

Background: The regulation of glucose metabolism by estradiol is poorly defined.
Results: We find that estradiol stimulates glucose metabolism in part by stimulating the production of fructose 2,6-bisphosphate by PFKFB3.
Conclusion: PFKFB3 is a downstream target of estradiol required to stimulate glucose metabolism.
Significance: Combined targeting of PFKFB3 and the estrogen receptor may prove beneficial to ER+ stage IV breast cancer patients.
Keywords: Breast Cancer, Estrogen, Estrogen Receptor, Glycolysis, Metabolism, PFKFB3
Abstract
Estradiol (E2) administered to estrogen receptor-positive (ER+) breast cancer patients stimulates glucose uptake by tumors. Importantly, this E2-induced metabolic flare is predictive of the clinical effectiveness of anti-estrogens and, as a result, downstream metabolic regulators of E2 are expected to have utility as targets for the development of anti-breast cancer agents. The family of 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatases (PFKFB1–4) control glycolytic flux via their product, fructose-2,6-bisphosphate (F26BP), which activates 6-phosphofructo-1-kinase (PFK-1). We postulated that E2 might promote PFKFB3 expression, resulting in increased F26BP and glucose uptake. We demonstrate that PFKFB3 expression is highest in stage III lymph node metastases relative to normal breast tissues and that exposure of human MCF-7 breast cancer cells to E2 causes a rapid increase in [14C]glucose uptake and glycolysis that is coincident with an induction of PFKFB3 mRNA (via ER binding to its promoter), protein expression and the intracellular concentration of its product, F26BP. Importantly, selective inhibition of PFKFB3 expression and activity using siRNA or a PFKFB3 inhibitor markedly reduces the E2-mediated increase in F26BP, [14C]glucose uptake, and glycolysis. Furthermore, co-treatment of MCF-7 cells with the PFKFB3 inhibitor and the anti-estrogen ICI 182,780 synergistically induces apoptotic cell death. These findings demonstrate for the first time that the estrogen receptor directly promotes PFKFB3 mRNA transcription which, in turn, is required for the glucose metabolism and survival of breast cancer cells. Importantly, these results provide essential preclinical information that may allow for the ultimate design of combinatorial trials of PFKFB3 antagonists with anti-estrogen therapies in ER+ stage IV breast cancer patients.
Introduction
Increased uptake and metabolism of glucose as a source of energy and biosynthetic precursors are common features of primary and metastatic breast adenocarcinomas. As evidence, positron emission tomographic studies with 2-[18F]fluoro-2-deoxyglucose (FDG-PET)3 have consistently demonstrated that human breast tumors display a marked increase in glucose uptake in vivo relative to adjacent normal breast tissues (1–5). Consequently, FDG-PET scans have become widely used by medical oncologists to assess the extent of disease and clinical responses to chemotherapies in breast cancer patients. Importantly, objective responses to aromatase inhibitors or ER antagonists in ER+ breast cancer patients can be predicted by the magnitude of E2-stimulated FDG uptake suggesting that glucose metabolism is an essential clinical target for anti-estrogen therapies (6). Although the stimulation of glucose metabolism by E2 has recently been found to be dependent on the PI3K-AKT pathway (7), relatively little is known about the precise downstream effectors required for E2 to stimulate glucose metabolism in breast cancer cells.
6-Phosphofructo-1-kinase (PFK-1) is the first committed step of glycolysis and a key metabolic sensor of the entire pathway because this enzyme is allosterically inhibited by several products of glucose metabolism including ATP, citrate, and H+ ions. The most potent activator of PFK-1 is a shunt product of glycolysis, fructose-2,6-bisphosphate (F26BP), which can override the allosteric inhibition by ATP and stimulate PFK-1, glucose uptake, and flux through the entire glycolytic pathway (8, 9). The intracellular concentration of F26BP is maintained by a family of four bifunctional enzymes (PFKFB1–4) with fructose-6-phosphate kinase and F26BP phosphatase activities (10). The PFKFB3 family member is of particular interest because this enzyme (i) has a very high kinase:bisphosphatase ratio, thus overwhelmingly favoring F26BP synthesis and activation of glycolysis; and (ii) is overexpressed in breast tumors relative to normal mammary epithelial cells in situ (10–12).
Although the precise mechanisms for increased PFKFB3 expression in breast cancers are not well understood, PFKFB3 mRNA transcription is promoted by hypoxia inducible factor-1α (13, 14) and by the progesterone receptor (15). Additionally, loss of the tumor suppressor PTEN has recently been found to reduce APC/Cdh1-mediated degradation of PFKFB3 (16). The observation that the PFKFB3 promoter contains putative ER response elements (ERE) (14) prompted us to determine whether PFKFB3 is a direct transcriptional target of ER. Considering previous research showing that E2 increases glucose uptake in vivo and in vitro (6, 7), together with the glycolysis-activating function of PFKFB3, we postulated that PFKFB3 may be a downstream effector of E2 that serves to activate the energetic and anabolic pathways required for the survival and growth of breast cancer cells.
EXPERIMENTAL PROCEDURES
Chemicals
E2, cycloheximide, and actinomycin D were purchased from Sigma-Aldrich. ICI 182,780 (Fulvestrant) was purchased from Tocris. PFK158 was provided by Advanced Cancer Therapeutics. The compound consists of (E)-l-(pyridyn-4-yl)-3-(7-(trifluoromethyl)quinolin-2-yl)-prop-2-en-l-one.
Antibodies and Western Blotting
Antibodies against PFKFB3 and β-actin were purchased from Proteintech and Sigma-Aldrich, respectively. Whole cell lysates were harvested using Pierce lysis buffer supplemented with protease inhibitors. Protein concentration was determined by the BCA method (Pierce). Proteins were separated on 10% Mini PROTEAN TGX gels (Bio-Rad) and transferred to PVDF membranes (Millipore). The membranes were blocked with 5% nonfat milk in TBS-T (0.1% Tween 20) and immunoblotted with the indicated antibodies. Amersham Biosciences ECL Western blotting detection reagent (GE Healthcare) was used to detect protein bands. The membranes were visualized on Biomax ML film (Kodak).
