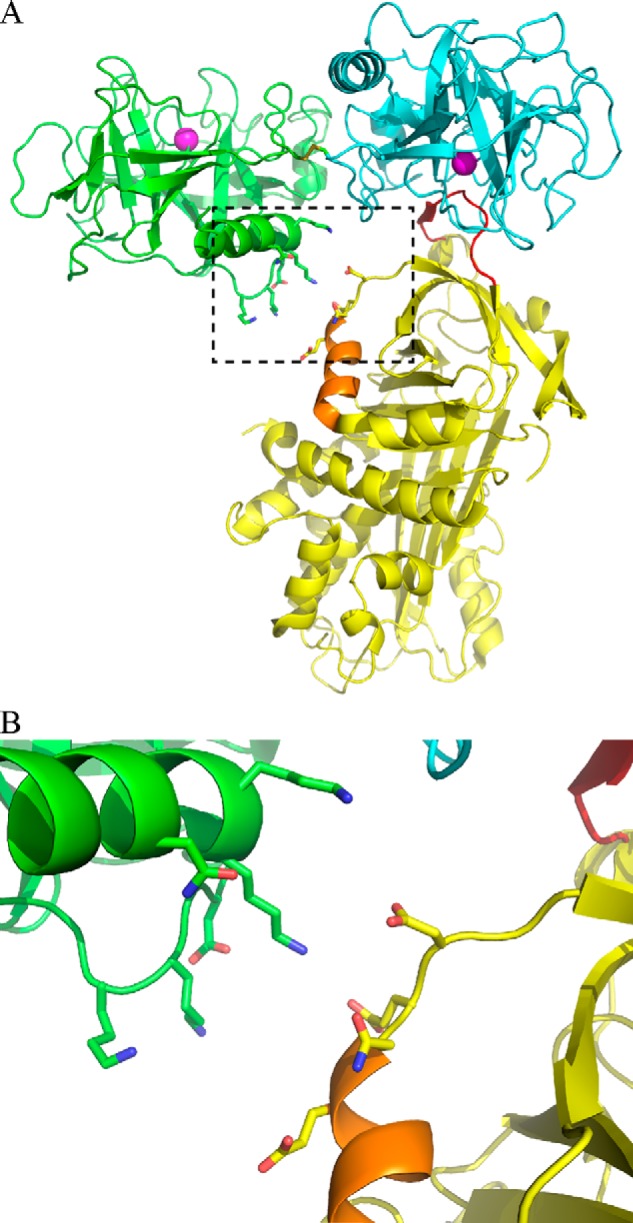
Model of the mouse granzyme A and Serpinb6b interaction. A, crystal structure of dimeric human GzmA (monomers are colored in green and cyan; PDB accession 1ORF) was aligned with the Michaelis complex (PDB accession 1OPH) between S195A trypsin (yellow) and M358R antitrypsin (aligned to the cyan GzmA monomer, not shown). Helix H is shown in orange. B, expanded view of the potential exosite interaction between mGzmA and Serpinb6b.
Serpinb6b Protects Cells from mGzmA-induced Cell Death
Clade B serpins are intracellular proteins lacking N-terminal secretion signals (31, 42), and human SERPINB6 cannot transit the endoplasmic reticulum even when fused to an efficient secretion signal (43). As such, Serpinb6b is most likely an intracellular protein, and therefore we next tested its ability to inhibit the actions of mGzmA in a cellular environment. To accomplish this, we transfected P815 cells with pIRES-GFP/Serpinb6b and pEF-puro or pEF-puro alone and confirmed by immunofluorescence that it was expressed in both the cytoplasm and nucleus (data not shown). GzmA was then delivered into the cytoplasm using streptolysin-O. Cell survival was then measured 24 h later by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay.
Mouse GzmA induced death in pEF-puro-transfected cells with an EC50 of 0.7 ± 0.5 μm, whereas transfection with Serpinb6b increased this 13-fold to 9.1 ± 3.0 μm (Fig. 7). Human GzmA has been shown to be less cytotoxic than mGzmA (5). We confirmed this by delivering hGzmA into P815 cells and deriving an EC50 of 3.5 ± 1.8 μm (Fig. 7). By contrast to mGzmA, hGzmA was not affected by the presence of Serpinb6b (EC50 4.1 ± 2.3), in agreement with the kinetic data, and suggesting that Serpinb6b is a biologically relevant species-specific inhibitor.
FIGURE 7.
