

Background: Endothelia express NEU1 sialidase and undergo changes in sialylation during angiogenesis.
Results: CD31 is a NEU1 substrate, and NEU1 disrupts endothelial cell capillary-like tube formation.
Conclusion: NEU1 works through its substrate, CD31, to dysregulate angiogenesis.
Significance: Human NEU1 is the first sialidase found to regulate angiogenesis, and the CD31 sialylation state dictates its ability to influence endothelial cell differentiation and tube formation.
Keywords: Angiogenesis, Endothelial Cell, Glycobiology, Sialic Acid, Sialyltransferase, CD31/PECAM-1, Neuraminidase, Sialidase, Sialoproteins
Abstract
The highly sialylated vascular endothelial surface undergoes changes in sialylation upon adopting the migratory/angiogenic phenotype. We recently established endothelial cell (EC) expression of NEU1 sialidase (Cross, A. S., Hyun, S. W., Miranda-Ribera, A., Feng, C., Liu, A., Nguyen, C., Zhang, L., Luzina, I. G., Atamas, S. P., Twaddell, W. S., Guang, W., Lillehoj, E. P., Puché, A. C., Huang, W., Wang, L. X., Passaniti, A., and Goldblum, S. E. (2012) NEU1 and NEU3 sialidase activity expressed in human lung microvascular endothelia. NEU1 restrains endothelial cell migration whereas NEU3 does not. J. Biol. Chem. 287, 15966–15980). We asked whether NEU1 might regulate EC capillary-like tube formation on a Matrigel substrate. In human pulmonary microvascular ECs (HPMECs), prior silencing of NEU1 did not alter tube formation. Infection of HPMECs with increasing multiplicities of infection of an adenovirus encoding for catalytically active WT NEU1 dose-dependently impaired tube formation, whereas overexpression of either a catalytically dead NEU1 mutant, NEU1-G68V, or another human sialidase, NEU3, did not. NEU1 overexpression also diminished EC adhesion to the Matrigel substrate and restrained EC migration in a wounding assay. In HPMECs, the adhesion molecule, CD31, also known as platelet endothelial cell adhesion molecule-1, was sialylated via α2,6-linkages, as shown by Sambucus nigra agglutinin lectin blotting. NEU1 overexpression increased CD31 binding to Arachis hypogaea or peanut agglutinin lectin, indicating CD31 desialylation. In the postconfluent state, when CD31 ectodomains are homophilically engaged, NEU1 was recruited to and desialylated CD31. In postconfluent ECs, CD31 was desialylated compared with subconfluent cells, and prior NEU1 silencing completely protected against CD31 desialylation. Prior CD31 silencing and the use of CD31-null ECs each abrogated the NEU1 inhibitory effect on EC tube formation. Sialyltransferase 6 GAL-I overexpression increased α2,6-linked CD31 sialylation and dose-dependently counteracted NEU1-mediated inhibition of EC tube formation. These combined data indicate that catalytically active NEU1 inhibits in vitro angiogenesis through desialylation of its substrate, CD31.
Introduction
Angiogenesis is a tightly orchestrated process in which proangiogenic and antiangiogenic factors are released to bind to their cognate endothelial cell (EC)2 surface receptors (1, 2). The proangiogenic receptor-ligand interactions are coupled to EC disengagement from neighboring ECs and protease-mediated degradation of the underlying EC extracellular matrix (ECM). These signaling and proteolytic events permit ECs to migrate through the ECM toward the proangiogenic stimulus. Proliferative signals expand the EC population, which, coupled with ECM remodeling, lead to EC-EC re-engagement with capillary-like tube formation. A number of these proangiogenic processes involve altered glycosylation patterns and specific carbohydrate-mediated recognition events (3–6).
The EC surface is highly sialylated (7–9). The sialic acid (SA)-bearing surface structures on endothelia, together with other membrane-bound polyanionic macromolecules, constitute the EC glycocalyx (10, 11). This negatively charged zone can be extended through hydration and acquisition of plasma proteins. The glycocalyx might nonspecifically diminish cell-cell adhesion through either negative charge-mediated repulsion or the masking of specific binding sites. In fact, we found that prior neuraminidase treatment of pulmonary vascular endothelia increases their adhesiveness for resting polymorphonuclear leukocytes >3-fold, independent of E-selectin or intercellular adhesion molecule (ICAM)-1 expression (12). However, SA residues also can be intrinsic to EC surface recognition motifs that participate in specific intermolecular interactions. For example, increased α2,6-sialylation of the EC surface increases its adhesiveness for CD22-bearing B cells (13) and lymphocyte function-associated antigen (LFA)-1 expressing T cells (14). Migrating bovine aortic ECs display a distinct phenotype that includes hyperglycosylation and surface expression of specific glycoproteins compared with contact-inhibited ECs (3). After neuraminidase treatment of these migrating ECs, increased terminal sialylation coupled to subterminal galactose residues was revealed through increased binding of Arachis hypogaea or peanut agglutinin (PNA) lectin (15). In another study, pharmacologic blockade of synthesis of hybrid and complex-type oligosaccharides, including sialyl Lewis-X determinants, inhibited capillary tube formation of bovine capillary ECs (4). In still another study, 48% of the 432 glycan-specific genes profiled in human bone marrow-derived ECs stimulated by the proangiogenic factor, vascular endothelial growth factor (VEGF), were expressed (6). After VEGF stimulation, expression of several sialyltransferases (STs), including ST6GAL-I, were increased. Multiple galectins, endogenous lectins that bind galactose residues and regulate angiogenesis (16), were also up-regulated. Finally, a number of EC sialoproteins directly participate in the angiogenic process, including vascular endothelial cadherin (17), selectins and other adhesion molecules (18, 19), CD31 (20), CD44 (21), fibroblast growth factor receptor (22), and αvβ3 integrin (23). Taken together, these combined studies establish a central role for glycan structures (and more specifically, sialylation) as intrinsic to the angiogenic process.
SAs comprise a family of 9-carbon sugars, each carboxylated on the C1 position (24, 25). These SA residues are almost always positioned at the terminus of glycan chains. The subterminal sugars to which SA is usually coupled are galactose and N-acetylgalactosamine. In most sialoproteins, the C2 position of SA is coupled to underlying galactose via α2,3- or α2,6-linkage and to N-acetylgalactosamine via α2,6-linkage. For only a few sialoproteins, SA is attached to another SA via α2,8-linkage, generating homopolymers of polysialic acid. The sialylation state of a specific glycoprotein is dynamically and coordinately regulated through the opposing catalytic activities of STs (26, 27) and neuraminidase/sialidase (NEU) (28–30). STs use cytidine monophosphate (CMP)-SA as an activated donor to catalyze transfer of SA residues to terminal positions on glycan chains (26, 27). More than 15 distinct human ST cDNAs have been cloned and characterized (26, 27). These STs can be classified based on 1) the specific SA linkage they synthesize and 2) the sugar to which they transfer the SA. In contrast, NEUs counterregulate sialylation through hydrolysis of the linkage between terminal SAs and their subterminal sugars (28–30). Although the numerous prokaryote and eukaryote members of the NEU superfamily share little amino acid (aa) sequence homology, they contain conserved motifs, including the Asp box (an aa sequence composed of SXDXGXTW) and the (F/Y)RIP motif (28). More recently, bioinformatic analysis has revealed high conservation of 6 residues essential for catalytic activity, including an Arg triad (Arg-21, Arg-237, and Arg-304), a Tyr/Glu nucleophile pair (Tyr-334 and Glu-218), and an Asp that functions as an acid/base catalyst (Asp-46) (31). Four putative phosphorylation sites at positions 384, 390, 569, and 865 were >90% conserved in all sialidases. More specific to NEU1 in higher mammals, lysosomal localization signals and the residues that participate in NEU1-protective protein/cathepsin A (PPCA) interaction were conserved. These highly dissimilar NEUs do share a tertiary conformation in which six antiparallel β-sheets are arranged into a six-blade propeller-like configuration (28–30). Four human sialidases have been identified, NEU1, -2, -3, and -4 (28–30). We recently established that vascular endothelia predominantly express NEU1 (32).