Cell Culture
MCF-7 and T-47D cells were purchased from American Type Culture Collection (ATCC). MCF-7 cells were maintained in Improved Minimum Essential Medium supplemented with 10% fetal bovine serum (FBS) and Pen-Strep (Invitrogen). T-47D cells were maintained in RPMI 1640 medium supplemented with 10% FBS and 0.2 unit/ml bovine insulin. Cells were serum-starved (5% dextran-coated charcoal-stripped FBS) in phenol red-free media for 72 h prior to E2 treatments.
RNA Extraction and Real-time Quantitative RT-PCR
Total RNA was extracted using RNeasy spin columns (Qiagen). Reverse transcription was performed by using random primers and the High Capacity cDNA Archive Kit (Applied Biosystems). Quantitative RT-PCR was performed with the ABI PRISM 7500 using TaqMan primers and probes for PFKFB3 and control gene β-actin purchased as Assays-on-DemandTM Gene Expression Products (Applied Biosystems). Analysis and -fold differences were determined using the comparative CT method. Unless otherwise indicated, -fold change was calculated from the ΔΔCT values with the formula 2-ΔΔCT. Experiments were repeated at least three times.
Immunohistochemistry
Five-μm sections of formalin-fixed and paraffin-embedded tumor tissue were mounted on charged glass slides and dried at 58 °C for 60 min. Slides were first deparaffinized with xylene, and then epitope retrieval was carried out using citrate buffer in a 2100 Retriever (PickCell Laboratories). The sections were blocked with 10% goat serum for 1 h, then incubated with primary antibody (anti-PFKFB3 1:50; Proteintech) overnight, followed by an HRP-linked goat anti-rabbit secondary antibody (1:300; Pierce). The sections were developed with 3,3′-diaminobenzidine tetrahydrochloride (DAB; Vector Laboratories) for 2 min, and nuclei were counterstained with hematoxylin (Sigma-Aldrich) for 2 min. PBS washes were performed between all steps. The slides were dehydrated in graded alcohols (100, 95, and 80% ethanol (v/v) in H2O) and cleared in xylene, and coverslips were attached with Permount (Fisher Scientific). Slides were scanned using a ScanScope XT Digital Slide Scanner (Aperio) and data analyzed with the positive pixel count algorithm (ImageScope; Aperio).
Chromatin Immunoprecipitation (ChIP) Assay
ChIP experiments were performed using the simpleChIP kit (Cell Signaling) and following the manufacture's instructions. In brief, 107 MCF-7 cells/immunoprecipitation were treated with vehicle control EtOH or 10 nm E2 for 1 h. Chromatin was cross-linked by incubating the cells with 1% formaldehyde for 10 min. Immunoprecipitation of the lysed extracts was performed using the following ChIP grade antibodies: anti-ERα (HC-20), anti-ERβ (H-150) from Santa Cruz Biotechnology, or normal rabbit IgG (Cell Signaling). The primers used for PCR of the PFKFB3 promoter region containing an ERE were: (−2023 to −2198), CGCGTTATTGACAGACAGGA (FWD)/AGCTCACAGGGAGAGCAGAG (REV), and (−2982 to −3308), CTCCTGAGCTCAAGCAATCC (FWD)/GGGTGCTGAGTGGAATCCTA (REV). As a positive control, primers flanking the established ERE in the human pS2 (trefoil factor 1 (TFF1)) gene promoter were used (17). Primers to a distal region (−9196 to −9432), AGTTGAATCGCTGCAAAGGT (FWD)/ACCCAGGTGAAATCAACAGC (REV) of the PFKFB3 promoter were used as a negative control.
Measurement of F26BP
MCF-7 cells were treated with EtOH vehicle control, 10 or 100 nm E2 for the indicated times. Cells were washed in PBS and lysed in NaOH/Tris acetate by heating at 80 °C for 5 min. Lysates were neutralized to pH 7.2 by adding ice-cold acetic acid and HEPES. F26BP concentration was determined using the method described by Van Schaftingen et al. (9).
Glucose Uptake
MCF-7 cells were treated with EtOH vehicle control, 10 or 100 nm E2 for the indicated time periods and subsequently placed in glucose-free RPMI 1640 medium for 30 min. 2-[14C]Deoxyglucose (0.25 μCi/ml; PerkinElmer Life Sciences) was added for an additional 15 min, and cells were then washed three times with ice-cold RPMI 1640 medium containing no glucose. Cell lysates were collected in 500 μl of 0.1% SDS, and scintillation counts (counts/min) were measured using 350 μl of lysate. Protein concentration was determined using the BCA assay according to the manufacturer's instructions and measured on a Powerwave XS plate reader (Biotek). Counts were normalized to protein concentration.
Glycolysis Assay
MCF-7 cells growing in 6-well plates were incubated in 500 μl of complete medium containing 1 μCi of 5-[3H]glucose for 60 min in 5% CO2 at 37 °C. The medium was then collected and centrifuged for 5 min at 8000 rpm to pellet any cells. To separate the 3H2O formed via glycolysis from the 5-[3H]glucose added to the medium, an evaporation technique in a sealed system was utilized. Briefly, 150 μl of medium aliquots were added in open tubes that were placed upright inside scintillation vials containing 1 ml of H2O. The scintillation vials were sealed, and the 3H2O produced by glycolysis through enolase and released to the medium was allowed to equilibrate with the H2O in the outer vial for 48 h at 37 °C. The amounts of 3H2O that had diffused into the surrounding H2O was measured on a Tri-Carb 2910 liquid scintillation analyzer (PerkinElmer Life Sciences) and compared with 3H2O and 5-[3H]glucose standards. Protein concentration was determined using the BCA assay according to the manufacturer's instructions and measured on a Powerwave XS plate reader. Counts were normalized to protein concentration.