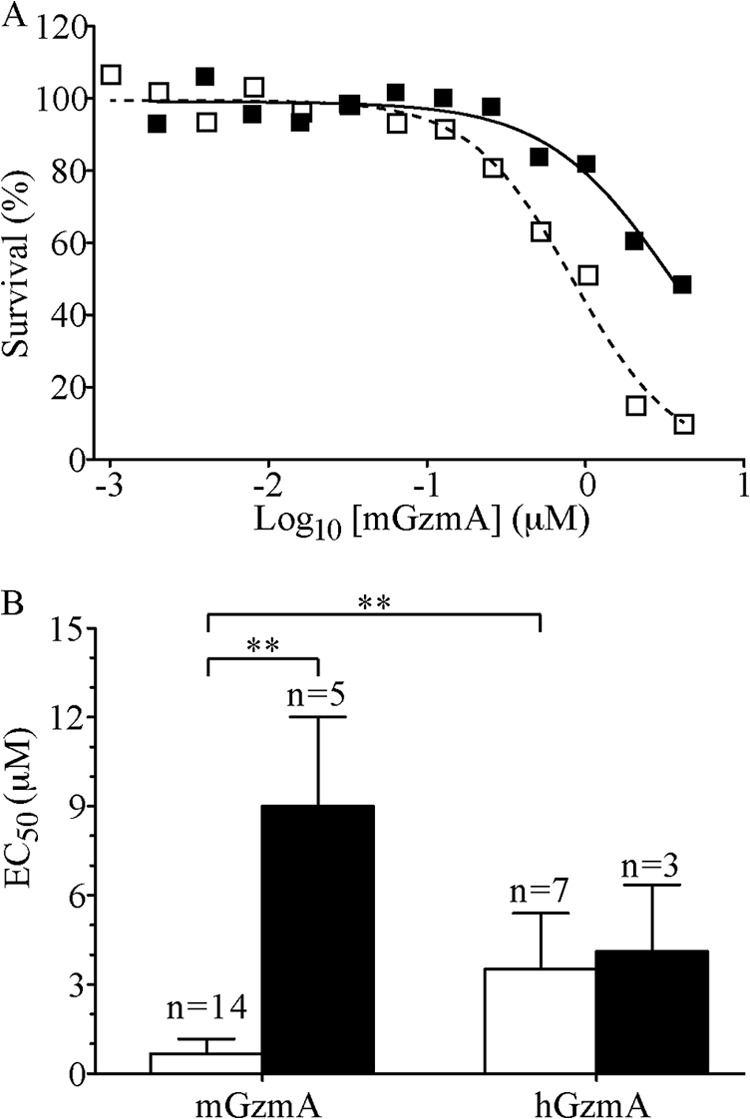
Serpinb6b protects target cells from mouse, but not human, granzyme A-induced death. A, survival curve of P815 cells transfected with pEF-puro (open squares) or pEF-puro and pEF-IRES-GFP/Serpinb6b (closed squares) exposed to a sublytic concentration of streptolysin-O and a serial dilution series of mGzmA for 1 h at 37 °C, with survival measured by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay 18 h later. EC50 values were derived by fitting a sigmoidal dose-response equation by nonlinear regression. B, bar graph summarizing all experiments indicating means ± S.D. **, p < 0.01.
DISCUSSION
The body of literature on GzmA function is confounding and often contradictory. We have addressed this controversy starting from the basic assumption that a protease capable of killing cells drives the evolution of stringent protective mechanisms. For granzymes, this includes storage within cytotoxic granules and regulation by cognate serpins (2, 15, 16, 32, 33). Here, we have identified Serpinb6b as a fast and efficient inhibitor of mGzmA, and we demonstrated that none of the human clade B serpins efficiently form complexes with hGzmA. We have reproduced previous results (5) showing a difference in cytotoxicity between human and mouse GzmA. Given that Serpinb6b was originally identified in natural killer cells (18), and it is a nucleocytoplasmic protein that protects against mGzmA-induced death in vitro (Fig. 7), we conclude it is a physiologically relevant granzyme regulator. Our results therefore support the view that mouse but not human GzmA has cytotoxic potential in vivo.
We used phage display and positional proteomics to probe and compare the substrate specificities of human and mouse GzmA. The phage display results agree with those determined by PS-SCL (27, 44) and the hGzmA pattern derived by van Damme et al. (28) by proteomics, while extending these cleavage motifs to the prime side (C-terminal to the cleavage point). These profiles show significant differences between human and mouse GzmA and were confirmed by assessing cleavage efficiencies of synthetic peptide substrates.
