

Background: AMD and STGD1 are blinding diseases with similar clinical presentations but unrelated genetic causes.
Results: Bisretinoid-dependent complement reactivity on RPE cells involves the alternative pathway and depends on the CFH haplotype.
Conclusion: Inefficient CFH synthesis because of either Y402H and I62V substitutions or bisretinoid accumulation predisposes RPE cells to disease.
Significance: These results suggest a common inflammatory etiology for AMD and STGD1.
Keywords: Complement System, Epithelium, Inflammation, Retinal Degeneration, Retinoid, Age-related Macular Degeneration (AMD), Bisretinoids, Complement Factor H (CFH), Recessive Stargardt Macular Degeneration (STGD1), Retinal Pigment Epithelium
Abstract
Age-related macular degeneration (AMD) is a common central blinding disease of the elderly. Homozygosity for a sequence variant causing Y402H and I62V substitutions in the gene for complement factor H (CFH) is strongly associated with risk of AMD. CFH, secreted by many cell types, including those of the retinal pigment epithelium (RPE), is a regulatory protein that inhibits complement activation. Recessive Stargardt maculopathy is another central blinding disease caused by mutations in the gene for ABCA4, a transporter in photoreceptor outer segments (OS) that clears retinaldehyde and prevents formation of toxic bisretinoids. Photoreceptors daily shed their distal OS, which are phagocytosed by the RPE cells. Here, we investigated the relationship between the CFH haplotype of human RPE (hRPE) cells, exposure to OS containing bisretinoids, and complement activation. We show that hRPE cells of the AMD-predisposing CFH haplotype (HH402/VV62) are attacked by complement following exposure to bisretinoid-containing Abca4−/− OS. This activation was dependent on factor B, indicating involvement of the alternative pathway. In contrast, hRPE cells of the AMD-protective CFH haplotype (YY402/II62) showed no complement activation following exposure to either Abca4−/− or wild-type OS. The AMD-protective YY402/II62 hRPE cells were more resistant to the membrane attack complex, whereas HH402/VV62 hRPE cells showed significant membrane attack complex deposition following ingestion of Abca4−/− OS. These results suggest that bisretinoid accumulation in hRPE cells stimulates activation and dysregulation of complement. Cells with an intact complement negative regulatory system are protected from complement attack, whereas cells with reduced CFH synthesis because of the Y402H and I62V substitutions are vulnerable to disease.
Introduction
Age-related macular degeneration (AMD)3 is a common cause of visual loss in the elderly (1, 2). Inflammation, oxidative stress, high-fat diets, light exposure, and genetic factors all contribute to the pathogenesis of AMD (3–12). Among the genetic determinants for AMD, the gene for complement factor H (CFH) is a strong susceptibility locus (4–7). A common AMD-associated haplotype in the CFH gene is a Y402H and I62V substitution (6). Y402H-substituted CFH has been shown recently to bind malondialdehyde with greatly reduced affinity (13). Malondialdehyde is a natural byproduct of lipid peroxidation, suggesting that oxidative stress and complement dysregulation may act through a common mechanism. The association between AMD and CFH was further strengthened with the demonstration that a rare allele causing a R1210C substitution in CFH is a highly penetrant cause of AMD (3).
The complement cascade is a component of the innate immune system and is activated via four pathways (classical, lectin, alternative, and intrinsic) with different initiating triggers (14). These pathways converge with activation of C3 and culminate by assembly of the membrane attack complex (MAC or C5b-9) (15). MAC deposition generates pores in cell membranes that efficiently destroy invading pathogens. However, inappropriate activation of the complement system can do damage to cells of the host. For this reason, the complement system includes a set of complement negative regulatory proteins (CRPs) to prevent spontaneous activation (16). One example is CFH, which is secreted by many cell types, including cells of the retinal pigment epithelium (RPE), as local protection against complement activation. Other CRPs include membrane cofactor protein (MCP or CD46), decay-accelerating factor (DAF or CD55), and membrane inhibitor of reactive lysis (MIRL or CD59). These proteins are expressed in cell membranes of peripheral tissues, including RPE, and prevent deposition of the MAC (17). Beside CFH, the genes for other complement-related genes, including complement factor H-related 1 and 3 (CFHR1 and CFHR3), complement factor B (CFB), complement factor 2 (C2), and complement factor 3 (C3), are less common susceptibility loci for AMD (8–10, 18, 19).
AMD is thought to include primary dysfunction of the choriocapillaris and RPE. Degeneration of macular photoreceptors probably results from loss of the critical RPE support role. Accumulation of fluorescent bisretinoid pigments in cells of the RPE is a pathologic hallmark of the related disease recessive Stargardt macular degeneration (STGD1) (20). In contrast to AMD, the onset of symptoms in STGD1 patients may occur as early as the first or second decade of life. STGD1 is caused exclusively by mutations in the ABCA4 gene, which encodes an ATP-dependent transporter for phosphatidylethanolamine conjugated to retinaldehyde (N-ret-PE) in photoreceptor outer segments (OS) (21, 22). Photoreceptors daily shed the distal ∼10% of their OS, which are phagocytosed by cells of the adjacent RPE (23). Thus, toxic compounds in the OS, such as bisretinoid condensation products of retinaldehyde (N-ret-PE), must be processed by the RPE.
Similar to STGD1 patients, mice with a knockout mutation in the Abca4 gene accumulate fluorescent lipofuscin granules and bisretinoids, such as A2E, in the RPE (24, 25). A2E exhibits several mechanisms of cytotoxicity and is implicated in the pathogenesis of STGD1 (26–32). Interestingly, Abca4−/− mice exhibit dysregulation of the complement system and signs of complement-mediated inflammation in the RPE before the onset of photoreceptor degeneration (25, 33). These observations establish a connection between bisretinoid accumulation and complement activation in vivo. Together, these findings suggest common etiologies for STGD1 and “dry” AMD (11). This study investigates the effects of AMD-protective and AMD-predisposing CFH haplotypes on complement activation by bisretinoids in RPE cells.
EXPERIMENTAL PROCEDURES
Cultured Fetal Human Retinal Pigment Epithelial Cells
Fetal hRPE cells were collected from the eyes of aborted fetuses of 18–23 weeks of gestation (Advanced Bioscience Resources, Alameda, CA). The hRPE cells were then grown and maintained in Chee's essential replacement medium containing 1% calf serum and minimal essential medium containing 1.8 mm CaCl2 (Sigma-Aldrich) for two months before use. A detailed protocol of our cultured hRPE cells has been presented previously by Hu and Bok (34, 35). For each experiment, unless specified otherwise, we used a minimum of three donor hRPE cell cultures of each CFH haplotype.