Transfection and siRNA
Cells were seeded in 6-well plates at a density of 1 × 105 cells/well. Cells were transfected with control siRNA or PFKFB3 siRNA (Invitrogen) using Lipofectamine RNAiMAX reagent (Invitrogen) and allowed to incubate for 48 h prior to E2 treatment.
PFKFB3 Adenovirus Expression
pAdenoG-HA (vector) and PFKFB3-HA adenovirus (human) were purchased from Applied Biological Materials. For infection experiments, MCF-7 cells were seeded in 6-well plates at a density of 1 × 105 cells/well and allowed to attach overnight. The cells were then placed in 1 ml of medium containing 1 × 106 viral particles and allowed to incubate for 48 h.
Annexin V/Propidium Iodide (PI) Staining
MCF-7 cells were seeded in 6-well plates at a density of 1 × 105 cells/well and starved for 72 h prior to treatment. Cells were incubated with 10 nm E2 and dimethyl sulfoxide, 100 nm ICI 182,780, 5 μm PFK158 or the combination of ICI 182,780 and PFK158 for 24 h. Cells were washed with PBS and stained with Annexin V and PI (BD Pharmingen). Fluorescence was measured using a FACSCalibur (BD Biosciences) and analyzed using FloJo (Tree Star). Annexin V−/PI − (live) and Annexin V+/PI + (late apoptotic) cells were quantified by the frequency of fluorescently labeled cells, and statistical significance was assessed by the two-sample t test (independent variable).
RESULTS
High PFKFB3 Protein Expression in Lymph Node Metastases
Previously, PFKFB3 expression was found to be elevated in primary breast adenocarcinomas (12). We confirmed high PFKFB3 expression in stage II breast adenocarcinomas relative to normal breast tissues using immunohistochemical analyses (Fig. 1A). We also examined the metastatic breast adenocarcinoma cells in lymph nodes resected from patients suffering from stage III breast cancer and observed an incremental increase in PFKFB3 protein expression (Fig. 1A). This increase in PFKFB3 expression in stage II and lymph node metastases then was statistically confirmed using digital immunohistochemical image analysis (Fig. 1B).
FIGURE 1.
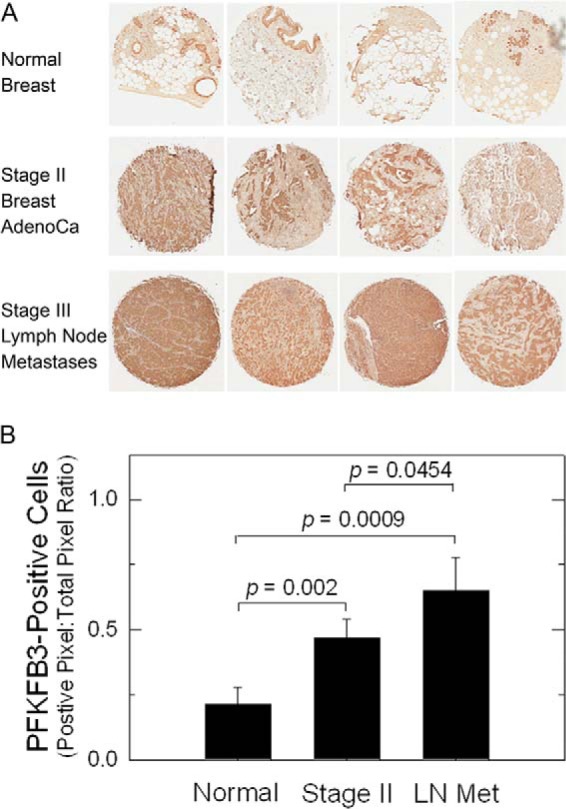
High expression of PFKFB3 by metastatic breast adenocarcinoma cells in lymph nodes. A, four normal breast tissues, stage II primary breast adenocarcinomas, and stage III lymph node metastases were analyzed for PFKFB3 expression using immunohistochemistry. B, digital immunohistochemical image analysis was performed, and the ratio of positive pixels to total pixels was quantified for each of the tissue specimens. p values comparing normal mammary tissues with stage II and stage III lymph node metastases and between stage II and III lymph node metastases are included in the figure.
PFKFB3 Expression, F26BP, Glucose Uptake, and Glycolysis Are Induced by E2 in MCF-7 Cells
Given that E2 increases the glucose uptake of ER+ breast cancer cells in vitro and in vivo (6, 7) and that the PFKFB3 promoter has putative EREs (14), we postulated that ER and its ligand, E2, might regulate PFKFB3 mRNA expression. Initially, we examined PFKFB3 mRNA expression by real-time RT-PCR after exposure to EtOH (vehicle control), or 10 or 100 nm E2 for 1, 3, 6, and 24 h. We observed a marked increase in PFKFB3 mRNA levels after only 1 h of exposure to either 10 or 100 nm E2 (Fig. 2A). PFKFB3 mRNA returned to near basal levels after 3 and 6 h of incubation with E2 but was still statistically increased relative to the vehicle control (Fig. 2A). Importantly, co-culture with the anti-estrogen ICI 182,780 (ICI; 100 nm) blocked the induction of PFKFB3 mRNA indicating that the ER is required for the up-regulation of PFKFB3 mRNA expression (Fig. 2A). In addition, pretreatment of MCF-7 cells with the transcription inhibitor actinomycin D, suppressed the increase in PFKFB3 mRNA indicating that new mRNA synthesis is required whereas pretreatment with the protein synthesis inhibitor cycloheximide, had no effect (Fig. 2A). We next examined endogenous PFKFB3 protein expression in response to E2 and found that PFKFB3 protein significantly increased after 3 h and 6 h of exposure to 10 or 100 nm E2 but returned to baseline after 24 h (Fig. 2, C and D). Similar to PFKFB3 protein, the steady-state concentration of its product, F26BP, significantly increased after 3 h and 6 h of exposure to 10 and 100 nm E2 (Fig. 2B). To then monitor the effect of E2 on glucose uptake, MCF-7 cells were placed in phenol red-free medium supplemented with 5% dextran-coated charcoal-stripped serum for 72 h prior to the addition of E2 and 2-[1-14C]deoxy-d-glucose (2-[14C]DG). We observed a time-dependent increase in glucose uptake in MCF-7 cells exposed to 10 and 100 nm E2 for 3–24 h (Fig. 2E). We next examined glycolysis by measuring the release of 3H2O from 5-[3H]glucose via enolase which is downstream of PFK-1 in the glycolytic pathway. We found that E2 also caused a marked increase in glycolysis by MCF-7 cells that coincided with the observed increase in glucose uptake (Fig. 2F). Given that the F26BP concentration returns to near baseline 6 h after stimulation by E2, we suspect that the maintenance of increased glucose metabolism observed at 24 h is mediated by secondary events that regulate glucose transporters and/or glycolytic enzymes. Importantly, we did not observe an increase in the mRNA expression of several key control steps in glycolysis including GLUT1–4, hexokinase, PFK-1, and pyruvate kinase at 24 h (data not shown). We also assessed PFKFB3 expression after 10 nm E2 exposure in a second ER+ breast cancer cell line (T-47D) and found that PFKFB3 mRNA and protein were increased 3.9-fold (1 h) and 3.0-fold (3 h), respectively (data not shown).