NEU1 is a ∼45.5-kDa (415-aa) protein that contains three conserved and two degenerate Asp boxes and one (F/Y)RIP motif (28). In human tissues, it is the most widely expressed and most abundant NEU (29). Among the four human NEUs, NEU1 shares the least homology (29) and displays a substrate preference for glycoproteins over other glycoconjugates, including the SA-containing glycolipids or gangliosides (28–30). NEU1 is a lysosomal enzyme that resides in a multienzyme complex composed of NEU1, PPCA, and β-galactosidase (33, 34). PPCA is a multifunctional enzyme that functions as an intracellular chaperone and transport protein that targets NEU1 to the lysosome and is absolutely required for proper folding, stability, oligomerization, and activation of NEU1 (34–36). Although NEU1 had been considered primarily a lysosomal enzyme (34, 35), it is now known to associate with several surface multireceptor signaling complexes, including epidermal growth factor receptor/mucin 1 (MUC1) (37), Toll-like receptor 4 (TLR4)/CD14/MD2 (38), and the elastin receptor complex (39). Although the mechanisms through which NEU1 might be translocated to the plasma membrane are poorly understood (40), it has been detected on the surface of multiple human cell types, including activated lymphocytes (40, 41), phorbol 12-myristate 13-acetate differentiated monocytes (42), fibroblasts (43), and, most recently, erythrocytes (44). NEU1 is also surface-expressed on human lung microvascular endothelia, where it restrains EC migration into a wound (32). In the current studies, we have asked whether NEU1 might also regulate EC capillary-like tube formation and whether the sialylated proangiogenic molecule, CD31, also known as platelet endothelial cell adhesion molecule (PECAM)-1 (45–48), might serve as an in vivo NEU1 substrate.
EXPERIMENTAL PROCEDURES
EC Culture
Human pulmonary microvascular ECs (HPMECs) (Promocell, Heidelberg, Germany) and human pulmonary artery ECs (HPAECs) (Lonza, Walkersville, MD) were cultured in EC growth medium (MV-2, Promocell) containing EC growth medium supplement mix (Promocell) as described (32). HPMECs and HPAECs were studied in passages 4–7. In selected experiments, immortalized CD31-null and CD31 reconstituted (CD31-RC) mouse lung microvascular ECs were cultured in Dulbecco's modified Eagle's medium enriched with 10% FBS, l-glutamine, nonessential amino acids, sodium pyruvate, HEPES, and β-mercaptoethanol as described (49). These ECs were established through retroviral transduction of primary lung ECs derived from the CD31 knock-out mouse with the polyoma virus middle T oncogene. These CD31-null ECs were retrovirally transduced with full-length murine CD31 cDNA to generate CD31-RC ECs. During passage, the CD31-RC ECs were selected with puromycin, but prior to and during experiments, puromycin was removed.
Manipulation of NEU1, CD31, and ST6GAL-I Expression in HPMECs
To overexpress NEU1, HPMECs and HPAECs were transiently infected with increasing multiplicities of infection (MOIs) of packaged adenovirus (Ad) encoding for human FLAG-tagged wild-type NEU1 (Ad-NEU1-FLAG) or Ad encoding green fluorescent protein (Ad-GFP) as an irrelevant vector control, as described (32, 37). After 48 h, the NEU1-overexpressing and control ECs were studied for NEU1 immunoblotting, EC tube formation, adhesion, migration, chemotaxis, and lectin blotting. A plasmid encoding a catalytically dead NEU1 mutant, NEU1-G68V, was kindly provided by Dr. L. Debelle (Université de Reims) (39). The NEU1-G68V mutant, which exhibits no sialidase activity when transfected in COS-7 cells or human fibroblasts (50), was subcloned into an Ad vector, as described for other gene products (32, 37, 51). An Ad encoding for hemagglutinin (HA)-tagged human ST6GAL-I (Ad-ST6GAL-I-HA) was purchased (Applied Biological Materials Inc., Richmond, Canada) for ST6GAL-I overexpression. To silence either NEU1 or CD31 expression, HPMECs were transfected with siRNA duplexes designed to specifically target NEU1 or CD31 or irrelevant control siRNA duplexes not corresponding to any known sequence in the human genome (Dharmacon, Lafayette, CO), as described (32, 37). For transfection, 5.0 × 105 ECs were centrifuged (200 × g for 10 min), and the cell pellet was resuspended in 100 μl of Amaxa Nucleofactor solution (Lonza) with 2.7 μg of siRNA duplexes. The EC-siRNA mixture was transferred to an Amaxa-certified cuvette and subjected to programmed electroporation (program S-005; Amaxa Biosystems). The transfected ECs were cultured for 48 h, after which they were lysed, and the lysates were processed for NEU1 or CD31 immunoblotting. In other experiments, HPMECs were transiently infected with Ad-NEU1-FLAG at an MOI of 100. After 48 h, NEU1-targeting or control siRNAs were transfected into these HPMECs overexpressing FLAG-tagged NEU1. After 48 h, the transfected cells were lysed, and the lysates were processed for FLAG tag immunoblotting. To control for protein loading and transfer, blots were stripped and reprobed for β-tubulin. Transfected cells were studied for EC tube formation.
In Vitro EC Capillary-like Tube Formation Assay
Each well of a 96-well plate was coated with 50 μl of Matrigel (10 mg/ml; BD Biosciences) as described with modifications (52, 53). Briefly, Matrigel (50 μl on ice) was allowed to polymerize for 1 h at room temperature in each of the wells, followed by 1 h at 37 °C in a humidified atmosphere of 5% CO2. HPMECs, HPAECs, and CD31-null and CD31-RC mouse lung microvascular ECs were seeded at 1.5 × 104 cells/well onto the Matrigel-coated wells and observed for tube formation. At 6 h, tubular structures were photographed through a Nikon inverted microscope, and branches of capillary-like tubes per high power field (HPF) were counted.
EC Proliferation and Viability
HPMECs seeded into 60-mm dishes at 105 cells/dish were cultured for 3 h, after which they were infected with Ad-GFP, Ad-NEU1, or Ad-NEU3, each at MOI = 100. Cells were collected at 24, 48, and 72 h and manually counted with trypan blue exclusion.
EC Adhesion Assay
The wells of 96-well plates were each incubated with Matrigel (50 μl/well) for 1 h at room temperature, followed by 1 h at 37 °C. The wells were washed with DMEM, blocked (3% BSA in DMEM, 1 h, 37 °C), and again washed (0.1% BSA in DMEM). HPMECs were seeded in the Matrigel-coated wells at 104 cells/well and incubated for 10–60 min at 37 °C in 0.1% BSA, DMEM. After washing twice, adherent cells were photographed through a microscope, and the adherent cell area was quantified using ImageJ software (National Institutes of Health, Bethesda, MD), as described (32, 51).
Migration in a Wounding Assay
HPMECs were seeded at 2 × 105 cells/well in the wells of a 24-well plate (Corning, Inc.), and after 3 h, they were infected with Ad-GFP, Ad-NEU1, or Ad-NEU1-G68V (MOI = 100). The HPMECs were cultured for 48 h to confluence. Using a sterile 200-μl pipette tip, a single wound was made across the diameter of each monolayer, after which cell debris was removed by washing with HEPES, as described (32, 51). The wounded monolayers were then incubated for increasing times, and photomicrographs of each well were taken at 0, 2, 4, 6, and 24 h with a Nikon inverted microscope. At each time point, HPMEC area was calculated using ImageJ software for comparison with that observed in the same wounded monolayer at 0 h, as a measure of migration into the wound.
EC Chemotaxis Assay
Gelatin-impregnated polycarbonate filters (13-mm diameter, 8.0-μm pore size; Nucleopore Inc., Pleasanton, CA) were mounted in chemotactic chambers (Neuroprobe Inc., Gaithersburg, MD) as described (51). HPMECs (5 × 104 cells in 100 μl) were added to the upper compartment of each assay chamber. Each lower compartment contained 0.5% FBS as the chemoattractant. After a 5-h incubation (37 °C, 5% CO2), filters were removed, fixed (3.7% formaldehyde in H2O, 30 min), washed, stained with 0.5% crystal violet in 25% ethanol, and washed, and the top surface of each filter was scraped free of cells. The crystal violet was then extracted from each filter with 0.1 m citric acid in 50% ethanol for 30 min, and the A560 nm of extracts was measured.
Lectin Blotting for Sialylated and Desialylated Molecules
HPMECs were infected with Ad-NEU1, Ad-ST6GAL-I, or Ad-GFP, each at MOI = 100, and incubated for 48 h. The cells were solubilized with lysis buffer as described (32, 37), and 1.0-mg aliquots were immunoprecipitated with a murine monoclonal antibody raised against human CD31 (89C2, Cell Signaling Technology, Danvers, MA). Immunoprecipitated proteins were resolved by SDS-PAGE and transferred to a PVDF membrane. The blots were incubated for 1 h with Tris-buffered saline-Tween 20 (TBS-T) and probed with biotinylated Maackia amurensis lectin II (MAL), Sambucus nigra agglutinin (SNA), or PNA (EY Labs, San Mateo, CA), as described (32, 37). The blots were washed with TBS-T, incubated with HRP-conjugated streptavidin, and developed with enhanced chemiluminescence (ECL) reagents. Fetuin (1.0 μg) was used as the positive control for MAL- and SNA-binding proteins, whereas asialofetuin (1.0 μg) was used as the positive control for PNA-binding proteins. To confirm equivalent protein loading and transfer, blots were stripped and reprobed with the immunoprecipitating anti-CD31 antibody followed by HRP-conjugated secondary antibody and ECL reagents. Densitometric quantitation of each MAL/SNA/PNA signal was normalized to total CD31 signal in the same lane on the same stripped and reprobed blot. In selected experiments, lysates were first incubated with PNA immobilized on Sepharose beads (Vector Labs, Burlingame, CA) to selectively enrich for PNA-binding proteins, which, in turn, were processed for CD31 immunoblotting.