Complement Factor H Single Nucleotide Polymorphism Analysis
Fetal hRPE cells from 84 donors were genotyped at the Y402H (rs1061170) and V62I (rs800292) alleles of CFH using TaqMan® SNP genotyping assays. The V62I allele was detected using Applied Biosystems predesigned assays C_2530382_10 and C_2530274_1 at the recommended primer-probe dilution. The Y402H genotype was determined using a custom-designed assay (probes 6FAM-TTCTTCCATAATTTTG (Y402) and VIC-TTTCTTCCATGATTTTG (H 402) and primers GGATGGCAGGCAACGTCTATAGAT and CTTTATTTATTTATCATTGTTATGGTCCTTAGGAAA).Y402H SNP analysis was carried out using primer and probe concentrations of 900 nm and 100 nm, respectively. Five-microliter PCR reactions were carried out in triplicate using ∼30 ng of genomic DNA and the following conditions: 50 mm KCl, 10 mm Tris-HCl (pH 9.0 at 25 °C), 5 mm MgCl2, 0.2 mm dNTPs, and 0.0125 units of Platinum Taq polymerase (Invitrogen) in a thermal cycle profile of 3 min at 95 °C, followed by 45 cycles of 15 s at 93 °C and 60 s at 60 °C. All genotyping calls were made by manual examination of the data.
Mouse Photoreceptor Outer Segment Preparation
Pooled retinas of 3- to 6-month-old Abca4−/− and corresponding age-matched wild-type (BALB/c and 129/Sv) mice were collected in 45% sucrose in Hanks' balanced salt solution (Invitrogen). Crude OS samples were obtained by gentle vortexing followed by sedimentation at 3000 × g for 10 min at 4 °C. The pellet was discarded, and the supernatant was diluted 1:4 (v/v) with Hanks' balanced salt solution. Samples were pelleted down after spinning at 10,000 × g for 10 min at 4 °C. The OS pellet was washed tree times with Hanks' balanced salt solution and resuspended in DMEM (Sigma). Wild-type and Abca4−/− OS samples were prepared freshly as a bulk, ensuring that both YY402 and HH402 received the same amount of bisretinoids for each experiment (average of four mouse retinas in 250 μl of DMEM per transwell). Qualitative analysis of the mouse OS samples was performed by high-performance liquid chromatography using methods published previously to evaluate the bisretinoid content (33). Fetal hRPE cells were then incubated with wild-type and Abca4−/− OS for 2 h (RNA) or overnight (protein). The cells were then harvested for analysis of RNA by quantitative real-time PCR and protein by immunoblotting or immunohistochemistry.
Transepithelial Resistance Measurements
Fetal hRPE cells were grown on culture wells for 2 months until they reached confluence as a monolayer. Using an epithelial volt Ohm meter (EVOM, World Precision Instruments, New Haven, CT), experimental hRPE transepithelial resistance was measured before and after exposure of the cells to mouse OS and serum.
Complement Activation Assay
Two groups of hRPE cells with genotype of YY402/II62 (YY402), and HH402/VV62 (HH402) were exposed overnight to mouse OS (wild-type and Abca4−/−) either as a single feeding or twice per week. Cells were washed gently with warm DMEM, followed by exposure for 2 h at 37 °C in a 5% CO2 incubator to complete or complement-depleted human serum on both sides of the wells. The sera were collected, and the cells were washed three times in Hanks' balanced salt solution medium and collected for further analysis.
Quantitative Real-time PCR
Total RNA was extracted from wild-type and Abca4−/− OS-treated hRPE cells using an Absolutely RNA Miniprep kit (Stratagene) with DNase treatment and was reverse-transcribed to cDNA using SuperScript III (Invitrogen) according to the instructions of the manufacturer. Quantitative real-time PCR (qRT-PCR) was done using a two-step kit with SYBR Green (Invitrogen) and human gene-specific primer sets for CD46, CD55, CD59, and CFH. Primers sequences and qRT-PCR conditions have been reported previously by our laboratory (33). Data were normalized to 18 S rRNA Ct values for each sample. Relative mRNA levels for each gene were determined from the ΔCt values. The results are presented as means ± S.D.
CFH Immunoblot Analysis
Total protein concentrations in the media and cell homogenates were determined by BCA protein assay reaction (Micro BCATM protein assay kit, Pierce). Media samples were concentrated using a 100,000 molecular weight cutoff centrifuge column (Millipore). 10 μg of medium and 30 μg of cell homogenate protein samples were separated using a 6% Tris-glycine gel in Tris-glycine running buffer and then transferred onto a polyvinylidene difluoride membrane (Millipore). Membranes were blocked overnight at 4 °C with Odyssey blocking buffer (LI-COR Biosciences, Lincoln, NE) followed by incubation at room temperature for 1 h with goat anti-human CFH (1:2000, Quidel Corp., San Diego, CA) and monoclonal anti-mouse α-tubulin (1:2000, Sigma) in 0.5% normal donkey serum. Bands were detected with fluorescently labeled donkey anti-goat 680 (1:15,000, LI-COR Biosciences) and goat anti-mouse 800 (1:15,000, LI-COR Biosciences) using the Odyssey infrared imaging system (LI-COR Biosciences). CFH band intensities from the cell homogenate immunoblot analysis were then quantified and normalized to α-tubulin. Quantification was done in three independent experiments using three donor hRPE cultures for each genotype group (YY402 and HH402), and data are mean ± S.D. Statistical analyses were done using Student's t test.
Antibodies and Human Sera
All human sera (complete or C1q-depleted, factor B-depleted, C5, and CFH-depleted) were obtained from Quidel Corp. Monoclonal mouse antibody to human CD46 protein was from Biolegend (1:100, San Diego, CA). Monoclonal mouse antibodies to human CD55 and CD59 proteins were obtained from Abcam (1:150, Cambridge, MA). Monoclonal mouse antibody to human C5b-9 (or MAC, 1:50) was obtained from Dako Cytomation (Denmark).
Immunofluorescence Analysis
hRPE cells along with their support were fixed in 4% formaldehyde in 0.1 m phosphate buffer for 30 min at room temperature and then embedded in agarose (type XI low gelling temperature, Sigma-Aldrich). 100-μm sections, cut on a vibratome (VT1000s, Leica Microsystems, Germany), were blocked with goat serum (0.5%, Sigma) and 1% BSA in PBS for 1 h, followed by incubation with primary antibodies. The sections were rinsed three times with PBS (Sigma) containing 0.1% Tween 20 (PBS/T) and then incubated in goat anti-mouse IgG secondary antibodies conjugated with Alexa Fluor 488 or 568 (1:400, Invitrogen) for 1 h, followed by rinsing with PBS/T. The sections were stained with DAPI (Invitrogen) and then mounted with 5% n-propylgallate in 100% glycerol mounting medium. The sections were imaged with an Olympus FluoView FV1000 confocal laser-scanning microscope under a ×60 oil objective with excitation wavelengths of 488 nm and 559 nm and emission wavelengths of 500–545 nm and 575–675 nm, respectively. The average fluorescence pixel intensity of each section was counted from a minimum of 10 sections for each donor hRPE cell of both haplotypes. The results are presented as the mean ± S.D. Statistical analysis was performed using Student's t test.