FIGURE 2.

Regulation of PFKFB3, F26BP, glucose uptake, and glycolysis by E2 in MCF-7 cells. MCF-7 cells were stimulated with 10 nm and 100 nm E2 for the indicated times and analyzed for PFKFB3 mRNA by real-time PCR (A), PFKFB3 protein expression by Western blot analysis and densitometry (C and D), F26BP using an enzyme-coupled reaction dependent on NADH oxidation (B), 2-[14C]DG uptake (E) and glycolysis as measured by the release of 3H2O by enolase (F). Values are the mean ± S.E. (error bars) of three experiments; *, p < 0.01.
ERα Is Recruited to the PFKFB3 Gene upon E2 Stimulation
We next analyzed the 5′ PFKFB3 promoter sequence using the TRANSFAC software (Biobase) to identify putative transcription factors binding sites, including the ER. Our analysis revealed four putative half-site ERE sites located around the −2000 and −3000 regions along with several SP1 sites. To investigate whether the E2-induced increase in PFKFB3 mRNA involves recruitment of the ER to the PFKFB3 promoter we performed ChIP assays using primers that could amplify the −2000, −3000, and a distal region of the PFKFB3 promoter. We used antibodies against ERα and ERβ to immunoprecipitate the chromatin collected from MCF-7 cells treated with 10 nm E2 for 1 h and then performed real-time quantitative PCR to determine recruitment to the PFKFB3 promoter. Our results demonstrate a 4-fold enrichment in the recruitment of the ERα to the −2000 region of the PFKFB3 promoter after 1 h of E2 exposure (Fig. 3A (real-time PCR) and Fig. 3B (2% agarose gel as a representative example)). We did not observe a significant recruitment of the ERα to the −3000 region or a distal region of the PFKFB3 gene which is devoid of putative EREs. We also observed no recruitment of the ERβ to the PFKFB3 gene and found that primers recognizing the pS2 promoter, a previously described ERα target (18), amplified the pS2 promoter sequence as a positive control. In summary, these studies demonstrate that stimulation of MCF-7 cells with 10 nm E2 for 1 h causes an increase in ERα recruitment to the PFKFB3 promoter.
FIGURE 3.
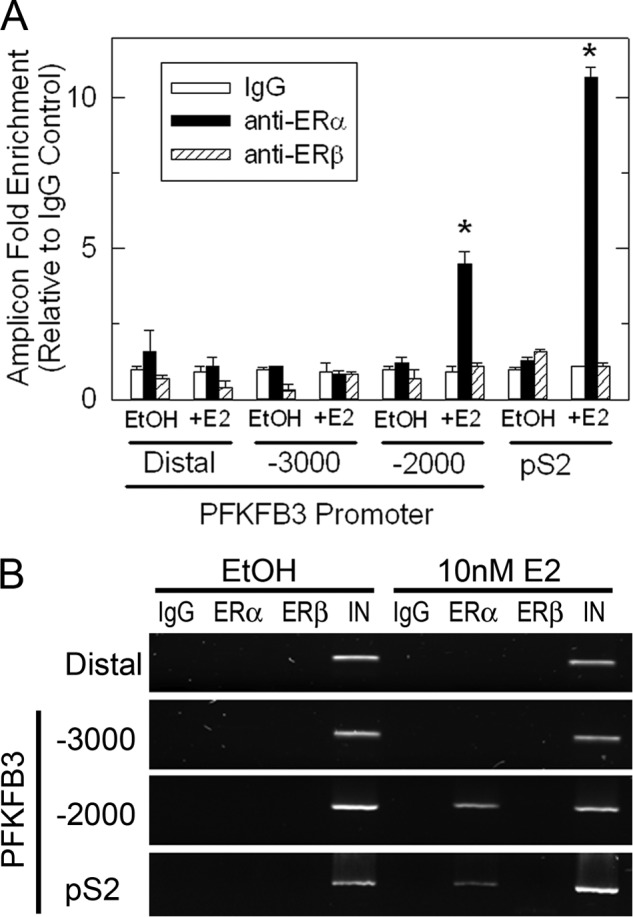
E2 causes ERα recruitment to the PFKFB3 promoter. A, ChIP assay experiments were performed with MCF-7 cells treated with EtOH or 10 nm E2 for 1 h using antibodies to ERα (HC20) or ERβ (H150) or with rabbit preimmune serum (IgG) and primers flanking the −2000, −3000, and distal regions (lacking ERE sites) of the PKFKB3 promoter. Real-time PCR -fold results were normalized to input and are shown as -fold enrichment over IgG immunoprecipitation in EtOH-treated cells from three independent ChIP experiments. B, representative 2% agarose gel using similar conditions and primers is also shown. *, p < 0.01 compared with vehicle control.