By contrast, the proteomic data show little difference in the overall specificity profiles of human and mouse GzmA. This contrasts to experiments with GzmB in which species-specific substrate patterns observed by phage display (5) were recapitulated in proteomic analyses (6) and PS-SCL (44). The discrepancy between methods for profiling GzmA substrate specificity can be explained by the presence of exosites formed by the dimer (27, 40). In both phage display and PS-SCL, the use of short peptides, which are not capable of extended surface interactions, precludes the assessment of exosite effects. However, the gentle lysis procedure utilized for proteomics presents fully folded proteins able to interact with these exosites and effectively overrides all but the strongest active site preferences. As such, we would predict that a proteomic analysis using monomeric GzmA would yield a pattern similar to that derived by phage display, and it would be interesting to note whether substrates are conserved between monomeric and dimeric GzmA.
Our data indicate that both the exosite interaction and a favorable RCL sequence are required for efficient inhibition of mGzmA by Serpinb6b. Serpinb6b cannot inhibit monomeric mGzmA, in which the exosite interaction is lost, and Serpinb6a, which is 82% identical to Serpinb6b and retains the exosite but has a different RCL sequence (4/14 residues conserved between Serpinb6a and Serpinb6b RCLs at P7, P1, P1′, and P2′), does not inhibit monomeric or dimeric mGzmA. The latter point differs from many other serpin exosites in which the exosite interaction itself is sufficient to drive reactions when the RCL sequence is not efficiently cleavable. The inability of Serpinb6b to inhibit hGzmA is curious as phage display suggests that the RCL should be cleaved by hGzmA, and the exosite interaction should also be present. However, the observation that kcat is dramatically reduced although Km is less affected for the GLFR substrate compared with the VANR substrate suggests that the extended incubation time used in phage display (overnight) may allow selection of kinetically unfavorable sequences. By contrast, serpin inhibition requires efficient cleavage of the RCL.
Previous studies have identified inhibitors of hGzmA in human plasma (21). Antithrombin III (SERPINC1) inhibits hGzmA in plasma with a ka of 4 × 105 m−1 s−1, similar to that we observed for Serpinb6b and mGzmA. Inhibition of both active sites of dimeric hGzmA proceeds independent of each other, allowing the formation of inhibitory complexes with either one or two SERPINC1 molecules; however, the SI was not determined (21). The RCL sequences of mouse (TSVVITGR↓SLNPNRVT) and human (TAVVIAGR↓SLNPNRVT) SERPINC1 are almost identical, and as such we would expect mGzmA to be inhibited by mouse SERPINC1 in vivo. This leads to a situation in which hGzmA has an efficient extracellular inhibitor and no intracellular inhibitor, whereas mGzmA has evolved both an extracellular and intracellular inhibitor, pointing to a gain of function in mGzmA, relative to hGzmA.
We have demonstrated a 5-fold increase of cytotoxicity of mGzmA compared with hGzmA (Fig. 7), yet the EC50 (680 nm) is still an order of magnitude higher than that of mGzmB and 2 orders of magnitude higher than that of hGzmB (5). With these figures, it is difficult to see how GzmA could function in its own right as a dedicated cytotoxin or as a backup if targets express a GzmB inhibitor. Given earlier studies implicating GzmA in extracellular processes (45) and that both human and mouse produce an effective extracellular GzmA inhibitor, a more likely scenario is that GzmA has a conserved extracellular role, perhaps in the regulation of inflammation (13). During evolution, the mGzmA active site has co-evolved with its natural extracellular substrates (and inhibitors), but this has coincidentally increased its cytotoxicity if inadvertently released into the cytoplasm of the host cell by stress. This relatively small increase in mGzmA cytotoxicity has been enough to drive the evolution of a specific intracellular inhibitor.
In closing, we have identified Serpinb6b as a specific intracellular inhibitor of mGzmA, but not hGzmA. Serpinb6b utilizes an unusual exosite interaction dependent on the dimeric nature of GzmA in its inhibitory pathway. The identification of Serpinb6b indicates a divergence in cytotoxic potential, and perhaps physiological function, between human and mouse GzmA.
Supplementary Material

This article contains supplemental Table S1.
- Gzm
- granzyme
- h
- human
- m
- mouse
- RCL
- reactive center loop
- PDB
- Protein Data Bank
- SI
- stoichiometry of inhibition
- K-dnp
- lysine-2,4-dinitrophenyl
- Abz
- o-aminobenzoic acid.