ELISA Analysis
Following the mouse OS and serum incubation, the MBL oligomers, MASP-1, and MASP-2 levels in the normal human serum were determined using MBL Oligomer (BIOPORT Diagnostics, Denmark) and MASP-1 and MASP-2 ELISA kits (Uscn Life Science Inc., Hubei, China) according to the protocol of the manufacturer. The serum was diluted 1:50 with “sample diluent” for MBL Oligomer and 1:5000 for MASP-1 and MASP-2. Serum samples and cell homogenates of single-donor human RPE cells of both genotypes (YY402 and HH402) along with standard samples were run in duplicate for the ELISA reading plate. Each experiment was repeated three times, and the data are presented as the means ± S.D. Statistical analysis was performed using Student's t test.
RESULTS
CFH Expression and Membrane-bound Complement Negative Regulatory Proteins Are Similar in Confluent hRPE Cells of AMD-protective and AMD-predisposing Haplotypes
Fetal hRPE cells from 84 unrelated donors were analyzed for the genotype at codons 402 and 62 in CFH. We identified and expanded 10 hRPE cell lines of the AMD-protective haplotype, YY402/II62 (hereafter YY402), and eight lines of the AMD-predisposing haplotype, HH402/VV62 (hereafter HH402). By immunoblotting, we observed similar levels of CFH in the apical media of both HH402 and YY402 cultured cell lines (Fig. 1A). We also measured levels of the CRP mRNAs CFH, CD46, CD55, and CD59 in HH402 and YY402 hRPE cells by qRT-PCR. Expression levels of these mRNAs were comparable in cells of both haplotypes (Fig. 1B). Consistently, unchallenged hRPE cells of the YY402 and HH402 haplotypes showed similar levels of CD46 and CD59 protein by immunohistochemistry (Fig. 1C).
FIGURE 1.
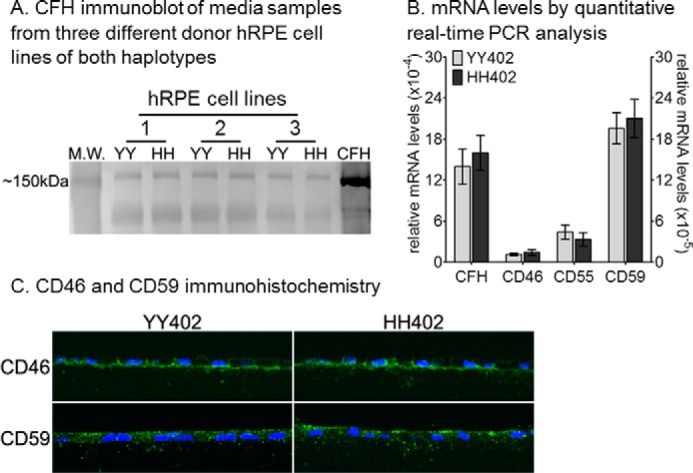
CFH expression and membrane-bound complement negative regulatory proteins are similar in confluent hRPE cells with AMD-protective and AMD-predisposing haplotypes. A, representative immunoblot analysis of CFH in the growth media of the AMD-protective (YY402, YY) and AMD-predisposing (HH402, HH) hRPE cells (10 μg of protein/lane). M.W., molecular weight. B, relative CD46, CD55, CD59, and CFH mRNA levels by qRT-PCR from YY402 and HH402 hRPE cells. Each mRNA level was normalized to 18 S rRNA (n = 9). The experiment was done in triplicate for three different donor cell lines for each haplotype. C, representative confocal images of CD46 (top row) and CD59 (bottom row) immunohistochemistry of YY402 (left column) and HH402 hRPE cells (right column). Each experiment was repeated three times with three different donor hRPE cell lines for each haplotype.
AMD-predisposing hRPE Cells Have Reduced Complement Factor H Synthesis following Ingestion of Abca4−/− Outer Segments
OS from Abca4−/− mice contain much higher levels of the bisretinoid, A2PE-H2 (dihydro-A2PE or A500 nm) than wild-type OS (Fig. 2A) (24, 25, 36). Previously, we showed increased expression of CRP-mRNAs in hRPE cells fed Abca4−/− but not wild-type OS (33). This presumably represents an adaptive response that protects the RPE from attack by the complement system because bisretinoids in the RPE stimulate complement activation (37, 38). We tested for possible differences in this response by hRPE cells of AMD-protective and AMD-predisposing haplotypes by incubating YY402 and HH402 hRPE cells with both wild-type and Abca4−/− OS. After growing to confluence, YY402 and HH402 hRPE cultures showed similar transepithelial resistances of 600–800 ohm/cm2 (Fig. 2B). Incubation with mouse OS and serum led to 50% reduction in the transepithelial resistance measurements in both YY402 and HH402 hRPE cultures irrespective of the bisretinoid content (Fig. 2B). Although the expression of CRP elicited by the wild-type OS was comparable in the YY402 and HH402 hRPE cells, significant differences were observed between the two haplotypes when the cells were incubated with Abca4−/− OS (Fig. 2C). In particular, we observed ∼10-fold reduced mRNA levels of CFH, the major fluid-phase complement negative regulator, in HH402 versus YY402 hRPE cells (Fig. 2C). Furthermore, we compared CFH protein expression in cell homogenates and culture media of HH402 and YY402 hRPE cells following incubation with Abca4−/− OS. CFH was reduced ∼2-fold in both samples of HH402 versus YY402 hRPE cells (Fig. 2D). By immunohistochemistry, we observed a modest reduction (20–25%) of CD46 and CD59 protein levels in the AMD-predisposed cells compared with the AMD-protective cells after incubation with Abca4−/− OS (data not shown). Consistent with the qRT-PCR results, there was no significant difference in the protein levels for CFH (Fig. 2D), CD46, and CD59 (data not shown) when cells were incubated with the wild-type OS. Thus, the CFH haplotype influences CFH expression following a challenge with bisretinoid-containing OS only.
FIGURE 2.