E2 Increases Glucose Uptake and Glycolysis via PFKFB3 in MCF-7 Cells
To determine whether the stimulation of glucose uptake by E2 requires PFKFB3 expression, we next analyzed E2-stimulated glucose uptake by MCF-7 cells transfected with a control or PFKFB3-specific siRNA. Briefly, MCF-7 cells were transiently transfected with either control siRNA or PFKFB3 siRNA, and, 48 h later the cells were exposed to vehicle or the physiologically relevant 10 nm E2 for 3 h followed by incubation with 2-[14C]DG. We confirmed that PFKFB3 protein expression was decreased by transfection with PFKFB3 siRNA relative to the control siRNA and found that the PFKFB3 siRNA completely blocked the E2-mediated up-regulation of PFKFB3 expression (Fig. 4, A and B). PFKFB3 has the highest kinase:bisphosphatase ratio of the PFKFB family, and we postulated that inhibition of PFKFB3 expression would suppress the intracellular F26BP concentration. We found that transfection with the PFKFB3 siRNA (which does not affect the other family members) markedly reduced the F26BP concentration in vehicle- and E2-stimulated MCF-7 cells after 3 h (Fig. 4C). To test the hypothesis that PFKFB3/F26BP is required for the stimulation of glucose uptake and glycolysis by E2, we examined the effect of PFKFB3 siRNA on the 2-[14C]DG uptake and 3H2O release (i.e. glycolysis) stimulated by E2. We found that transfection with the PFKFB3 siRNA reduced basal glucose uptake and glycolysis and completely blocked the stimulation of glucose uptake and glycolysis caused by E2 at 3 h (Fig. 4, D and E). Taken together, these data indicate that E2 rapidly stimulates glucose uptake in part by increasing PFKFB3 expression and F26BP.
FIGURE 4.

PFKFB3 is required for E2-induced glucose uptake and glycolysis. MCF-7 cells were transfected with PFKFB3 siRNA and allowed to incubate for 48 h prior to stimulation with 10 nm E2. Shown are PFKFB3 protein expression by Western blot analysis after 3-h stimulation with E2 (A), densitometry analysis of relative PFKFB3/β-actin (B), F26BP concentration (C), [1-14C]DG uptake (D), and glycolysis as measured by the release of 3H2O by enolase (E). Values represent three independent experiments. *, p < 0.01 relative to control siRNA.
A Small Molecule Inhibitor of PFKFB3 Blocks the E2-mediated Increase in Glucose Uptake and Glycolysis in MCF-7 Cells
We next postulated that inhibition of the kinase activity of PFKFB3 by a clinical-grade PFKFB3 inhibitor, PFK158, would, like the PFKFB3 siRNA, attenuate the E2-mediated stimulation of F26BP and glucose uptake and glycolysis. Initially, we identified a concentration of PFK158 that does not affect viability after 3 h of exposure (3 μm; data not shown). We then examined the effect of PFK158 on PFKFB3 protein expression in the absence and presence of 10 nm E2 and observed relatively little effect on the E2-induced expression of PFKFB3 (Fig. 5, A and B). However, we did observe a marked decrease in the E2-stimulated F26BP (Fig. 5C), 2-[14C]DG uptake (Fig. 5D) and glycolysis (Fig. 5E). These data indicate that selective inhibition of the substrate-binding domain of PFKFB3 is sufficient to completely block the ability of E2 to stimulate glucose uptake. Of note, whereas transfection of unstimulated MCF-7 cells with the PFKFB3 siRNA significantly reduced glucose uptake and glycolysis (Fig. 4, D and E), exposure of unstimulated MCF-7 cells to PFK158 did not reduce glucose uptake or glycolysis (Fig. 5D, 5E). We believe that this disparity may be because of the relative potency of PFK158 which competitively but not completely inhibits PFKFB3 (unlike the PFKFB3 siRNA which essentially depletes all PFKFB3 protein).
FIGURE 5.

PFK158 inhibits E2-induced glucose uptake and glycolysis. MCF-7 cells were simultaneously stimulated with E2 and exposed to 3 μm PFK158 for 3 h and then analyzed for PFKFB3 protein expression (A), densitometry analysis of relative PFKFB3/β-actin (B), F26BP (C), 2-[14C]DG uptake (D), and glycolysis as measured by the release of 3H2O by enolase (E). Values represent three independent experiments. *, p < 0.001 relative to vehicle.
Forced Expression of PFKFB3 Increases F26BP, Glucose Uptake, and Glycolysis in MCF-7 Cells
Given that both PFKFB3 siRNA and PFK158 can suppress the E2-stimulated increase in F26BP, glucose uptake, and glycolysis, we sought to determine whether PFKFB3 expression by itself would result in similar biochemical changes. We transiently expressed PFKFB3 in MCF-7 cells and found that forced expression of PFKFB3 mRNA (Fig. 6A) and protein (Fig. 6, C and D) can increase the steady-state concentration of F26BP (Fig. 6B), glucose uptake (Fig. 6E), and glycolysis (Fig. 6F) in MCF-7 cells.
FIGURE 6.

Forced expression of PFKFB3 increases glucose uptake and glycolysis. MCF-7 cells were transduced with adenovirus-encoded PFKFB3 and then analyzed for PFKFB3 mRNA (A), F26BP (B), PFKFB3 protein expression (C), densitometry analysis of relative PFKFB3/β-actin (D), 2-[14C]DG uptake (E), and glycolysis as measured by the release of 3H2O by enolase (F). Values represent three independent experiments. *, p < 0.001 relative to vehicle.