NEU1/CD31 and PPCA/CD31 Coimmunoprecipitation Assays
For the NEU1/CD31 coimmunoprecipitation assays, HPMECs infected with Ad-NEU1-FLAG (MOI = 100) were cultured to subconfluent and postconfluent states and lysed, and the lysates were immunoprecipitated with anti-CD31 antibodies. The CD31 immunoprecipitates were processed for FLAG (NEU1) immunoblotting, as described (37). To control for protein loading and transfer, the blots were stripped and reprobed for CD31. For the PPCA/CD31 coimmunoprecipitation assays, lysates of subconfluent and postconfluent HPMECs were immunoprecipitated with murine monoclonal anti-human PPCA antibodies (Santa Cruz Biotechnology, Inc.), and PPCA immunoprecipitates were processed for CD31 immunoblotting. To control for protein loading and transfer, the blots were stripped and reprobed with rabbit polyclonal anti-human PPCA antibody (Abcam Inc.). For the NEU1/CD31 coimmunoprecipitation assays, densitometric quantitation of FLAG (NEU1) signal was normalized to CD31 signal, and for the PPCA/CD31 coimmunoprecipitation assays, CD31 signal was normalized to PPCA signal, each in the same lane on the same stripped and reprobed blot.
Confocal Microscopy
HPMECs were fixed (4% paraformaldehyde), permeabilized (0.1% Triton X-100), blocked (PBS in 5% FBS), incubated with primary antibody (1:1000 for mouse anti-CD31, Cell Signaling and 1:400 for rabbit anti-FLAG, Cell Signaling), washed in PBS, and incubated with Alexa-labeled secondary antibody (1:100 Alexa-568 goat-anti-mouse and 1:100 Alexa-350 goat anti-rabbit). Cells were visualized by confocal laser-scanning microscopy (Zeiss-510 meta) using a ×63 oil immersion lens (numerical aperture 1.40).
Statistical Methods
Student's t test, with a two-tailed distribution and a homoscedastic variance was used for all statistical analyses (Microsoft Office Excel 2003). Significance was accepted for p values <0.05.
RESULTS
NEU1 Impairs EC Capillary-like Tube Formation
Because a number of EC sialoproteins participate in the angiogenic response (17–23) and the angiogenic phenotype is associated with altered sialylation patterns (3–6), we asked whether NEU1 might influence EC capillary-like tube formation in a Matrigel system as a measure of in vitro angiogenesis (Fig. 1). When HPMECs were seeded on Matrigel at 1.5 × 104 cells/well, the cells formed tubes within 6 h with a mean ± S.E.) of 91.2 ± 5.5 branches/HPF (Fig. 1, A and F). In HPMECs, NEU1 protein was detected at its anticipated gel mobility (Fig. 1B, lane 1). In HPMECs transfected with NEU1-targeting siRNAs, NEU1 protein was reduced ∼95% relative to control siRNA-transfected cells (Fig. 1B, lane 2 versus lane 1), but EC tube formation was not different from either nontransfected ECs or control siRNA-transfected ECs (Fig. 1C). In HPMECs infected with increasing MOIs of Ad-NEU1-FLAG, NEU1 was dose-dependently overexpressed (Fig. 1D). The resolved proteins were first probed with anti-FLAG antibody to reveal ectopically expressed FLAG-tagged NEU1 (top blot). The same blot was stripped and reprobed with anti-NEU1 antibody to reveal total NEU1 (i.e. both endogenous and ectopically expressed NEU1) (middle blot). After HPMECs were infected with Ad-NEU1 (MOI = 10, 50, and 100) or Ad-GFP as the control (MOI = 100), Ad-NEU1 at MOI ≥10 reduced tube formation compared with Ad-GFP-infected cells (Fig. 1, E and F). Ad-NEU1 at MOI = 100 inhibited tube formation ∼60% compared with Ad-GFP (MOI = 100) (Fig. 1, E (ii versus i) and F). In contrast, overexpression of NEU3 (MOI = 200), the second most abundantly expressed NEU in human ECs (32), did not alter tube formation (Fig. 1F). To exclude any NEU1 effect on HPMEC proliferation or viability, noninfected HPMECs and HPMECs infected with Ad-GFP, Ad-NEU1, or Ad-NEU3, each at MOI = 100, were cultured at equivalent seeding densities and manually counted with trypan blue exclusion every 24 h for a total of 72 h (Fig. 1G). No differences in cell proliferation or viability were detected. To establish whether NEU1 overexpression might similarly impair capillary-like tube formation in pulmonary endothelia from a macrovascular source, HPAECs and HPAECs infected with Ad-GFP (MOI = 200) or Ad-NEU1 (MOI = 100 or 200) were incubated for 6 h on Matrigel, and tubes were quantified (Fig. 1H). NEU1 overexpression in HPAECs dramatically reduced their ability to form capillary-like tubes. Therefore, NEU1 disrupts in vitro angiogenesis in both microvascular and macrovascular lung endothelia.
FIGURE 1.

NEU1 inhibits EC capillary-like tube formation. A, photomicrograph of noninfected HPMECs at 6 h after seeding on Matrigel at 1.5 × 104 ECs/well. Magnification is ×10. B, HPMECs infected with Ad-NEU1-FLAG were transfected with NEU1-targeting or control siRNAs and lysed, and the lysates were processed for FLAG tag (NEU1) immunoblotting. To control for protein loading and transfer, blots were stripped and reprobed for β-tubulin. C, HPMECs and HPMECs transfected with either NEU1-targeting or control siRNAs were seeded on Matrigel, incubated for 6 h, and photographed, and branches of capillary-like tubes were counted. For each condition, n = 7. D, HPMECs were infected with increasing MOIs of Ad-NEU1-FLAG and lysed, and the lysates were processed for FLAG tag immunoblotting. The blots were stripped and reprobed for endogenous NEU1, and to control for protein loading and transfer, they were stripped and reprobed for β-tubulin. IB, immunoblot; IB*, immunoblot after stripping. Molecular mass in kDa is indicated on the left. Each blot is representative of at least two independent experiments. E, photomicrographs of Ad-GFP-infected (i) and Ad-NEU1-infected (ii) HPMECs at 6 h after incubation on Matrigel (MOI = 100). Magnification is ×10. F, HPMECs and HPMECs infected with Ad-GFP (MOI = 100), Ad-NEU1 (MOI = 10, 50, and 100), or Ad-NEU3 (MOI = 200) were incubated for 6 h on Matrigel, at which time they were photographed, and branches of capillary-like tubes were counted. G, HPMECs and HPMECs infected with Ad-GFP, Ad-NEU1, or Ad-NEU3, each at MOI = 100, were cultured at equivalent seeding densities (105 ECs/well) and manually counted with trypan blue exclusion every 24 h for a total of 72 h. Each symbol represents mean ECs/ml. n = 2. H, HPAECs and HPAECs infected with Ad-GFP (MOI = 100) or Ad-NEU1 (MOI = 100 or 200) were incubated for 6 h on Matrigel, at which time they were photographed, and branches of capillary-like tubes were counted. In C, F, and H, each vertical bar represents mean ± S.E. (error bars) branches/HPF. In F and H, the MOI and n for each condition are indicated below each bar. *, significantly decreased compared with the simultaneous Ad-GFP-infected control at p < 0.05.
To determine whether NEU1 sialidase inhibits EC tube formation through its catalytic activity, we studied tube formation in HPMECs infected with increasing MOIs of an Ad encoding for the catalytically dead NEU1 mutant, Ad-NEU1-G68V (39). First, infection of HPMECs with increasing MOIs of Ad-NEU1-G68V dose-dependently increased its expression (Fig. 2A). As anticipated, overexpression of wild-type NEU1 (MOI = 100) disrupted tube formation >50% compared with Ad-GFP (MOI = 100)-infected cells, whereas infection with Ad-NEU1-G68V at MOIs up to 200 did not (Fig. 2B). These combined data indicate that NEU1 sialidase activity specifically and dose-dependently impairs HPMEC capillary-like tube formation. To be catalytically active, NEU1 must operate within a multiprotein complex comprising NEU1, PPCA, and β-galactosidase (33–36). In those experiments where NEU1 alone was overexpressed, our results may be understated.
FIGURE 2.