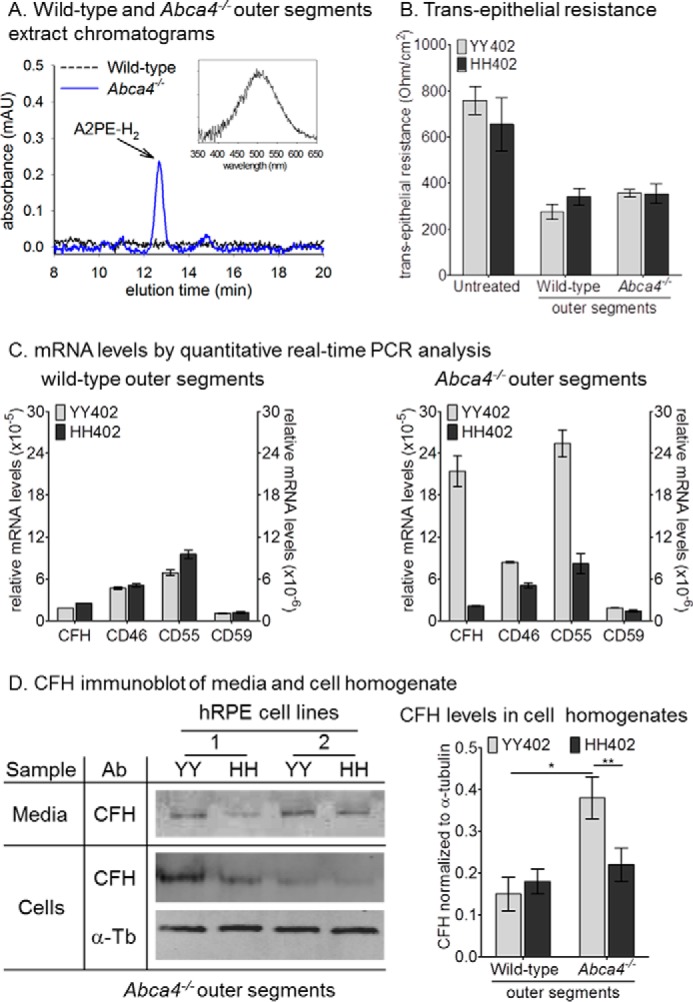
AMD-predisposing hRPE cells have reduced complement factor H synthesis following ingestion of Abca4−/− outer segments. A, high-performance liquid chromatography chromatograms of wild-type (black trace) and Abca4−/− OS (blue trace) chloroform extracts at 500-nm wavelength. The inset shows the maximum absorbance spectra of A2PE-H2 (dihydro-A2PE) at 502 nm. mAU, milliabsorbance units. B, transepithelial resistance measurements of six donor hRPE cell lines of each haplotype (YY402 and HH402) under normal growth conditions (untreated) and after incubation with wild-type and Abca4−/− OS and serum. C, relative CFH, CD46, CD55, and CD59 mRNA levels by quantitative real-time PCR from YY402 and HH402 hRPE cells fed with wild-type (left panel) and Abca4−/− (right panel) OS. The right y axes correspond to CD46, CD55, and CD59. Each mRNA level was normalized to 18 S rRNA (n = 9). The experiment was done in triplicate for three different donor cell lines for each haplotype. D, representative immunoblot analyses of CFH for YY402 and HH402 hRPE cells incubated with Abca4−/− OS (10 and 30 μg of protein/lane for media (top row) and hRPE cell homogenate (center and bottom rows), respectively). CFH band intensity (center column) in cell homogenate samples was normalized to α-tubulin (α-Tb) and the data are presented in the right panel. Note the 2-fold reduction in the CFH levels in the HH402 hRPE cells after incubation with Abca4−/− OS (n = 6). *, p < 0.005; **, p < 0.0005. The experiment was done in duplicate for three different donor cell lines for each haplotype.
Elevated C3/C3b Immunoreactivity in AMD-predisposing hRPE Cells after Challenging with Abca4−/− OS
To assay for complement activation, we performed immunohistochemistry using an antibody against C3/C3b in hRPE cells of both CFH haplotypes following incubation with wild-type or Abca4−/− OS. In this experiment, the OS were added in the presence of human serum depleted of CFH. Levels of C3/C3b immunofluorescence were similar in YY402 and HH402 cells after incubation with wild-type mouse OS (Fig. 3). In contrast, we observed ∼1.5-fold higher C3/C3b immunofluorescence in HH402 versus YY402 hRPE cells following incubation with Abca4−/− OS (Fig. 3). This observation suggests that the bisretinoids containing OS promotes elevated CFH-dependent C3 deposition on hRPE cells.
FIGURE 3.
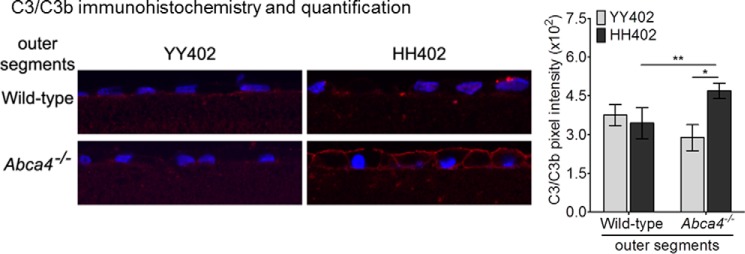
Elevated C3/C3b immunoreactivity in AMD-predisposing hRPE cells after challenge with Abca4−/− OS. Representative confocal images of C3/C3b immunoreactivity of YY402 (left panel) and HH402 (right panel) hRPE cells incubated with wild-type (top row) and Abca4−/− (bottom row) OS and for 2 hours with CFH-depleted serum. The histogram in the right panel shows 1.5-fold higher C3/C3b immunoreactivity on HH402 hRPE cells following Abca4−/− OS incubation compared with the YY402 hRPE cells (n = 6 for wild-type OS and n = 8 for Abca4−/− OS). *, p < 10−6; **, p < 10−4). The experiment was repeated three times with two different donor hRPE cell lines of the YY402 and HH402 haplotypes.
The Complement Alternative Pathway Is Responsible for Bisretinoid-mediated Complement Activation on hRPE Cells
To test for bisretinoid-stimulated MAC formation, we added complete human serum or serum lacking C1q (classical pathway), factor B (alternative pathway), or C5 (MAC component) to the hRPE culture medium 1 day after incubating the cells with Abca4−/− mouse OS. Then, we assayed for C5b-9 (MAC) deposition on the cells by immunohistochemistry. In the presence of C1q-depleted serum, HH402 hRPE cells showed 1.5-fold higher bisretinoid-dependent C5b-9 deposition versus YY402 cells (Fig. 4B). Increased MAC deposition was also seen in HH402 cells exposed to Abca4−/− OS following incubation with complete human serum (39). MAC deposition in the absence of component C1q, which is required in the classical pathway, suggests complement activation via the alternative pathway. To confirm this possibility, we repeated the experiment using human serum depleted of factor B, which is required for the alternative pathway. Eliminating factor B or C5 resulted in no deposition of C5b-9 on hRPE cells of either CFH haplotype (Fig. 4 and data not shown). Also, wild-type OS led to similar MAC immunoreactivity irrespective of the hRPE CFH haplotype (Fig. 4A). However, using factor B-depleted serum, we did observe C1q-dependent deposition of C5b-9 within the culture insert filters below the hRPE cells (Fig. 4), as described previously (40). Deposition of C5b-9 within the nitrocellulose filters was seen in hRPE cultured cells of both CFH haplotypes regardless of the OS bisretinoid content (Fig. 4, A and B). These observations indicate that bisretinoids activate the complement system through the alternative pathway.
FIGURE 4.