Effect of ICI 182,780 on PFK158-induced Apoptotic Death
To determine the utility of combining anti-estrogens with PFKFB3 inhibitors, we next examined the effects of 100 nm ICI 182,780, a commonly prescribed pure anti-estrogen compound, a pro-apoptotic concentration of PFK158 (5 μm at 24 h), or combined 100 nm ICI 182,780 + 5 μm PFK158 on apoptosis using Annexin V/PI staining by flow cytometry. We observed that the addition of ICI 182,780 caused a synergistic increase in late apoptotic MCF-7 cells relative to that caused by PFK158 alone after 24 h of exposure (Fig. 7, A and B). These data suggest that the addition of PFK158 to anti-estrogen therapeutic agents may increase response rates and perhaps the durability of responses in preclinical and clinical studies.
FIGURE 7.
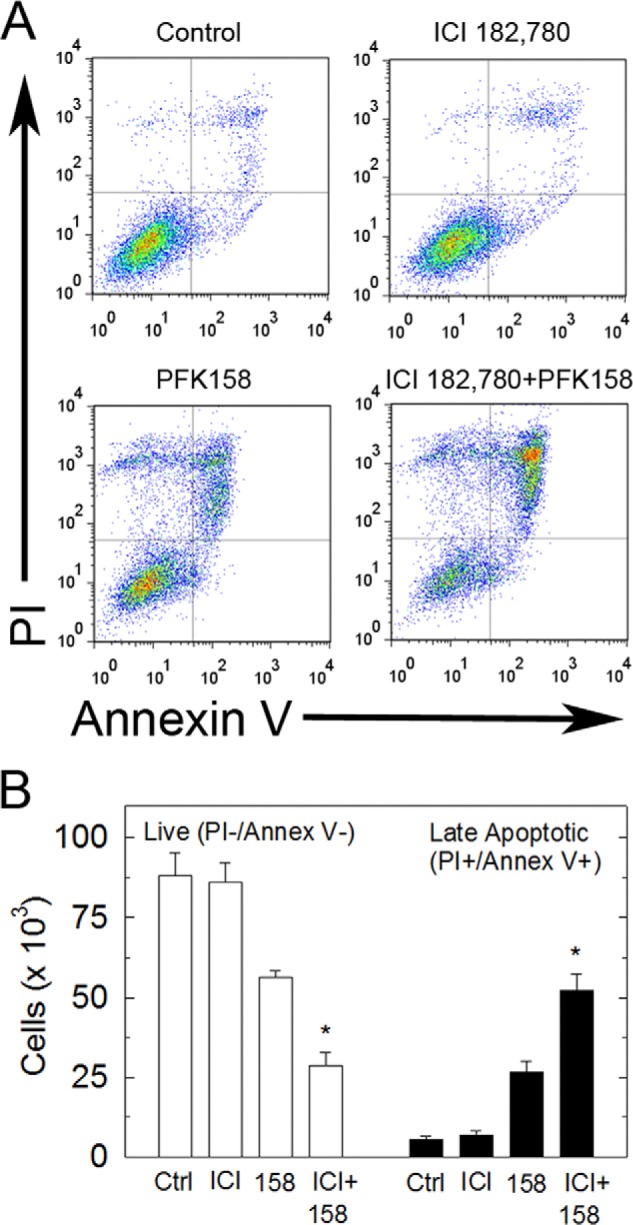
Synergistic pro-apoptotic effect of combined treatment with ICI 182,780/PFK158. A, MCF-7 cells were exposed to 100 nm ICI 182,780, 5 μm PFK158, or both agents for 24 h and analyzed for Annexin V/PI staining by flow cytometry. B, absolute cell numbers for double negative and double positive cells were quantified.
DISCUSSION
In this study, we describe PFKFB3 as an early molecular target of estradiol via direct binding of the ER to the PFKFB3 promoter. We also show that E2 up-regulates PFKFB3 expression in ER-responsive MCF-7 cells, increasing the steady-state concentration of F26BP, glucose uptake, and glycolysis. Although our study does not characterize all of the potential mechanisms by which E2 increases glucose metabolism, our results do demonstrate that PFKFB3 expression and activity are required to mediate E2-mediated glucose uptake and glycolysis. We postulate that the increase in F26BP by E2 leads to the stimulation of PFK-1 which, in turn, may cause a reduction in the PFK-1 substrate Fru-6-P, which is in equilibrium with Glc-6-P, an allosteric inhibitor of hexokinase, which itself is required for glucose uptake (19–21). Importantly, the ability of PFKFB3 to regulate glucose uptake has been demonstrated previously by work showing that genetic ablation of the pfkfb3 gene reduces F26BP and glucose uptake in transformed cells in vivo and in vitro (22).
The therapeutic implications of our observation that E2 requires PFKFB3 to stimulate glucose metabolism in vitro are 2-fold: (i) F26BP may be required for the metabolic flare induced by E2 in breast cancer patients; and (ii) combined inhibition of PFKFB3 and ER functions may be an effective therapeutic strategy for ER+ stage IV breast cancer patients.
The activation of the ER by estrogens serves a critical role in cancer initiation and progression, and their antagonists have proven efficacy in the treatment of breast cancers. Anti-estrogen therapies include selective ER modulators such as Tamoxifen that act as partial receptor agonists, estrogen synthesis inhibitors such as aromatase inhibitors, and receptor expression inhibitors such as the anti-estrogen ICI 182,780. Each of these modalities is effective in 50–70% of breast cancer patients initially, but recurrent ER+ breast tumors respond to such anti-estrogen treatments in only ∼30% of patients, and durable responses are even rarer (23). Consequently, there is an important need to identify novel ER signaling targets, such as PFKFB3, to guide the development of therapeutic strategies to maximize endocrine responses.