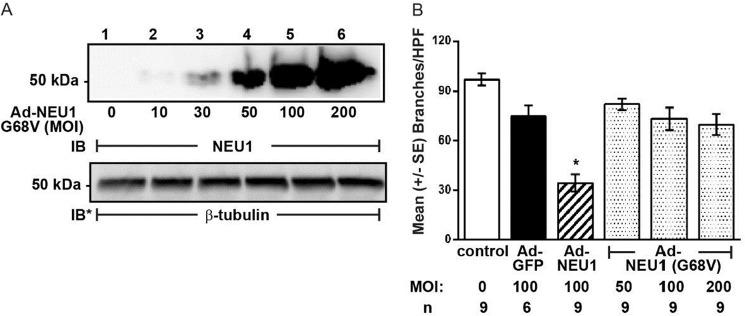
NEU1 catalytic activity required for inhibition of tube formation. A, HPMECs were infected with increasing MOIs of Ad-NEU1-G68V, an Ad encoding for a catalytically dead NEU1 mutant, and lysed, and the lysates were processed for NEU1 immunoblotting. To control for protein loading and transfer, the blots were stripped and reprobed for β-tubulin. IB, immunoblot; IB*, immunoblot after stripping. Molecular mass in kDa is indicated on left. The blot is representative of two independent experiments. B, HPMECs and HPMECs infected with Ad-GFP (MOI = 100), Ad-NEU1 (MOI = 100), or Ad-NEU1-G68V (MOI = 50, 100, and 200), at 1.5 × 104 ECs/well, were incubated for 6 h on Matrigel, at which time they were photographed, and branches of capillary-like tubes were counted. Each bar represents mean ± S.E. (error bars) branches/HPF. The MOI and n for each condition are indicated below each bar. *, significantly decreased compared with the simultaneous Ad-GFP-infected control at p < 0.05.
NEU1 Inhibits EC Adhesion to the Matrigel Substrate
We asked whether the ability of NEU1 to impair tube formation might be explained through altered EC adhesion to the same substrate. Noninfected HPMECs and HPMECs infected at MOI = 100 with Ad-GFP, Ad-NEU1, or Ad-NEU1-G68V were incubated for 10–60 min on Matrigel-coated wells and washed, and adherent ECs were quantified. Adhesion of Ad-GFP-infected cells was comparable with either noninfected control cells or Ad-NEU1-G68V-infected cells (Fig. 3, A (i versus ii) and B). After 10 min of incubation, NEU1 overexpression diminished EC adhesion to Matrigel by ∼60%, at 20 min by ∼50%, at 30 min by ∼25%, and at 1 h by ∼20%, compared with either Ad-GFP-infected or Ad-NEU1-G68V-infected cells (Fig. 3, A (iii versus ii) and B). Over the 60-min study period, EC adhesion increased, whereas NEU1-mediated inhibition decreased. At >1 h incubation on Matrigel, HPMECs begin to organize into capillary-like tubes, making adhesion studies unreliable. This NEU1-mediated inhibition of HPMEC adhesion to Matrigel may, in part, explain NEU1-mediated impairment of EC capillary-like tube formation.
FIGURE 3.
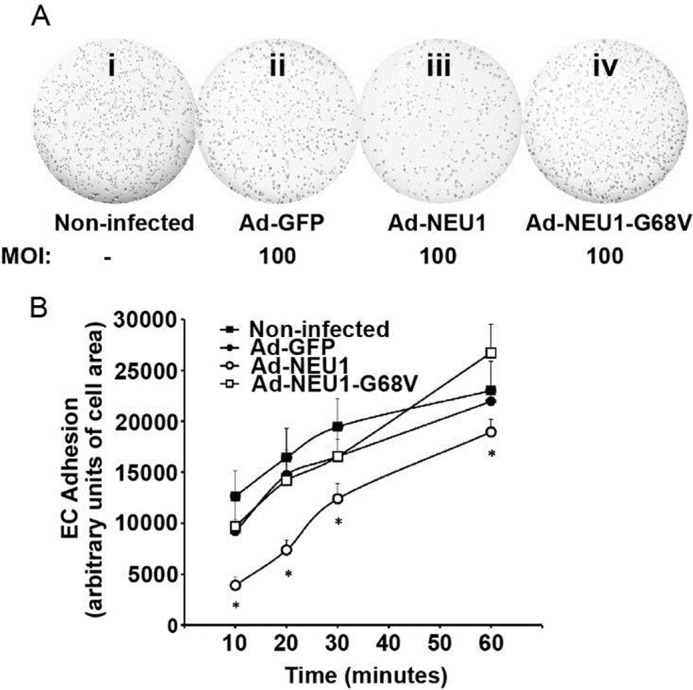
NEU1 decreases EC adhesion to the Matrigel substrate. A, HPMECs (i) and HPMECs infected with Ad-GFP (ii), Ad-NEU1 (iii), or Ad-NEU1-G68V (iv), each at MOI = 100, were incubated for 10, 20, 30, or 60 min in Matrigel-coated wells of 96-well plates (104 cells/well) and washed, and adherent cells were photographed. B, adherent cells were photographed, and adherent EC area was quantified, using ImageJ software, as a measure of EC adhesion to the Matrigel substrate. Each symbol represents mean ± S.E. (error bars) EC adhesion. For each condition, n = 9. *, significantly decreased compared with the simultaneous Ad-GFP- or Ad-NEU1-G68V-infected controls at p < 0.05.
NEU1 Inhibits EC Migration in a Wounding Assay
NEU1-mediated inhibition of EC tube formation might also be explained through diminished EC migration over this same time frame. HPMECs and HPMECs infected at MOI = 100 with Ad-GFP, Ad-NEU1, or Ad-NEU1-G68V were cultured to confluence and wounded (Fig. 4). At 2 h, migration of Ad-NEU1-infected HPMECs into a wound was no different from that seen for Ad-GFP-infected HPMECs (Fig. 4B). At 4 h, NEU1 overexpression inhibited HPMEC migration by 27%, at 6 h by 31%, and at 24 h by 38%, compared with the simultaneous Ad-GFP-infected controls (Fig. 4, A and B). In contrast, at no time did overexpression of the catalytically dead NEU1 mutant, NEU1-G68V, inhibit EC migration compared with the same controls. These combined data indicate that, within ≤4 h, overexpression of catalytically active NEU1 restrains HPMEC migration into a wound. This early NEU1-mediated inhibition of HPMEC migration may, in part, explain the ability of NEU1 to inhibit EC capillary-like tube formation.
FIGURE 4.

NEU1 inhibits EC migration into a wound but not chemotactic responsiveness. A, HPMECs (i) and HPMECs infected with Ad-GFP (ii), Ad-NEU1 (iii), or Ad-NEU1-G68V (iv), each at MOI = 100, were cultured to confluence in the wells of 24-well plates, wounded, and, after 24 h, photographed. Magnification is ×4. B, HPMECs and HPMECs infected with Ad-GFP, Ad-NEU1, or Ad-NEU1-G68V, each at MOI = 100, were cultured to confluence in the wells of 24-well plates, after which a single wound was placed across the diameter of each monolayer. At 0, 2, 4, 6, and 24 h, images of each monolayer were captured, and EC migration into the wound was calculated relative to that observed at 0 h in the same wounded monolayer. Vertical bars, mean ± S.E. (error bars) percentage migration into the wound. n for each condition is indicated below each bar. *, significantly decreased compared with the simultaneous Ad-GFP-infected controls at p < 0.05. C, HPMECs or HPMECs infected with either Ad-GFP or Ad-NEU1, each at MOI = 100, were seeded at 5 × 104 cells/chamber in the upper compartments of modified Boyden chambers in the presence of 0.5% FBS in each lower compartment as the chemoattractant. Each vertical bar represents mean ± S.E. EC chemotaxis measured as A560 nm. n for each condition is indicated below each bar.
Effect of NEU1 on EC Motility
EC migration into a wound requires both disengagement from adjacent ECs at the wound margin and EC motility (1, 2). Because NEU1 overexpression diminishes EC migration into a wound (Fig. 4, A and B), we asked whether NEU1 might directly inhibit EC motility. Suspended, unattached noninfected HPMECs and HPMECs infected with either Ad-GFP or Ad-NEU1, each at MOI = 100, were placed in the upper compartments of modified Boyden chambers, whereas FBS was placed in the lower compartments to generate a chemotactic gradient, as described (51). No differences between the chemotactic indices for noninfected, Ad-GFP-infected, and Ad-NEU1-infected ECs were detected (Fig. 4C). These data suggest that the ability of NEU1 to inhibit angiogenesis cannot be explained through inhibition of EC chemotactic responsiveness or motility.