The complement alternative pathway is responsible for bisretinoid-mediated complement activation in hRPE cells. Representative confocal images of MAC (C5b-9) immunoreactivity of the YY402 (left panels, top rows) and HH402 (left panels, bottom rows) cells incubated with wild-type (A) and Abca4−/− (B) OS using C1q-depleted (left panels, left columns) and factor B-depleted (left panels, right columns) serum. Note that significant C5b-9 deposition on hRPE appears in the C1q-depleted serum only (B, left panel, bottom row). The absence of factor B in the serum prevents deposition of MAC on hRPE cells (A and B, right panels). Sub-RPE deposits because of activation of the classical pathway are present in filter substrate for both YY402 and HH402 hRPE cells in the factor B-depleted serum (fB-, A and B, right panels). Average of MAC pixel intensity measurements from five different donor hRPE cell lines of both YY402 and HH402 haplotype exposed to wild-type (A) and Abca4−/− (B) OS in the presence of C1q- and fB-depleted serum are presented in the right panels (C1q-depleted serum, n = 10 sections; fB-depleted serum, n = 7). *, p < 0.0001.
The Lectin Pathway Does Not Contribute to Bisretinoid-dependent Complement Reactivity on hRPE Cells
A recent study suggested that a hydrogen peroxide oxidatively stressed RPE cell line (ARPE-19) is initially attacked by complement through the lectin pathway, with subsequent amplification of this effect through activation of the alternative pathway (41). The lectin pathway is initiated by the binding of mannose-binding lectin (MBL) or ficolins to carbohydrates on the surfaces of pathogens (42). This initial step leads to MBL oligomerization and formation of complexes between the lectins and various MBL-associated serine proteases (MASPs), such as MASP-1 and MASP-2. The MBL oligomers and lectin-MASP complexes then bind to pathogens, resulting in the activation of C4 and C2 to generate a C3 convertase capable of activating C3 (43–45). To test whether the lectin pathway is activated by bisretinoids, we incubated hRPE cells with complete human serum after exposure to wild-type or Abca4−/− OS. We then quantitated MBL oligomers in the serum and cell homogenates by ELISA. MBL oligomers were similar in YY402 and HH402 cells following exposure to wild-type or Abca4−/− OS (Fig. 5, A and B). Importantly, the level of MBL oligomers in the serum above the cells was within the normal range (approximately 1 μg/ml) (43, 46) (Fig. 5B). Levels of the MBL activation products MASP-1 and MASP-2 were also similar in homogenates and sera from hRPE cells of both haplotypes and following incubation with wild-type or Abca4−/− OS (Fig. 5, C and D). These results suggest that bisretinoids do not activate the lectin pathway.
FIGURE 5.
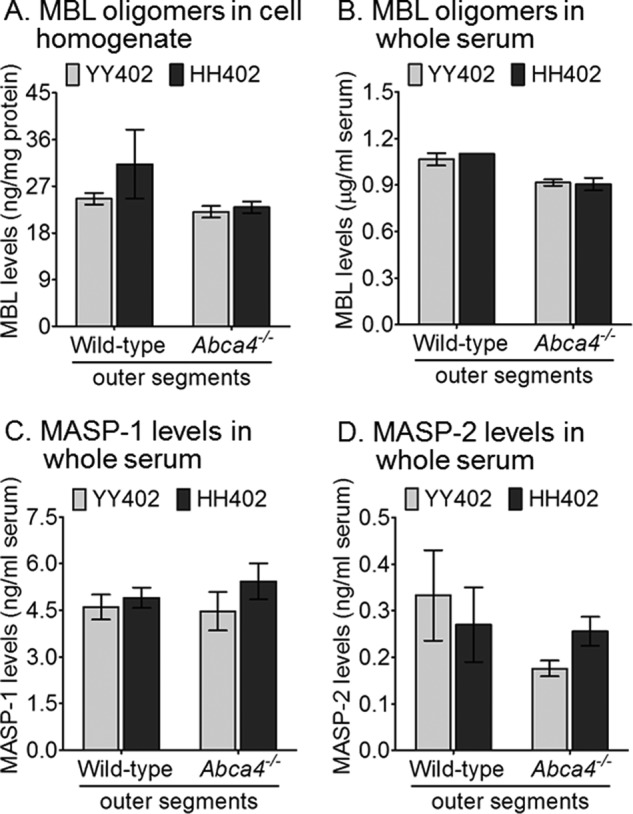
The lectin pathway does not contribute to bisretinoid-dependent complement reactivity. Representative data of a single hRPE cell donor of each haplotype (YY402 and HH402) following overnight incubation with OS of wild-type and Abca4−/− and 2-hour exposure to whole serum. Histograms show the average data for MBL oligomers in the hRPE cell homogenate (A) and MBL oligomer (B) MASP-1 (C) and MASP-2 (D) levels in the serum quantified using ELISA kits. Regardless of the mouse OS bisretinoid content, both YY402 and HH402 hRPE have similar levels of the MBL oligomers MASP-1 and MASP-2 (n = 3). The experiment was done in triplicate for each YY402 and HH402 hRPE haplotype.
Bisretinoid-mediated MAC Formation Is Dependent on the CFH Haplotype of hRPE Cells
To assess the role of CFH produced by hRPE cells on MAC deposition, we incubated hRPE cells with CFH-depleted serum after exposure to wild-type or Abca4−/− OS. We observed a minor deposition of C5b-9 on hRPE cells of both haplotypes by immunohistochemistry after exposure to wild-type OS (Fig. 6). C5b-9 immunofluorescence was slightly higher in YY402 hRPE cells fed Abca4−/− OS compared with the wild-type OS. In contrast, we observed 4-fold higher C5b-9 immunofluorescent labeling of HH402 hRPE cells following exposure to Abca4−/− OS (Fig. 6). This result suggests that locally secreted CFH inhibits MAC deposition and that Y402H/I62V-substituted CFH is much less active than “normal” (YY402) CFH at suppressing MAC deposition.
FIGURE 6.
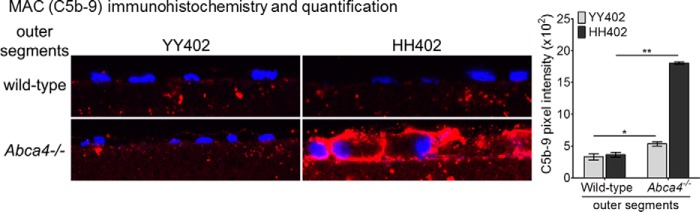
Bisretinoid-mediated MAC formation is dependent on the CFH haplotype of hRPE cells. Representative confocal images of MAC (C5b-9) immunoreactivity on YY402 (left panel, left column) and HH402 (left panel, right column) hRPE cells incubated with wild-type (left panel, top row) and Abca4−/− (left panel, bottom row) OS using CFH-depleted serum. The histogram in the right panel shows 3-fold higher MAC immunoreactivity on HH402 hRPE cells following Abca4−/− OS incubation compared with the YY402 hRPE cells when the cellular response relies only on endogenous CFH release. Similar MAC immunoreactivity levels are seen when both YY402 and HH402 hRPE cells are incubated with wild-type OS (n = 7). *, p < 0.003; **, p < 10−5. Data are representative of a single hRPE donor cell line of YY402 and HH402 of experiments done in triplicate.