Our observation that E2 increases PFKFB3 expression and F26BP synthesis is the first report that ER signaling regulates PFKFB3 expression and activity. PFKFB3 is a key regulator of glucose metabolism and has proven to be an important target for antineoplastic therapy (24). Additionally, PFKFB3 ChIP experiments demonstrate interaction of the ERα at the half-site EREs located on the −2000 region of the PFKFB3 promoter. Based on these results, we envision the following series of events mediated by exposure of ER+ breast cancer cells to E2: (i) E2 increases PFKFB3 transcription by a mechanism involving ERα occupancy of the PFKFB3 promoter; (ii) the increase in PFKFB3 mRNA translates into an increase in protein and more importantly, an increase in the product, F26BP; and (iii) F26BP activates PFK-1, an essential control enzyme for the entire glycolytic pathway, which reduces Fru-6-P and Glc-6-P, resulting in less allosteric inhibition of hexokinase and increased glucose uptake (Fig. 8).
FIGURE 8.
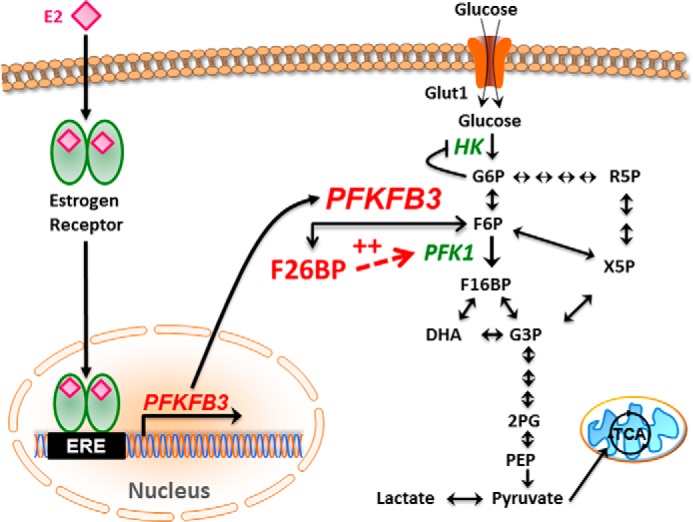
Model of the regulation of PFKFB3 by E2. In this schematic, we summarize our model whereby E2 binds and activates ER, translocates to the nucleus, and promotes PFKFB3 mRNA transcription. Translated PFKFB3 produces F26BP which allosterically activates PFK-1 and stimulates glucose uptake and the entire glycolytic pathway.
CONCLUSIONS
In this study we have found that PFKFB3 and its product, F26BP, are induced by E2 and required for the E2-mediated stimulation of glucose uptake and glycolysis. We also have found that a PFKFB3 inhibitor undergoing phase I trial testing in solid tumor patients, PFK158, causes a synergistic increase in apoptosis by ER+ breast cancer cells when combined with the widely used anti-estrogen ICI 182,780. These observations support the conduct of Investigational New Drug-enabling preclinical toxicity and efficacy studies of combinations of PFK158 and ICI 182,780 to facilitate the eventual development of phase I/II trials in ER+ stage IV breast cancer patients.
Acknowledgments
We thank Drs. John Eaton and Otto Grubraw for assisting with the interpretation of our data.
This work was supported, in whole or in part, by NCI/National Institutes of Health Grants 1R01CA140991 (to S. T.) and 1R01CA149438 (to J. C.). This work was also supported by Congressionally Directed Medical Research Program post-doctoral fellowships (to Y. I.-F. and J. O.) and American Cancer Society RSG-10-021-01CNE (to S. T.).
- FDG-PET
- positron emission tomographic studies with 2-[18F]-fluoro-2-deoxyglucose
- 2-[14C]DG
- 2-[1-14C]-deoxy-d-glucose
- E2
- estradiol
- ER
- estrogen receptor
- ERE
- estrogen receptor response element
- F26BP
- fructose-2,6-bisphosphate
- Fru-6-P
- fructose 6-phosphate
- Glu-6-P
- glucose 6-phosphate
- PFK
- 1–6-phosphofructo-1-kinase
- PFKFB
- 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase
- PI
- propidium iodide.
REFERENCES
- 1. Constantinidou A., Martin A., Sharma B., Johnston S. R. (2011) Positron emission tomography/computed tomography in the management of recurrent/metastatic breast cancer: a large retrospective study from the Royal Marsden Hospital. Ann. Oncol. 22, 307–314 [DOI] [PubMed] [Google Scholar]
- 2. Martoni A. A., Zamagni C., Quercia S., Rosati M., Cacciari N., Bernardi A., Musto A., Fanti S., Santini D., Taffurelli M. (2010) Early 18F-2-fluoro-2-deoxy-d-glucose positron emission tomography may identify a subset of patients with estrogen receptor-positive breast cancer who will not respond optimally to preoperative chemotherapy. Cancer 116, 805–813 [DOI] [PubMed] [Google Scholar]
- 3. Niikura N., Ueno N. T. (2010) The role of F-FDG-positron emission tomography/computed tomography in staging primary breast cancer. J. Cancer 1, 51–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pennant M., Takwoingi Y., Pennant L., Davenport C., Fry-Smith A., Eisinga A., Andronis L., Arvanitis T., Deeks J., Hyde C. (2010) A systematic review of positron emission tomography (PET) and positron emission tomography/computed tomography (PET/CT) for the diagnosis of breast cancer recurrence. Health Technol. Assess. 14, 1–103 [DOI] [PubMed] [Google Scholar]
- 5. Specht J. M., Kurland B. F., Montgomery S. K., Dunnwald L. K., Doot R. K., Gralow J. R., Ellis G. K., Linden H. M., Livingston R. B., Allison K. H., Schubert E. K., Mankoff D. A. (2010) Tumor metabolism and blood flow as assessed by positron emission tomography varies by tumor subtype in locally advanced breast cancer. Clin. Cancer Res. 16, 2803–2810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dehdashti F., Mortimer J. E., Trinkaus K., Naughton M. J., Ellis M., Katzenellenbogen J. A., Welch M. J., Siegel B. A. (2009) PET-based estradiol challenge as a predictive biomarker of response to endocrine therapy in women with estrogen-receptor-positive breast cancer. Breast Cancer Res. Treat. 113, 509–517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ko B. H., Paik J. Y., Jung K. H., Lee K. H. (2010) 17β-Estradiol augments 18F-FDG uptake and glycolysis of T-47D breast cancer cells via membrane-initiated rapid PI3K-AKT activation. J. Nucl. Med. 51, 1740–1747 [DOI] [PubMed] [Google Scholar]