REFERENCES
- 1. Voskoboinik I., Dunstone M. A., Baran K., Whisstock J. C., Trapani J. A. (2010) Perforin: structure, function, and role in human immunopathology. Immunol. Rev. 235, 35–54 [DOI] [PubMed] [Google Scholar]
- 2. Bird C. H., Sutton V. R., Sun J., Hirst C. E., Novak A., Kumar S., Trapani J. A., Bird P. I. (1998) Selective regulation of apoptosis: the cytotoxic lymphocyte serpin proteinase inhibitor 9 protects against granzyme B-mediated apoptosis without perturbing the Fas cell death pathway. Mol. Cell. Biol. 18, 6387–6398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Joeckel L. T., Bird P. I. (2014) Are all granzymes cytotoxic in vivo? Biol. Chem. 395, 181–202 [DOI] [PubMed] [Google Scholar]
- 4. Casciola-Rosen L., Garcia-Calvo M., Bull H. G., Becker J. W., Hines T., Thornberry N. A., Rosen A. (2007) Mouse and human granzyme B have distinct tetrapeptide specificities and abilities to recruit the bid pathway. J. Biol. Chem. 282, 4545–4552 [DOI] [PubMed] [Google Scholar]
- 5. Kaiserman D., Bird C. H., Sun J., Matthews A., Ung K., Whisstock J. C., Thompson P. E., Trapani J. A., Bird P. I. (2006) The major human and mouse granzymes are structurally and functionally divergent. J. Cell Biol. 175, 619–630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Van Damme P., Maurer-Stroh S., Plasman K., Van Durme J., Colaert N., Timmerman E., De Bock P. J., Goethals M., Rousseau F., Schymkowitz J., Vandekerckhove J., Gevaert K. (2009) Analysis of protein processing by N-terminal proteomics reveals novel species-specific substrate determinants of granzyme B orthologs. Mol. Cell. Proteomics 8, 258–272 [DOI] [PubMed] [Google Scholar]
- 7. Cullen S. P., Adrain C., Lüthi A. U., Duriez P. J., Martin S. J. (2007) Human and murine granzyme B exhibit divergent substrate preferences. J. Cell Biol. 176, 435–444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Adrain C., Murphy B. M., Martin S. J. (2005) Molecular ordering of the caspase activation cascade initiated by the cytotoxic T lymphocyte/natural killer (CTL/NK) protease granzyme B. J. Biol. Chem. 280, 4663–4673 [DOI] [PubMed] [Google Scholar]
- 9. Shi L., Kam C. M., Powers J. C., Aebersold R., Greenberg A. H. (1992) Purification of three cytotoxic lymphocyte granule serine proteases that induce apoptosis through distinct substrate and target cell interactions. J. Exp. Med. 176, 1521–1529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Shresta S., Graubert T. A., Thomas D. A., Raptis S. Z., Ley T. J. (1999) Granzyme A initiates an alternative pathway for granule-mediated apoptosis. Immunity 10, 595–605 [DOI] [PubMed] [Google Scholar]
- 11. Lieberman J. (2010) Granzyme A activates another way to die. Immunol. Rev. 235, 93–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Susanto O., Stewart S. E., Voskoboinik I., Brasacchio D., Hagn M., Ellis S., Asquith S., Sedelies K. A., Bird P. I., Waterhouse N. J., Trapani J. A. (2013) Mouse granzyme A induces a novel death with writhing morphology that is mechanistically distinct from granzyme B-induced apoptosis. Cell Death Differ. 20, 1183–1193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Metkar S. S., Menaa C., Pardo J., Wang B., Wallich R., Freudenberg M., Kim S., Raja S. M., Shi L., Simon M. M., Froelich C. J. (2008) Human and mouse granzyme A induce a proinflammatory cytokine response. Immunity 29, 720–733 [DOI] [PubMed] [Google Scholar]
- 14. Sower L. E., Klimpel G. R., Hanna W., Froelich C. J. (1996) Extracellular activities of human granzymes. I. Granzyme A induces IL6 and IL8 production in fibroblast and epithelial cell lines. Cell. Immunol. 171, 159–163 [DOI] [PubMed] [Google Scholar]