DISCUSSION
CFH is an important inhibitor of complement activation through the alternative pathway (47). The results of human genetic studies have established the importance of the CFH gene in the pathogenesis of AMD (4–7). Although the bisretinoids, A2E, and all trans-retinaldehyde dimers have been shown to activate the complement system in RPE cell culture (37, 38), the effect of the CFH haplotype on CRP expression and complement activation have never been studied. This was our focus in this work.
Cultured hRPE lines of various CFH haplotypes, including YY402/II62 and HH402/VV62, exhibit similar function and morphology. Here, we show that unchallenged YY402 and HH402 hRPE cells express similar levels of CD46, CD55, CD59, and CFH (Fig. 1). Differences between cells of these two haplotypes were uncovered following the ingestion of Abca4−/− OS. Although cells with the AMD-protective haplotype showed increased expression of CFH and other CRPs following exposure to Abca4−/− OS, no increased CRP expression was seen in cells with the AMD-predisposing haplotype (Fig. 2 and data not shown). Increased expression of CRPs by YY402 hRPE cells represents an adaptive response, protecting the cells against complement attack. This protective response was missing in the HH402 hRPE cells, resulting in accumulation of C3/C3b (Fig. 3). Previously, we observed significant complement activation and other signs of complement-mediated inflammation in the RPE of Abca4−/− mice (33). Interestingly, CRPs, including CFH, were low in Abca4−/− RPE, suggesting a direct connection between bisretinoid buildup and complement activation (33).
Consistent with defective regulation of the complement cascade, hRPE cells of the HH402 haplotype showed significant C5b-9 (MAC) deposition following an acute challenge when incubated with C1q-depleted serum (Fig. 4). This response was not observed in factor B- or C5-depleted serum (Fig. 4 and data not shown). Component C1q is required for complement activation through the classical pathway, whereas factor B is an essential component of the alternative pathway (14). The absence of MAC deposition from serum lacking factor B suggests that bisretinoid-dependent complement activation occurs via the alternative pathway. Much greater MAC deposition was observed on HH402 cells following incubation with CFH-depleted serum (Fig. 6). In this experimental system, the hRPE cells were entirely dependent on endogenous CFH secretion for protection against complement activation. C5b-9 deposition was observed within the nitrocellulose filters below the hRPE cells independently of the challenge with bisretinoid-containing OS or the CFH haplotype (Fig. 4). Further, this sub-RPE deposition was only seen following incubation with serum containing factor C1q, which is required for activation via the classical pathway. This deposition of C5b-9 within the filters may arise through interactions between RPE- and serum-derived proteins, as proposed in Ref. 40, and is likely unrelated to the MAC deposition on hRPE cells reported here.
Various complement activators (triggers) have been proposed, but none have been tested in the context of normal RPE function. A recent study by Berchuck et al. (48) evidenced cell death via the alternative pathway because of increased sensitization of RPE cells by all-trans-retinaldehyde. Another recent study by Joseph et al. (41) suggested that oxidatively stressed RPE cells have an amplified alternative pathway following an early activation of the lectin-pathway by IgM bound to phospholipids. Our observation that the levels of MBL oligomer, MASP-1, and MASP-2 were similar for cells of both CFH haplotypes (Fig. 5), and within the physiological range (43, 46), suggests that the lectin pathway is not involved in bisretinoid-mediated activation of complement.
The results of this study suggest a final common pathway in the etiologies of AMD and STGD1 despite the very different genetic causes underlying the two diseases. Both appear to involve chronic inflammation of the RPE through sublethal deposition of the MAC (49). In Abca4−/− mice and STGD1 patients, complement attack on RPE cells is stimulated by bisretinoid accumulation and subsequent oxidation. Presumably, bisretinoid oxidative products, which are more soluble, are exiting from the cell and activate complement in the extracellular fluid. The reduced expression of CRPs in Abca4−/− mice (33) is likely a maladaptive secondary response of the “sick” RPE cells. In AMD associated with CFH mutations, complement attack may result from insufficient secretion of CFH by RPE cells or a dysfunctional CFH protein. In both diseases, photoreceptor degeneration and visual impairment stem from loss of the RPE support role. Further evidence that STGD1 and AMD share a common etiology is the observed strong correlation between monoallelic sequence variants in the ABCA4 gene, without AMD-associated CFH variants, and a clinically distinct subset of dry AMD (11). These observations suggest that the progression of visual loss in both STGD1 and AMD patients may be slowed with a treatment strategy targeting complement activation by RPE cells.
Acknowledgments
We thank Dr. Lincoln Johnson and Dr. Monte J. Radeke (University of California, Santa Barbara) and Dr. Gregory Hageman (University of Utah) for genotyping our hRPE cell lines. We also thank Shannan Eddington for technical assistance and Drs. Gabe Travis and Jeremy Cook for reading and providing critical insights regarding the manuscript.
This work was supported by a Stein/Oppenheimer Endowment Award grant (to R. A. R.), by National Eye Institute/Jules Stein Eye Institute Core Grant EY000331 (to R. A. R. and D. B.), by Macula Vision Research Foundation Award 20081840 (to D. B.), and by Arnold and Mabel Beckman Initiative for Macular Degeneration Award 1102 (to D. B.).
- AMD
- age-related macular degeneration
- CFH
- complement factor H
- MAC
- membrane attack complex
- CRP
- complement negative regulatory protein
- RPE
- retinal pigment epithelium
- STGD1
- recessive Stargardt macular degeneration
- OS
- outer segment(s)
- hRPE
- human retinal pigment epithelium
- qRT-PCR
- quantitative RT-PCR
- MBL
- mannose-binding lectin
- MASP
- MBL-associated serine protease.