CD31 Is an in Vivo NEU1 Substrate
CD31 participates in both EC migration (52, 54) and angiogenesis (52, 55–57) and in human umbilical vein ECs (HUVECs) is sialylated via α2,6-linkage (20). We first asked whether in HPMECs, CD31 is similarly sialylated via α2,6-linkages. Lectin probes were validated using fetuin and asialofetuin as sialylated and nonsialylated positive controls, respectively (Fig. 5A). As anticipated, MAL and SNA each recognized fetuin (lanes 1 and 3), whereas PNA recognized asialofetuin (lane 6). CD31 immunoprecipitates from HPMECs were processed for blotting with MAL and SNA lectins, which recognized α2,3- and α2,6-linked SA residues, respectively (15) (Fig. 5B). SNA clearly recognized CD31 (lane 4), whereas MAL did not (lane 3), indicating α2,6- but not α2,3-linked SAs. We then asked whether CD31 might be an in vivo substrate for NEU1. CD31 immunoprecipitates obtained from HPMECs infected with either Ad-NEU1, Ad-NEU1-G68V, or Ad-GFP, each at MOI = 100, were processed for blotting with PNA, a lectin that recognizes subterminal galactose residues that only become accessible after removal of terminal SA (15, 32, 37), as a measure of desialylation (Fig. 5C). When each PNA signal was normalized to the total CD31 signal in the same lane in the same blot, NEU1 overexpression increased PNA signal by 1.9-fold (lane 2) compared with Ad-GFP-infected cells (lane 1), indicating NEU1-mediated CD31 desialylation (Fig. 5D). These results were confirmed in reciprocal coimmunoprecipitation assays using PNA immobilized on Sepharose to enrich for PNA-bound proteins, which, in turn, were probed for CD31 (Fig. 5E, lane 2 versus lane 1). In contrast, overexpression of the catalytically dead NEU1 mutant, NEU1-G68V, did not increase PNA signal compared with Ad-GFP-infected cells (Fig. 5, C (lanes 3 and 4) and E (lanes 3 and 4)). These combined data indicate that in HPMECs, catalytically active NEU1 removes SA residues from CD31.
FIGURE 5.

CD31 is an in vivo NEU1 substrate. A, the positive controls, fetuin and asialofetuin (1.0 μg of each), were processed for lectin blotting with biotinylated MAL (lanes 1 and 2), SNA (lanes 3 and 4), and PNA (lanes 5 and 6). B, HPMECs were lysed, and the lysates were immunoprecipitated with anti-CD31 antibody. Total cell lysates (lane 1) and the CD31 immunoprecipitates (lane 2) were processed for blotting with anti-CD31 antibody. The CD31 immunoprecipitates were also probed with the biotinylated lectins, MAL to detect α2,3-linked SA (lane 3) and SNA to detect α2,6-linked SA (lane 4). C, CD31 immunoprecipitates from lysates of HPMECs infected with Ad-GFP, Ad-NEU1, or Ad-NEU1-G68V, each at MOI = 100, were processed for PNA lectin blotting. In B (lanes 3 and 4) and C (lanes 1–4), to control for protein loading and transfer, blots were stripped and reprobed with anti-CD31 antibody. D, densitometric analyses of the blots in C. Vertical bars, mean ± S.E. (error bars) PNA signal normalized to CD31 signal in the same lane on the same blot. n for each condition is indicated below each bar. *, significantly increased PNA/CD31 densitometry of Ad-NEU1-infected ECs compared with Ad-GFP-infected control ECs at p < 0.05. E, lysates from HPMECs infected with Ad-GFP (lanes 1 and 3), Ad-NEU1 (lane 2), or Ad-NEU1-G68V (lane 4) were incubated with PNA immobilized on Sepharose beads, and the PNA-binding proteins were processed for CD31 immunoblotting. IP, immunoprecipitation; IB, immunoblot; IB*, immunoblot after stripping. Molecular mass in kDa is indicated on the left. Each blot is representative of at least three independent experiments.
NEU1 Is Recruited to and Desialylates CD31 in Postconfluent HPMECs
Forced overexpression of NEU1 clearly desialylates CD31 (Fig. 5, C–E). We previously found that NEU1 is recruited to epidermal growth factor receptor in human airway epithelia stimulated with its cognate ligand, EGF (37). Because an established ligand for CD31 is its own ectodomain (ED) expressed on adjacent cells (45–48), we compared both NEU1-CD31 and PPCA-CD31 association in coimmunoprecipitation assays in postconfluent HPMECs, where CD31 ectodomains (CD31-EDs) are homophilically engaged, versus subconfluent HPMECs, where they are not (Fig. 6A). The relative abundance of NEU1, PPCA, and CD31, each normalized to β-tubulin, was comparable in subconfluent and postconfluent ECs (Fig. 6B). For the NEU1-CD31 coimmunoprecipitation assays, HPMECs infected with Ad-NEU1-FLAG were cultured to subconfluent or postconfluent states and lysed, and the lysates were immunoprecipitated with anti-CD31 antibody. The CD31 immunoprecipitates were processed for FLAG tag immunoblotting. To control for loading and transfer, the blots were stripped and reprobed for CD31. FLAG (NEU1) signal was normalized to CD31 in the same lane in the same stripped and reprobed blot. NEU1 was recruited to CD31 in postconfluent cells compared with subconfluent cells (Fig. 6, C and D). Double-labeled, co-localization immunofluorescence microscopy was used to study HPMECs preinfected with Ad-NEU1-FLAG and probed with antibodies raised against CD31 and the FLAG epitope on NEU1 (Fig. 6E). The merged images revealed that CD31 and NEU1 co-localize at zones of cell-to-cell contact in postconfluent cells. For the PPCA-CD31 coimmunoprecipitation assays, lysates of subconfluent and postconfluent HPMECs were immunoprecipitated with anti-PPCA antibody. The PPCA immunoprecipitates were processed for CD31 and PPCA immunoblotting. Like NEU1, PPCA also was recruited to CD31 in postconfluent cells compared with subconfluent cells (Fig. 6, F and G). We then asked whether this CD31-associated NEU1 might desialylate CD31. CD31 immunoprecipitates harvested from subconfluent and postconfluent HPMECs were processed for PNA lectin blotting (Fig. 7A, lanes 1 and 2). Normalized PNA signals for CD31 immunoprecipitates from postconfluent cells (lane 2) were increased >2-fold relative to subconfluent cells (lane 1) (Fig. 7B). Further, prior silencing of NEU1 completely protected against CD31 desialylation (lane 4) compared with control siRNA-transfected cells (lane 3). That CD31 was desialylated in the postconfluent state was confirmed in reciprocal coimmunoprecipitation assays using PNA immobilized on Sepharose to enrich for PNA-bound proteins from lysates of subconfluent and postconfluent HPMECs, which, in turn, were probed for CD31 (Fig. 7C). In addition to the full-length 130 kDa band seen with postconfluence, CD31 immunoblotting also revealed another 100 kDa band. Whether this 100 kDa CD31-immunoreactive band represents a CD31 proteolytic cleavage product or a deglycosylated form is unclear. These combined data indicate that under postconfluent conditions, when CD31 is homophilically engaged, NEU1, accompanied by PPCA, is recruited to and desialylates CD31.
FIGURE 6.

NEU1 is recruited to CD31 in postconfluent ECs. A, photomicrographs of HPMECs cultured to subconfluent (i) and postconfluent (ii) states. Magnification is ×10. B, lysates from subconfluent and postconfluent HPMECs were processed for NEU1 (lanes 1 and 2), PPCA (lanes 3 and 4), and CD31 (lanes 5 and 6) immunoblotting. To control for protein loading and transfer, the blots were stripped and reprobed for β-tubulin. C, HPMECs infected with Ad-NEU1-FLAG (MOI = 100) were allowed to achieve subconfluent or postconfluent states and lysed, and the lysates were immunoprecipitated with anti-CD31 antibodies. The CD31 immunoprecipitates were processed for immunoblotting with anti-FLAG antibody. To control for protein loading and transfer, the blots were stripped and reprobed for CD31. E, after preinfection with Ad-NEU1-FLAG, HPMECs were cultured to postconfluence and probed for CD31 (i) and NEU1 (FLAG) (ii), after which the two images were merged (iii). A 20-μm scale bar is shown. Arrows indicate CD31 signal (i), NEU1 (FLAG) signal (ii), or NEU1/CD31 co-localization (iii). F, lysates of subconfluent and postconfluent HPMECs were immunoprecipitated with anti-PPCA antibodies, and the PPCA immunoprecipitates were processed for CD31 immunoblotting. To control for protein loading and transfer, the blots were stripped and reprobed for PPCA. IP, immunoprecipitation; IB, immunoblot; IB*, immunoblot after stripping. Molecular mass in kDa is indicated on left. Each blot is representative of three independent experiments. D, densitometric analyses of the blots in C. Vertical bars, mean ± S.E. (error bars) FLAG (NEU1) signal normalized to CD31 signal in the same lane on the same blot (n = 3). G, densitometric analyses of the blots in F. Vertical bars, mean ± S.E. CD31 signal normalized to PPCA signal in the same lane on the same blot (n = 3). *, significantly increased FLAG/CD31 or CD31/PPCA densitometry of postconfluent versus subconfluent cells at p < 0.05.