- 15. de Koning P. J., Kummer J. A., de Poot S. A., Quadir R., Broekhuizen R., McGettrick A. F., Higgins W. J., Devreese B., Worrall D. M., Bovenschen N. (2011) Intracellular serine protease inhibitor SERPINB4 inhibits granzyme M-induced cell death. PLoS One 6, e22645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wang L., Li Q., Wu L., Liu S., Zhang Y., Yang X., Zhu P., Zhang H., Zhang K., Lou J., Liu P., Tong L., Sun F., Fan Z. (2013) Identification of SERPINB1 as a physiological inhibitor of human granzyme H. J. Immunol. 190, 1319–1330 [DOI] [PubMed] [Google Scholar]
- 17. Gettins P. G. (2002) Serpin structure, mechanism, and function. Chem. Rev. 102, 4751–4804 [DOI] [PubMed] [Google Scholar]
- 18. Sun J., Ooms L., Bird C. H., Sutton V. R., Trapani J. A., Bird P. I. (1997) A new family of 10 murine ovalbumin serpins includes two homologs of proteinase inhibitor 8 and two homologs of the granzyme B inhibitor (proteinase inhibitor 9). J. Biol. Chem. 272, 15434–15441 [DOI] [PubMed] [Google Scholar]
- 19. Gettins P. G., Olson S. T. (2009) Exosite determinants of serpin specificity. J. Biol. Chem. 284, 20441–20445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tsuzuki S., Kokado Y., Satomi S., Yamasaki Y., Hirayasu H., Iwanaga T., Fushiki T. (2003) Purification and identification of a binding protein for pancreatic secretory trypsin inhibitor: a novel role of the inhibitor as an anti-granzyme A. Biochem. J. 372, 227–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Masson D., Tschopp J. (1988) Inhibition of lymphocyte protease granzyme A by antithrombin III. Mol. Immunol. 25, 1283–1289 [DOI] [PubMed] [Google Scholar]
- 22. Plasman K., Van Damme P., Kaiserman D., Impens F., Demeyer K., Helsens K., Goethals M., Bird P. I., Vandekerckhove J., Gevaert K. (2011) Probing the efficiency of proteolytic events by positional proteomics. Mol. Cell. Proteomics 10.1074/mcp.M110.003301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Staes A., Impens F., Van Damme P., Ruttens B., Goethals M., Demol H., Timmerman E., Vandekerckhove J., Gevaert K. (2011) Selecting protein N-terminal peptides by combined fractional diagonal chromatography. Nat. Protoc. 6, 1130–1141 [DOI] [PubMed] [Google Scholar]
- 24. Sun J., Bird C. H., Buzza M. S., McKee K. E., Whisstock J. C., Bird P. I. (1999) Expression and purification of recombinant human granzyme B from Pichia pastoris. Biochem. Biophys. Res. Commun. 261, 251–255 [DOI] [PubMed] [Google Scholar]
- 25. Sun J., Coughlin P., Salem H. H., Bird P. (1995) Production and characterization of recombinant human proteinase inhibitor 6 expressed in Pichia pastoris. Biochim. Biophys. Acta 1252, 28–34 [DOI] [PubMed] [Google Scholar]
- 26. Sun J., Whisstock J. C., Harriott P., Walker B., Novak A., Thompson P. E., Smith A. I., Bird P. I. (2001) Importance of the P4′ residue in human granzyme B inhibitors and substrates revealed by scanning mutagenesis of the proteinase inhibitor 9 reactive center loop. J. Biol. Chem. 276, 15177–15184 [DOI] [PubMed] [Google Scholar]
- 27. Bell J. K., Goetz D. H., Mahrus S., Harris J. L., Fletterick R. J., Craik C. S. (2003) The oligomeric structure of human granzyme A is a determinant of its extended substrate specificity. Nat. Struct. Biol. 10, 527–534 [DOI] [PubMed] [Google Scholar]
- 28. Van Damme P., Maurer-Stroh S., Hao H., Colaert N., Timmerman E., Eisenhaber F., Vandekerckhove J., Gevaert K. (2010) The substrate specificity profile of human granzyme A. Biol. Chem. 391, 983–997 [DOI] [PubMed] [Google Scholar]
- 29. Colaert N., Helsens K., Martens L., Vandekerckhove J., Gevaert K. (2009) Improved visualization of protein consensus sequences by iceLogo. Nat. Methods 6, 786–787 [DOI] [PubMed] [Google Scholar]