REFERENCES
- 1. Friedman D. S., O'Colmain B. J., Muñoz B., Tomany S. C., McCarty C., de Jong P. T., Nemesure B., Mitchell P., Kempen J., and Eye Diseases Prevalence Research Group (2004) Prevalence of age-related macular degeneration in the United States. Arch. Ophthalmol. 122, 564–572 [DOI] [PubMed] [Google Scholar]
- 2. Rein D. B., Wittenborn J. S., Zhang X., Honeycutt A. A., Lesesne S. B., Saaddine J., and Vision Health Cost-Effectiveness Study, G. (2009) Forecasting age-related macular degeneration through the year 2050. The potential impact of new treatments. Arch. Ophthalmol. 127, 533–540 [DOI] [PubMed] [Google Scholar]
- 3. Raychaudhuri S., Iartchouk O., Chin K., Tan P. L., Tai A. K., Ripke S., Gowrisankar S., Vemuri S., Montgomery K., Yu Y., Reynolds R., Zack D. J., Campochiaro B., Campochiaro P., Katsanis N., Daly M. J., Seddon J. M. (2011) A rare penetrant mutation in CFH confers high risk of age-related macular degeneration. Nat. Genet. 43, 1232–1236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Klein R. J., Zeiss C., Chew E. Y., Tsai J. Y., Sackler R. S., Haynes C., Henning A. K., SanGiovanni J. P., Mane S. M., Mayne S. T., Bracken M. B., Ferris F. L., Ott J., Barnstable C., Hoh J. (2005) Complement factor H polymorphism in age-related macular degeneration. Science 308, 385–389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Haines J. L., Hauser M. A., Schmidt S., Scott W. K., Olson L. M., Gallins P., Spencer K. L., Kwan S. Y., Noureddine M., Gilbert J. R., Schnetz-Boutaud N., Agarwal A., Postel E. A., Pericak-Vance M. A. (2005) Complement factor H variant increases the risk of age-related macular degeneration. Science 308, 419–421 [DOI] [PubMed] [Google Scholar]
- 6. Hageman G. S., Anderson D. H., Johnson L. V., Hancox L. S., Taiber A. J., Hardisty L. I., Hageman J. L., Stockman H. A., Borchardt J. D., Gehrs K. M., Smith R. J., Silvestri G., Russell S. R., Klaver C. C., Barbazetto I., Chang S., Yannuzzi L. A., Barile G. R., Merriam J. C., Smith R. T., Olsh A. K., Bergeron J., Zernant J., Merriam J. E., Gold B., Dean M., Allikmets R. (2005) A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age-related macular degeneration. Proc. Natl. Acad. Sci. U.S.A. 102, 7227–7232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Edwards A. O., Ritter R., 3rd, Abel K. J., Manning A., Panhuysen C., Farrer L. A. (2005) Complement factor H polymorphism and age-related macular degeneration. Science 308, 421–424 [DOI] [PubMed] [Google Scholar]
- 8. Yates J. R., Sepp T., Matharu B. K., Khan J. C., Thurlby D. A., Shahid H., Clayton D. G., Hayward C., Morgan J., Wright A. F., Armbrecht A. M., Dhillon B., Deary I. J., Redmond E., Bird A. C., Moore A. T., and Genetic Factors in AMD Study Group (2007) Complement C3 variant and the risk of age-related macular degeneration. N. Engl. J. Med. 357, 553–561 [DOI] [PubMed] [Google Scholar]
- 9. Maller J. B., Fagerness J. A., Reynolds R. C., Neale B. M., Daly M. J., Seddon J. M. (2007) Variation in complement factor 3 is associated with risk of age-related macular degeneration. Nat. Genet. 39, 1200–1201 [DOI] [PubMed] [Google Scholar]
- 10. Fritsche L. G., Lauer N., Hartmann A., Stippa S., Keilhauer C. N., Oppermann M., Pandey M. K., Köhl J., Zipfel P. F., Weber B. H., Skerka C. (2010) An imbalance of human complement regulatory proteins CFHR1, CFHR3 and factor H influences risk for age-related macular degeneration (AMD). Hum. Mol. Genet. 19, 4694–4704 [DOI] [PubMed] [Google Scholar]
- 11. Fritsche L. G., Fleckenstein M., Fiebig B. S., Schmitz-Valckenberg S., Bindewald-Wittich A., Keilhauer C. N., Renner A. B., Mackensen F., Mößner A., Pauleikhoff D., Adrion C., Mansmann U., Scholl H. P., Holz F. G., Weber B. H. (2012) A subgroup of age-related macular degeneration is associated with mono-allelic sequence variants in the ABCA4 gene. Invest. Ophthalmol. Vis. Sci. 53, 2112–2118 [DOI] [PubMed] [Google Scholar]
- 12. Johnson P. T., Betts K. E., Radeke M. J., Hageman G. S., Anderson D. H., Johnson L. V. (2006) Individuals homozygous for the age-related macular degeneration risk-conferring variant of complement factor H have elevated levels of CRP in the choroid. Proc. Natl. Acad. Sci. U.S.A. 103, 17456–17461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Weismann D., Hartvigsen K., Lauer N., Bennett K. L., Scholl H. P., Charbel Issa P., Cano M., Brandstätter H., Tsimikas S., Skerka C., Superti-Furga G., Handa J. T., Zipfel P. F., Witztum J. L., Binder C. J. (2011) Complement factor H binds malondialdehyde epitopes and protects from oxidative stress. Nature 478, 76–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Anderson D. H., Radeke M. J., Gallo N. B., Chapin E. A., Johnson P. T., Curletti C. R., Hancox L. S., Hu J., Ebright J. N., Malek G., Hauser M. A., Rickman C. B., Bok D., Hageman G. S., Johnson L. V. (2010) The pivotal role of the complement system in aging and age-related macular degeneration. Hypothesis re-visited. Prog. Retin. Eye Res. 29, 95–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Donoso L. A., Kim D., Frost A., Callahan A., Hageman G. (2006) The role of inflammation in the pathogenesis of age-related macular degeneration. Surv. Ophthalmol. 51, 137–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sohn J. H., Kaplan H. J., Suk H. J., Bora P. S., Bora N. S. (2000) Chronic low level complement activation within the eye is controlled by intraocular complement regulatory proteins. Invest. Ophthalmol. Vis. Sci. 41, 3492–3502 [PMC free article] [PubMed] [Google Scholar]
- 17. Yang P., Tyrrell J., Han I., Jaffe G. J. (2009) Expression and modulation of RPE cell membrane complement regulatory proteins. Invest. Ophthalmol. Vis. Sci. 50, 3473–3481 [DOI] [PubMed] [Google Scholar]
- 18. Hughes A. E., Orr N., Esfandiary H., Diaz-Torres M., Goodship T., Chakravarthy U. (2006) A common CFH haplotype, with deletion of CFHR1 and CFHR3, is associated with lower risk of age-related macular degeneration. Nat. Genet. 38, 1173–1177 [DOI] [PubMed] [Google Scholar]
- 19. Gold B., Merriam J. E., Zernant J., Hancox L. S., Taiber A. J., Gehrs K., Cramer K., Neel J., Bergeron J., Barile G. R., Smith R. T., AMD Genetics Clinical Study Group, Hageman G. S., Dean M., Allikmets R. (2006) Variation in factor B (BF) and complement component 2 (C2) genes is associated with age-related macular degeneration. Nat. Genet. 38, 458–462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mata N. L., Weng J., Travis G. H. (2000) Biosynthesis of a major lipofuscin fluorophore in mice and humans with ABCR-mediated retinal and macular degeneration. Proc. Natl. Acad. Sci. U.S.A. 97, 7154–7159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Allikmets R., Shroyer N. F., Singh N., Seddon J. M., Lewis R. A., Bernstein P. S., Peiffer A., Zabriskie N. A., Li Y., Hutchinson A., Dean M., Lupski J. R., Leppert M. (1997) Mutation of the Stargardt disease gene (ABCR) in age-related macular degeneration. Science 277, 1805–1807 [DOI] [PubMed] [Google Scholar]