FIGURE 7.
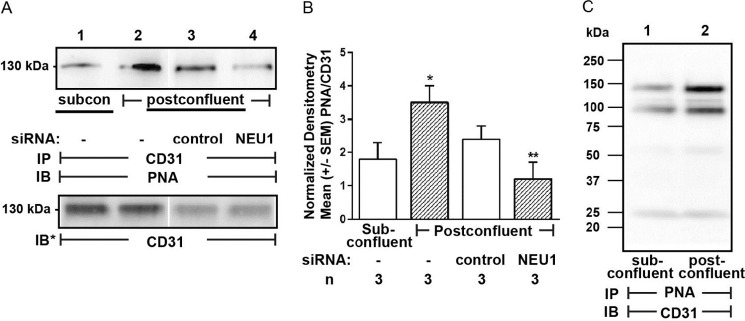
NEU1 desialylates CD31 in postconfluent ECs. A, HPMECs (lanes 1 and 2) or HPMECs transfected with NEU1-targeting (lane 4) or control siRNAs (lane 3) were allowed to achieve subconfluent (lane 1) or postconfluent (lanes 2–4) states and lysed, and the lysates were immunoprecipitated with anti-CD31 antibody. The CD31 immunoprecipitates were processed for PNA lectin blotting. To control for protein loading and transfer, the blots were stripped and reprobed for CD31. B, densitometric analyses of the blots in A. Vertical bars, mean ± S.E. (error bars) PNA signal normalized to CD31 signal in the same lane on the same blot (n = 3). *, significantly increased PNA/CD31 densitometry of postconfluent versus subconfluent cells at p < 0.05. **, significantly decreased PNA/CD31 densitometry compared with control siRNA-transfected cells at p < 0.05. C, lysates from subconfluent (lane 1) and postconfluent (lane 2) HPMECs were incubated with PNA immobilized on Sepharose beads, and the PNA-binding proteins were processed for CD31 immunoblotting. IP, immunoprecipitation; IB, immunoblot; IB*, immunoblot after stripping. Molecular mass in kDa is indicated on the left. Each blot is representative of three independent experiments.
NEU1 Inhibition of EC Tube Formation Requires CD31
NEU1 overexpression both desialylates CD31 (Fig. 5, C–E) and impairs EC tube formation (Fig. 1, E and F). Because CD31 is an established participant in angiogenesis (52, 55–57), we asked whether NEU1-induced inhibition of EC tube formation might be mediated through CD31. First, we established the ability to silence CD31 in HPMECs using siRNA technology (Fig. 8A). When HPMECs were transfected with CD31-targeting siRNAs, CD31 protein was reduced ≥95% relative to control siRNA-transfected cells (lane 2 versus lane 1). HPMECs infected with Ad-NEU1 or Ad-GFP, each at MOI = 100, were transfected with CD31-targeting or control siRNAs. After 48 h, the ECs were incubated for 6 h on Matrigel, and EC capillary-like tubes were quantified (Fig. 8B). As anticipated, Ad-GFP-infected HPMECs in which CD31 was silenced formed fewer tubes than did ECs transfected with control siRNAs. In HPMECs transfected with control siRNAs, NEU1 overexpression inhibited tube formation by ∼50% compared with Ad-GFP-infected cells. In HPMECs in which CD31 was silenced, NEU1 overexpression did not decrease tube formation compared with Ad-GFP-infected controls. To provide additional evidence that NEU1 inhibits EC tube formation through CD31, we compared the impact of NEU1 overexpression on tube formation in CD31-null murine lung microvascular ECs with tube formation in CD31-RC ECs. First, we established the relative abundance of CD31 protein in these two EC lines (Fig. 8C). CD31 protein could not be detected in the CD31-null cells (lane 1), whereas it was abundant in the CD31-RC cells (lane 2). CD31-null and CD31-RC ECs were each infected with Ad-NEU1 or Ad-GFP, each at MOI = 100. After 48 h, the ECs were incubated for 6 h on Matrigel, and EC capillary-like tubes were quantified (Fig. 8D). As anticipated, the Ad-GFP-infected CD31-null ECs formed fewer tubes than did Ad-GFP-infected CD31-RC ECs. In the CD31-RC ECs, NEU1 overexpression diminished tube formation by ∼60% compared with Ad-GFP-infected cells. In contrast, in the CD31-null ECs, not only did NEU1 overexpression fail to inhibit tube formation compared with Ad-GFP-infected cells; it enhanced it. These combined data indicate that NEU1-induced inhibition of EC capillary-like tube formation requires unperturbed EC levels of CD31.
FIGURE 8.

CD31 depletion abrogates the inhibitory effect of NEU1 on EC capillary-like tube formation. A, HPMECs transfected with CD31-targeting or control siRNAs were cultured for 48 h and lysed, and the lysates were processed for CD31 immunoblotting. B, HPMECs transfected with CD31-targeting or control siRNAs were infected with Ad-NEU1 or Ad-GFP, each at MOI = 100. After 48 h, the HPMECs were incubated for 6 h on Matrigel, at which time they were photographed, and branches of capillary-like tubes were counted. C, lysates of CD31-null and CD31-RC murine lung microvascular ECs were processed for CD31 immunoblotting. D and E, CD31-null and CD31-RC ECs were infected with Ad-NEU1 or Ad-GFP, each at MOI = 100. After 48 h, ECs were incubated for 6 h on Matrigel and photographed (magnification, ×10) (D), and branches of capillary-like tubes were counted (E). In A and C, to control for protein loading and transfer, blots were stripped and reprobed for β-tubulin. IB, immunoblot; IB*, immunoblot after stripping. Molecular mass in kDa is indicated on the left. Each blot is representative of two independent experiments. In B and E, each bar represents mean ± S.E. (error bars) branches/HPF. For each condition n = 9. *, significantly decreased compared with the simultaneous Ad-GFP-infected controls at p < 0.05.
ST6GAL-I Overexpression Increases α2,6-Linked Sialylation of CD31 and Counteracts NEU1-mediated Inhibition of EC Capillary-like Tube Formation
Because we detected α2,6-linked but not α2,3-linked SA residues on CD31 (Fig. 5B) and found that NEU1 overexpression increased CD31 recognition of the galactose-binding lectin, PNA (Fig. 5, C–E), we asked which of the two STs known to transfer SA to subterminal galactose residues in an α2,6-linkage, ST6GAL-I or ST6GAL-II (26, 27), might counterregulate the CD31 sialylation state. Upon screening HPMECs for expression of these two STs, ST6GAL-I was expressed, whereas ST6GAL-II was not (Fig. 9A), confirming a prior report (58). To begin to establish whether NEU1 influences CD31 function through its ability to remove α2,6-linked SA from CD31-expressed galactose residues, we asked whether ST6GAL-I might counteract this effect. HPMECs were infected with increasing MOIs of Ad encoding for HA-tagged ST6GAL-I, and after 48 h, they were lysed, and the lysates were processed for immunoblotting with anti-HA antibody (Fig. 9B). ST6GAL-I protein expression was dose-dependently increased. To establish whether ST6GAL-I overexpression might increase α2,6-linked sialylation of CD31, CD31 immunoprecipitates from both ST6GAL-I-overexpressing and Ad-GFP-infected HPMECs were processed for SNA lectin blotting (Fig. 9C). ST6GAL-I overexpression increased SNA binding to CD31 >2-fold compared with Ad-GFP-infected controls (Fig. 9, C and D). Finally, we asked if ST6GAL-I overexpression might counteract the NEU1 inhibitory effect on EC tube formation. HPMECs infected with Ad-GFP, Ad-ST6GAL-I, or Ad-NEU1 or co-infected with both Ad-NEU1 and Ad-ST6GAL-I were incubated for 6 h on Matrigel and photographed, and branches of capillary-like tubes were counted (Fig. 10). Infection of HPMECs with increasing MOIs (3–200) of Ad-ST6GAL-I did not alter tube formation compared with either noninfected or Ad-GFP-infected cells (Fig. 10A). As anticipated, NEU1 overexpression dramatically reduced tube formation (Fig. 10, B (ii versus i) and C). However, when Ad-NEU1-infected HPMECs were co-infected with increasing MOIs of Ad-ST6GAL-I, the NEU1 inhibitory effect on tube formation was dose-dependently counteracted (Fig. 10C). Co-infection of Ad-NEU1-infected HPMECs with Ad-ST6GAL-I at MOIs of ≥25 protected against loss of tube formation, and at MOI = 100, the Ad-ST6GAL-I co-infection provided almost complete protection (Fig. 10B, iii versus ii). Therefore, in HPMECs, ST6GAL-I overexpression increases CD31 α2,6-linked sialylation and abrogates NEU1-mediated inhibition of EC tube formation.
FIGURE 9.