- 30. Irving J. A., Pike R. N., Lesk A. M., Whisstock J. C. (2000) Phylogeny of the serpin superfamily: implications of patterns of amino acid conservation for structure and function. Genome Res. 10, 1845–1864 [DOI] [PubMed] [Google Scholar]
- 31. Remold-O'Donnell E. (1993) The ovalbumin family of serpin proteins. FEBS Lett. 315, 105–108 [DOI] [PubMed] [Google Scholar]
- 32. Kaiserman D., Bird P. I. (2010) Control of granzymes by serpins. Cell Death Differ. 17, 586–595 [DOI] [PubMed] [Google Scholar]
- 33. Bots M., Kolfschoten I. G., Bres S. A., Rademaker M. T., de Roo G. M., Krüse M., Franken K. L., Hahne M., Froelich C. J., Melief C. J., Offringa R., Medema J. P. (2005) SPI-CI and SPI-6 cooperate in the protection from effector cell-mediated cytotoxicity. Blood 105, 1153–1161 [DOI] [PubMed] [Google Scholar]
- 34. Masson D., Zamai M., Tschopp J. (1986) Identification of granzyme A isolated from cytotoxic T-lymphocyte-granules as one of the proteases encoded by CTL-specific genes. FEBS Lett. 208, 84–88 [DOI] [PubMed] [Google Scholar]
- 35. Travis J., Salvesen G. S. (1983) Human plasma proteinase inhibitors. Annu. Rev. Biochem. 52, 655–709 [DOI] [PubMed] [Google Scholar]
- 36. Kaiserman D., Knaggs S., Scarff K. L., Gillard A., Mirza G., Cadman M., McKeone R., Denny P., Cooley J., Benarafa C., Remold-O'Donnell E., Ragoussis J., Bird P. I. (2002) Comparison of human chromosome 6p25 with mouse chromosome 13 reveals a greatly expanded ov-serpin gene repertoire in the mouse. Genomics 79, 349–362 [DOI] [PubMed] [Google Scholar]
- 37. Pemberton P. A., Wong D. T., Gibson H. L., Kiefer M. C., Fitzpatrick P. A., Sager R., Barr P. J. (1995) The tumor suppressor maspin does not undergo the stressed to relaxed transition or inhibit trypsin-like serine proteases. Evidence that maspin is not a protease inhibitory serpin. J. Biol. Chem. 270, 15832–15837 [DOI] [PubMed] [Google Scholar]
- 38. Pike R. N., Buckle A. M., le Bonniec B. F., Church F. C. (2005) Control of the coagulation system by serpins. Getting by with a little help from glycosaminoglycans. FEBS J. 272, 4842–4851 [DOI] [PubMed] [Google Scholar]
- 39. Ibarra C. A., Blouse G. E., Christian T. D., Shore J. D. (2004) The contribution of the exosite residues of plasminogen activator inhibitor-1 to proteinase inhibition. J. Biol. Chem. 279, 3643–3650 [DOI] [PubMed] [Google Scholar]
- 40. Hink-Schauer C., Estébanez-Perpiñá E., Kurschus F. C., Bode W., Jenne D. E. (2003) Crystal structure of the apoptosis-inducing human granzyme A dimer. Nat. Struct. Biol. 10, 535–540 [DOI] [PubMed] [Google Scholar]
- 41. Chen V. C., Chao L., Chao J. (2000) A positively charged loop on the surface of kallistatin functions to enhance tissue kallikrein inhibition by acting as a secondary binding site for kallikrein. J. Biol. Chem. 275, 40371–40377 [DOI] [PubMed] [Google Scholar]
- 42. Silverman G. A., Whisstock J. C., Askew D. J., Pak S. C., Luke C. J., Cataltepe S., Irving J. A., Bird P. I. (2004) Human clade B serpins (ov-serpins) belong to a cohort of evolutionarily dispersed intracellular proteinase inhibitor clades that protect cells from promiscuous proteolysis. Cell. Mol. Life Sci. 61, 301–325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Scott F. L., Coughlin P. B., Bird C., Cerruti L., Hayman J. A., Bird P. (1996) Proteinase inhibitor 6 cannot be secreted, which suggests it is a new type of cellular serpin. J. Biol. Chem. 271, 1605–1612 [DOI] [PubMed] [Google Scholar]
- 44. Mahrus S., Craik C. S. (2005) Selective chemical functional probes of granzymes A and B reveal granzyme B is a major effector of natural killer cell-mediated lysis of target cells. Chem. Biol. 12, 567–577 [DOI] [PubMed] [Google Scholar]
- 45. Buzza M. S., Bird P. I. (2006) Extracellular granzymes: current perspectives. Biol. Chem. 387, 827–837 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.