- 22. Quazi F., Lenevich S., Molday R. S. (2012) ABCA4 is an N-retinylidene-phosphatidylethanolamine and phosphatidylethanolamine importer. Nat. Commun. 3, 925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Young R. W., Bok D. (1969) Participation of the retinal pigment epithelium in the rod outer segment renewal process. J. Cell Biol. 42, 392–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Weng J., Mata N. L., Azarian S. M., Tzekov R. T., Birch D. G., Travis G. H. (1999) Insights into the function of Rim protein in photoreceptors and etiology of Stargardt's disease from the phenotype in ABCR knockout mice. Cell 98, 13–23 [DOI] [PubMed] [Google Scholar]
- 25. Radu R. A., Yuan Q., Hu J., Peng J. H., Lloyd M., Nusinowitz S., Bok D., Travis G. H. (2008) Accelerated accumulation of lipofuscin pigments in the RPE of a mouse model for ABCA4-mediated retinal dystrophies following vitamin A supplementation. Invest. Ophthalmol. Vis. Sci. 49, 3821–3829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sparrow J. R., Zhou J., Cai B. (2003) DNA is a target of the photodynamic effects elicited in A2E-laden RPE by blue-light illumination. Invest. Ophthalmol. Vis. Sci. 44, 2245–2251 [DOI] [PubMed] [Google Scholar]
- 27. Sparrow J. R., Nakanishi K., Parish C. A. (2000) The lipofuscin fluorophore A2E mediates blue light-induced damage to retinal pigmented epithelial cells. Invest. Ophthalmol. Vis. Sci. 41, 1981–1989 [PubMed] [Google Scholar]
- 28. Schütt F., Davies S., Kopitz J., Holz F. G., Boulton M. E. (2000) Photodamage to human RPE cells by A2-E, a retinoid component of lipofuscin. Invest. Ophthalmol. Vis. Sci. 41, 2303–2308 [PubMed] [Google Scholar]
- 29. Finnemann S. C., Leung L. W., Rodriguez-Boulan E. (2002) The lipofuscin component A2E selectively inhibits phagolysosomal degradation of photoreceptor phospholipid by the retinal pigment epithelium. Proc. Natl. Acad. Sci. U.S.A. 99, 3842–3847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. De S., Sakmar T. P. (2002) Interaction of A2E with model membranes. Implications to the pathogenesis of age-related macular degeneration. J. Gen. Physiol. 120, 147–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sparrow J. R., Boulton M. (2005) RPE lipofuscin and its role in retinal pathobiology. Exp. Eye Res. 80, 595–606 [DOI] [PubMed] [Google Scholar]
- 32. Liu J., Itagaki Y., Ben-Shabat S., Nakanishi K., Sparrow J. R. (2000) The biosynthesis of A2E, a fluorophore of aging retina, involves the formation of the precursor, A2-PE, in the photoreceptor outer segment membrane. J. Biol. Chem. 275, 29354–29360 [DOI] [PubMed] [Google Scholar]
- 33. Radu R. A., Hu J., Yuan Q., Welch D. L., Makshanoff J., Lloyd M., McMullen S., Travis G. H., Bok D. (2011) Complement system dysregulation and inflammation in the retinal pigment epithelium of a mouse model for Stargardt macular degeneration. J. Biol. Chem. 286, 18593–18601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hu J., Bok D. (2001) A cell culture medium that supports the differentiation of human retinal pigment epithelium into functionally polarized monolayers. Mol. Vis. 7, 14–19 [PubMed] [Google Scholar]
- 35. Hu J., Bok D. (2010) Culture of highly differentiated human retinal pigment epithelium for analysis of the polarized uptake, processing, and secretion of retinoids. Methods Mol. Biol. 652, 55–73 [DOI] [PubMed] [Google Scholar]
- 36. Wu Y., Fishkin N. E., Pande A., Pande J., Sparrow J. R. (2009) Novel lipofuscin bisretinoids prominent in human retina and in a model of recessive Stargardt disease. J. Biol. Chem. 284, 20155–20166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhou J., Jang Y. P., Kim S. R., Sparrow J. R. (2006) Complement activation by photooxidation products of A2E, a lipofuscin constituent of the retinal pigment epithelium. Proc. Natl. Acad. Sci. U.S.A. 103, 16182–16187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhou J., Kim S. R., Westlund B. S., Sparrow J. R. (2009) Complement activation by bisretinoid constituents of RPE lipofuscin. Invest. Ophthalmol. Vis. Sci. 50, 1392–1399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hu J., Bok D. (2013) The use of cultured human fetal retinal pigment epithelium in studies of the classical retinoid visual cycle and retinoid-based disease processes. Exp. Eye Res. 10.1016/j.exer.2013.09.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Johnson L. V., Forest D. L., Banna C. D., Radeke C. M., Maloney M. A., Hu J., Spencer C. N., Walker A. M., Tsie M. S., Bok D., Radeke M. J., Anderson D. H. (2011) Cell culture model that mimics drusen formation and triggers complement activation associated with age-related macular degeneration. Proc. Natl. Acad. Sci. U.S.A. 108, 18277–18282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Joseph K., Kulik L., Coughlin B., Kunchithapautham K., Bandyopadhyay M., Thiel S., Thielens N. M., Holers V. M., Rohrer B. (2013) Oxidative stress sensitizes RPE cells to complement-mediated injury in a natural antibody-, lectin pathway- and phospholipid epitope-dependent manner. J. Biol. Chem. 288, 12753–12765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Turner M. W. (2003) The role of mannose-binding lectin in health and disease. Mol. Immunol. 40, 423–429 [DOI] [PubMed] [Google Scholar]
- 43. Turner M. W. (1998) Mannose-binding lectin (MBL) in health and disease. Immunobiology 199, 327–339 [DOI] [PubMed] [Google Scholar]
- 44. Endo Y., Matsushita M., Fujita T. (2011) The role of ficolins in the lectin pathway of innate immunity. Int. J. Biochem. Cell Biol. 43, 705–712 [DOI] [PubMed] [Google Scholar]
- 45. Kjaer T. R., Thiel S., Andersen G. R. (2013) Toward a structure-based comprehension of the lectin pathway of complement. Mol. Immunol. 56, 413–422 [DOI] [PubMed] [Google Scholar]
- 46. Dommett R. M., Klein N., Turner M. W. (2006) Mannose-binding lectin in innate immunity: past, present and future. Tissue Antigens 68, 193–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bradley D. T., Zipfel P. F., Hughes A. E. (2011) Complement in age-related macular degeneration. A focus on function. Eye 25, 683–693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Berchuck J. E., Yang P., Toimil B. A., Ma Z., Baciu P., Jaffe G. J. (2013) All-trans-retinal sensitizes human RPE cells to alternative complement pathway-induced cell death. Invest. Ophthalmol. Vis. Sci. 54, 2669–2677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lueck K., Wasmuth S., Williams J., Hughes T. R., Morgan B. P., Lommatzsch A., Greenwood J., Moss S. E., Pauleikhoff D. (2011) Sub-lytic C5b-9 induces functional changes in retinal pigment epithelial cells consistent with age-related macular degeneration. Eye 25, 1074–1082 [DOI] [PMC free article] [PubMed] [Google Scholar]