- 8. Van Schaftingen E., Hue L., Hers H. G. (1980) Fructose 2,6-bisphosphate, the probably structure of the glucose- and glucagon-sensitive stimulator of phosphofructokinase. Biochem. J. 192, 897–901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Van Schaftingen E., Hue L., Hers H. G. (1980) Control of the fructose-6-phosphate/fructose 1,6-bisphosphate cycle in isolated hepatocytes by glucose and glucagon: role of a low-molecular-weight stimulator of phosphofructokinase. Biochem. J. 192, 887–895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yalcin A., Telang S., Clem B., Chesney J. (2009) Regulation of glucose metabolism by 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatases in cancer. Exp. Mol. Pathol. 86, 174–179 [DOI] [PubMed] [Google Scholar]
- 11. Sakakibara R., Kato M., Okamura N., Nakagawa T., Komada Y., Tominaga N., Shimojo M., Fukasawa M. (1997) Characterization of a human placental fructose-6-phosphate, 2-kinase/fructose-2,6-bisphosphatase. J Biochem. 122, 122–128 [DOI] [PubMed] [Google Scholar]
- 12. Atsumi T., Chesney J., Metz C., Leng L., Donnelly S., Makita Z., Mitchell R., Bucala R. (2002) High expression of inducible 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase (iPFK-2; PFKFB3) in human cancers. Cancer Res. 62, 5881–5887 [PubMed] [Google Scholar]
- 13. Minchenko A., Leshchinsky I., Opentanova I., Sang N., Srinivas V., Armstead V., Caro J. (2002) Hypoxia-inducible factor-1-mediated expression of the 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase-3 (PFKFB3) gene: its possible role in the Warburg effect. J. Biol. Chem. 277, 6183–6187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Obach M., Navarro-Sabaté A., Caro J., Kong X., Duran J., Gómez M., Perales J. C., Ventura F., Rosa J. L., Bartrons R. (2004) 6-Phosphofructo-2-kinase (pfkfb3) gene promoter contains hypoxia-inducible factor-1 binding sites necessary for transactivation in response to hypoxia. J. Biol. Chem. 279, 53562–53570 [DOI] [PubMed] [Google Scholar]
- 15. Novellasdemunt L., Obach M., Millán-Arino L., Manzano A., Ventura F., Rosa J. L., Jordan A., Navarro-Sabate A., Bartrons R. (2012) Progestins activate 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 3 (PFKFB3) in breast cancer cells. Biochem. J. 442, 345–356 [DOI] [PubMed] [Google Scholar]
- 16. Garcia-Cao I., Song M. S., Hobbs R. M., Laurent G., Giorgi C., de Boer V. C., Anastasiou D., Ito K., Sasaki A. T., Rameh L., Carracedo A., Vander Heiden M. G., Cantley L. C., Pinton P., Haigis M. C., Pandolfi P. P. (2012) Systemic elevation of PTEN induces a tumor-suppressive metabolic state. Cell 149, 49–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bourdeau V., Deschênes J., Métivier R., Nagai Y., Nguyen D., Bretschneider N., Gannon F., White J. H., Mader S. (2004) Genome-wide identification of high-affinity estrogen response elements in human and mouse. Mol. Endocrinol. 18, 1411–1427 [DOI] [PubMed] [Google Scholar]
- 18. Liu X. F., Bagchi M. K. (2004) Recruitment of distinct chromatin-modifying complexes by tamoxifen-complexed estrogen receptor at natural target gene promoters in vivo. J. Biol. Chem. 279, 15050–15058 [DOI] [PubMed] [Google Scholar]
- 19. Gregoriou M., Cornish-Bowden A., Trayer I. P. (1981) Isotope-exchange evidence for allosteric regulation of hexokinase II by glucose 6-phosphate and for an obligatory addition of substrates. Biochem. Soc. Trans. 9, 62–63 [DOI] [PubMed] [Google Scholar]
- 20. Gregoriou M., Trayer I. P., Cornish-Bowden A. (1983) Isotope-exchange evidence that glucose 6-phosphate inhibits rat-muscle hexokinase II at an allosteric site. Eur. J. Biochem. 134, 283–288 [DOI] [PubMed] [Google Scholar]
- 21. Lazo P. A., Bosca L. (1982) Mitochondrial membrane-bound hexokinase of ascites tumor cells. Functional implications of lysine residues studied by modification with imidoesters. Hoppe Seylers Z. Physiol. Chem. 363, 635–641 [DOI] [PubMed] [Google Scholar]
- 22. Telang S., Yalcin A., Clem A. L., Bucala R., Lane A. N., Eaton J. W., Chesney J. (2006) Ras transformation requires metabolic control by 6-phosphofructo-2-kinase. Oncogene 25, 7225–7234 [DOI] [PubMed] [Google Scholar]
- 23. Ingle J. N. (2013) Pharmacogenomics of endocrine therapy in breast cancer. J. Hum. Genet. 58, 306–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Clem B. F., O'Neal J., Tapolsky G., Clem A. L., Imbert-Fernandez Y., Kerr D. A., 2nd, Klarer A. C., Redman R., Miller D. M., Trent J. O., Telang S., Chesney J. (2013) Targeting 6-phosphofructo-2-kinase (PFKFB3) as a therapeutic strategy against cancer. Mol. Cancer Ther. 12, 1461–1470 [DOI] [PMC free article] [PubMed] [Google Scholar]