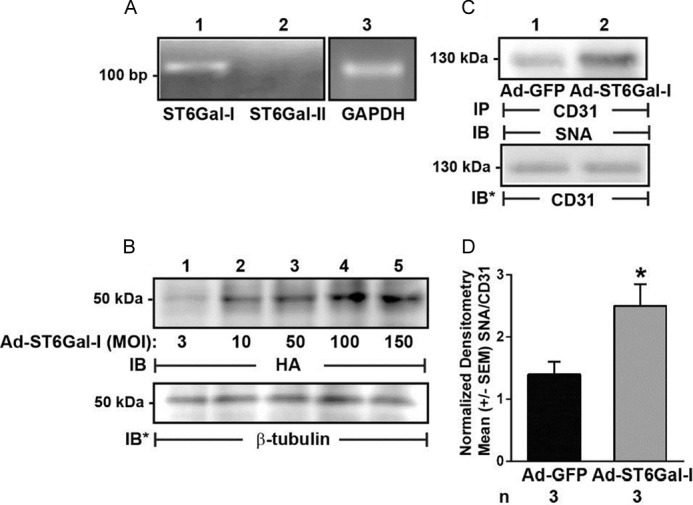
Increased ST6GAL-I expression in HPMECs increases α2,6-linked CD31 sialylation. A, total cellular RNA was extracted from HPMECs and processed for RT-PCR for ST6GAL-I and -II. GAPDH was run as an internal control. Base pairs (bp) are indicated on the left. B, HPMECs were infected with increasing MOIs of Ad encoding for HA-tagged ST6GAL-I, and after 48 h, they were lysed, and the lysates were processed for immunoblotting with anti-HA antibody. To control for protein loading and transfer, blots were stripped and reprobed for β-tubulin. C, HPMECs were infected with Ad-ST6GAL-I (lane 2) or Ad-GFP (lane 1), each at MOI = 200, and after 48 h, they were lysed, and the lysates were immunoprecipitated with anti-CD31 antibody. The CD31 immunoprecipitates were processed for SNA lectin blotting. To control for protein loading and transfer, blots were stripped and reprobed for CD31. For A–C, each blot is representative of three independent experiments. IP, immunoprecipitation; IB, immunoblot; IB*, immunoblot after stripping. Molecular mass in kDa is indicated on the left. D, densitometric analyses of blots in C. Vertical bars, mean ± S.E. (error bars) SNA signal normalized to CD31 signal in the same lane on the same blot (n = 3). *, significantly increased SNA/CD31 densitometry of Ad-ST6GAL-I-infected ECs compared with the Ad-GFP-infected control ECs at p < 0.05.
FIGURE 10.

ST6GAL-I overexpression counteracts NEU1-mediated inhibition of EC capillary-like tube formation. A, HPMECs, HPMECs infected with Ad-GFP (MOI = 100), and HPMECs infected with increasing MOIs of Ad-ST6GAL-I were seeded at 1.5 × 104 ECs/well and incubated for 6 h on Matrigel, at which time they were photographed, and branches of capillary-like tubes were counted. B, photomicrographs of HPMECs infected with Ad-GFP (i) or Ad-NEU1 (ii), each at MOI = 100, or co-infected with both Ad-NEU1 and Ad-ST6GAL-I, each at MOI = 100, after 6 h incubation on Matrigel. Magnification is ×10. C, HPMECs and HPMECs infected with Ad-GFP or Ad-NEU1, each at MOI = 100, or co-infected with Ad-NEU1 (MOI = 100) and increasing MOIs of Ad-ST6GAL-I were incubated for 6 h on Matrigel and photographed, and branches of capillary-like tubes were counted. In A and C, each vertical bar represents mean ± S.E. (error bars) branches/HPF. The MOI and n for each condition are indicated below each bar. *, significantly decreased compared with the simultaneous Ad-GFP-infected control at p < 0.05.
DISCUSSION
In these studies, we have found that catalytically active NEU1 dose-dependently impairs EC capillary-like tube formation without influencing EC proliferation or viability in both HPMECs and HPAECs (Fig. 1). Overexpression of a catalytically dead NEU1 mutant exerted no such effect (Fig. 2). NEU1 diminished EC adhesion to a Matrigel substrate (Fig. 3) and restrained EC migration in wounding but not in chemotaxis assays (Fig. 4). In HPMECs, CD31 was found to express α2,6-linked SA residues and to be a NEU1 substrate (Fig. 5). Upon achieving a postconfluent state, NEU1, accompanied by PPCA, associated with (Fig. 6) and desialylated (Fig. 7) CD31. Finally, prior silencing of CD31, the use of CD31-null ECs (Fig. 8), and increased α2,6-linked sialylation of CD31 (Figs. 9 and 10) each counteracted the ability of NEU1 to disrupt EC tube formation.
Our combined data provide evidence that NEU1, at least in part, regulates EC tube formation through CD31. First, the sialoprotein CD31 is a NEU1 substrate (Figs. 5 (C–E) and 7). Further, CD31 is central to at least two EC processes that NEU1 counterregulates, EC migration (52, 54) and angiogenesis (52, 55–57). Anti-CD31 antibodies were shown to restrain HUVEC migration in wounding assays and movement through Matrigel-coated transwell inserts (52). CD31 depletion in HUVECs, using antisense technology, decreased migration into a wound (54). In these same studies, murine CD31-null ECs displayed less migration compared with CD31-reconstituted ECs. Similarly, multiple studies have established a role for CD31 in both in vitro and in vivo angiogenesis (52, 55–57). Again, anti-CD31 antibodies impaired in vitro EC tube formation using HUVECs seeded on Matrigel (52) and rat microvascular ECs from epididymal fat pads in three-dimensional collagen gels (55). CD31 immunoblockade also inhibits in vivo angiogenesis in multiple experimental systems, including cytokine-induced neovascularization in a rat corneal micropocket model (55) and subcutaneous implantation of Matrigel plugs supplemented with basic fibroblast growth factor in C57BL/6 mice (55). In a model of foreign body-induced chronic inflammation, subcutaneous implantation of polyvinyl acetyl sponges in CD31-null mice elicited dramatically less angiogenesis than that seen in wild-type mice (57). In models of in vivo tumor angiogenesis, anti-CD31 antibodies diminished subcutaneous growth and vascularity of multiple human tumors implanted in various murine strains (52, 56). Taken together, these data establish a positive regulatory role for CD31 in EC migration and angiogenesis, the very same EC processes counterregulated by NEU1 (Figs. 1 and 4). Finally, prior silencing of CD31 and the use of CD31-null ECs each completely abrogated the inhibitory effect of NEU1 on EC tube formation (Fig. 8).
The early reports on the biochemical structure of CD31 recognized its extensive glycosylation (59, 60). In one study, the predicted molecular size of the mature 711-aa polypeptide was ∼80 kDa, whereas on immunoblot, the fully processed molecule appeared to be ∼130 kDa (59). Upon sequencing the CD31 polypeptide, nine predicted asparagine-linked glycosylation sites were identified, leading to the assumption that carbohydrate residues accounted for ∼40% of the 130-kDa molecular size. A second study was in complete agreement but also reported numerous serine/threonine-rich regions as potential sites for O-linked sugar attachment (60). In still another study of recombinant CD31 expressed in CHO-K1 cells, MALDI mass spectrometry of peptide:N-glycosidase F-released N-linked glycans revealed structures consistent with sialylated bi-, tri-, and tetra-antennary carbohydrates (61). Glycan release with application of Arthrobacter ureafaciens neuraminidase confirmed the presence of SA residues. More recently, lectin blotting with Sambucus sieboldiana agglutinin of CD31 expressed in HUVECs revealed SA residues α2,6-linked to galactose (20). In the current studies, we used combined SNA and PNA lectin blotting of CD31 expressed in HPMECs to show that it too, contains SA α2,6-linked to subterminal galactose (Fig. 5) and that CD31 sialylation could be increased with ST6GAL-I overexpression (Fig. 9, C and D), consistent with the previous report (20).
Our data not only implicate CD31 as a downstream intermediary to NEU1 sialidase in the regulation of EC migration and tube formation but, more specifically, indicate that changes in the CD31 sialylation state regulate these CD31-driven EC responses. First, in HPMECs, CD31 is a sialoprotein with α2,6-linked SA residues detected with SNA lectin blotting, whereas application of MAL lectin blotting failed to detect α2,3-linked sialylation (Fig. 5B). Further, the same NEU1 overexpression that restrains EC migration in a wounding assay (Fig. 4) and disrupts EC tube formation (Fig. 1) also desialylates CD31, as detected by PNA lectin blotting (Fig. 5, C–E). In contrast, overexpression of the catalytically dead NEU1 mutant, NEU1-G68V, to even higher levels of expression failed to inhibit EC tube formation (Fig. 2). Finally, overexpression of ST6GAL-I both increased α2,6-sialylation of CD31 (Fig. 9, C and D) and completely counteracted the inhibitory effect of NEU1 on EC capillary-like tube formation (Fig. 10, B and C). That the sialylation state of CD31 influences its function(s) is further supported by a previous report that presents evidence that α2,6-linked CD31 sialylation increased its cell surface residency, homophilic adhesion between its ectodomains, and cellular resistance to mitochondrion-dependent apoptosis (20). Any one of these properties imparted by α2,6-linked sialylation of CD31 could explain the dysregulated angiogenesis evident in our studies with NEU1 overexpression (Fig. 1) and NEU1-mediated CD31 desialylation (Fig. 5, C–E). However, other substrates for both NEU1 and ST6GAL-I probably exist in HPMECs, one or more of which may be operative during these NEU1/CD31-driven EC responses.
NEU1 also diminished EC adhesion to the underlying Matrigel ECM (Fig. 3). Although the ability of the CD31-ED to homophilically engage the identical molecule on adjacent cells is well established (20, 61, 62), its participation in heterotypic interactions is less clear. Multiple studies in a range of human cell types support the ability of CD31 to interact with and activate multiple integrins (63–68). Engagement of CD31 in human T cell subsets (64), neutrophils and monocytes (65), eosinophils (66), and NK cells (67), as well as murine lymphokine-activated killer cells (68), all reportedly increase the adhesive functions of β1/β2 integrins and adhesion to the endothelial surface. Ligation of CD31 in human CD34+ hematopoietic progenitor cells selectively increased adhesive activity of very late antigen 4 (VLA-4) (i.e. α4β1 integrin for vascular cell adhesion molecule (VCAM)-1 but not LFA-1 integrin for ICAM-1 expressed on CHO cells) (63). Finally, ligation of CD31 on HUVECs increased the adhesive function of αvβ3 integrin to its ligand, RGD peptide (66). Therefore, the sialylation state of the CD31 ectodomain influences its ability to engage integrins, which, in turn, interferes with integrin-ECM protein interactions and EC adhesion to the underlying ECM.
We speculate that during EC capillary-like tube formation, as ECs engage each other and the CD31-EDs homophilically interact, NEU1 sialidase, accompanied by PPCA, is recruited to CD31 to remove SA residues from the CD31-ED, either unmasking or disrupting surface recognition motifs. At the same time, ST6GAL-I is transferring SA to the same CD31-ED. The opposing catalytic activities of NEU1 and ST6GAL-I, possibly in concert with other NEUs and STs, impose a net sialylation state on CD31, which, in turn, dictates its impact on EC function. Whether such NEU1/ST6GAL-I-elicited changes in CD31 sialylation directly alter CD31-ED homophilic adhesion or indirectly influence interactions between the CD31 cytoplasmic domain and its cytosolic binding partners and whether such downstream events lead to dysregulated angiogenesis is the focus of ongoing studies in our laboratory.
Acknowledgement
We thank Shirley Taylor for excellent secretarial support.
This work was supported by National Institutes of Health Grant R01 HL 084223A (to S. E. G.) and the VA Research Service, Department of Veterans Affairs.
- EC
- endothelial cell
- Ad
- adenovirus
- HPMEC
- human pulmonary microvascular endothelial cell
- PECAM-1
- platelet endothelial cell adhesion molecule-1
- CD31-ED
- CD31 ectodomain
- CD31-RC
- CD31 reconstituted
- SNA
- S. nigra agglutinin
- PNA
- peanut agglutinin
- ST
- sialyltransferase
- ECM
- extracellular matrix
- ICAM-1
- intercellular adhesion molecule-1
- SA
- sialic acid
- LFA-1
- lymphocyte function-associated antigen-1
- NEU
- neuraminidase/sialidase
- PPCA
- protective protein/cathepsin A
- HPAEC
- human pulmonary artery endothelial cell
- MOI
- multiplicity of infection
- HPF
- high power field
- MAL
- M. amurensis lectin II
- TBS-T
- Tris-buffered saline-Tween 20
- HUVEC
- human umbilical vein endothelial cell
- VCAM-1
- vascular cell adhesion molecule-1
- aa
- amino acid.
REFERENCES
- 1. Carmeliet P. (2000) Mechanisms of angiogenesis and arteriogenesis. Nat. Med. 6, 389–395 [DOI] [PubMed] [Google Scholar]
- 2. Nguyen L. L., D'Amore P. A. (2001) Cellular interactions in vascular growth and differentiation. Int. Rev. Cytol. 204, 1–48 [DOI] [PubMed] [Google Scholar]
- 3. Augustin-Voss H. G., Pauli B. U. (1992) Migrating endothelial cells are distinctly hyperglycosylated and express specific migration-associated cell surface glycoproteins. J. Cell Biol. 119, 483–491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Nguyen M., Folkman J., Bischoff J. (1992) 1-Deoxymannojirimycin inhibits capillary tube formation in vitro. Analysis of N-linked oligosaccharides in bovine capillary endothelial cells. J. Biol. Chem. 267, 26157–26165 [PubMed] [Google Scholar]
- 5. Nangia-Makker P., Baccarini S., Raz A. (2000) Carbohydrate-recognition and angiogenesis. Cancer Metastasis Rev. 19, 51–77 [DOI] [PubMed] [Google Scholar]
- 6. Willhauck-Fleckenstein M., Moehler T. M., Merling A., Pusunc S., Goldschmidt H., Schwartz-Albiez R. (2010) Transcriptional regulation of the vascular endothelial glycome by angiogenic and inflammatory signalling. Angiogenesis 13, 25–42 [DOI] [PubMed] [Google Scholar]
- 7. Born G. V., Palinski W. (1985) Unusually high concentrations of sialic acids on the surface of vascular endothelia. Br. J. Exp. Pathol. 66, 543–549 [PMC free article] [PubMed] [Google Scholar]
- 8. Fischer E., Wagner M., Bertsch T. (2000) Cepaea hortensis agglutinin-I, specific for O-glycosidically linked sialic acids, selectively labels endothelial cells of distinct vascular beds. Histochem. J. 32, 105–109 [DOI] [PubMed] [Google Scholar]
- 9. Cioffi D. L., Pandey S., Alvarez D. F., Cioffi E. A. (2012) Terminal sialic acids are an important determinant of pulmonary endothelial barrier integrity. Am. J. Physiol. Lung Cell. Mol. Physiol. 302, L1067–L1077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Constantinescu A. A., Vink H., Spaan J. A. (2003) Endothelial cell glycocalyx modulates immobilization of leukocytes at the endothelial surface. Arterioscler. Thromb. Vasc. Biol. 23, 1541–1547 [DOI] [PubMed] [Google Scholar]
- 11. Pahakis M. Y., Kosky J. R., Dull R. O., Tarbell J. M. (2007) The role of endothelial glycocalyx components in mechanotransduction of fluid shear stress. Biochem. Biophys. Res. Commun. 355, 228–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sakarya S., Rifat S., Zhou J., Bannerman D. D., Stamatos N. M., Cross A. S., Goldblum S.E. (2004) Mobilization of neutrophil sialidase activity desialylates the pulmonary vascular endothelial surface and increases resting neutrophil adhesion to and migration across the endothelium. Glycobiology 14, 481–494 [DOI] [PubMed] [Google Scholar]
- 13. Hanasaki K., Varki A., Powell L. D. (1995) CD22-mediated cell adhesion to cytokine-activated human endothelial cells. Positive and negative regulation by α2–6-sialylation of cellular glycoproteins. J. Biol. Chem. 270, 7533–7542 [DOI] [PubMed] [Google Scholar]
- 14. Weber K. S., Alon R., Klickstein L. B. (2004) Sialylation of ICAM-2 on platelets impairs adhesion of leukocytes via LFA-1 and DC-SIGN. Inflammation 28, 177–188 [DOI] [PubMed] [Google Scholar]
- 15. Sharan N., Lis H. (2003) Lectins, 2nd Ed., Kluwer Academic Publishers, Dordrecht, The Netherlands [Google Scholar]
- 16. Nangia-Makker P., Honjo Y., Sarvis R., Akahani S., Hogan V., Pienta K. J., Raz A. (2000) Galectin-3 induces endothelial cell morphogenesis and angiogenesis. Am. J. Pathol. 156, 899–909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Geyer H., Geyer R., Odenthal-Schnittler M., Schnittler H. J. (1999) Characterization of human vascular endothelial cadherin glycans. Glycobiology 9, 915–925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Koch A. E., Halloran M. M., Haskell C. J., Shah M. R., Polverini P. J. (1995) Angiogenesis mediated by soluble forms of E-selectin and vascular cell adhesion molecule-1. Nature 376, 517–519 [DOI] [PubMed] [Google Scholar]
- 19. Hanasaki K., Varki A., Stamenkovic I., Bevilacqua M. P. (1994) Cytokine-induced β-galactoside α-2,6-sialyltransferase in human endothelial cells mediates α2,6-sialylation of adhesion molecules and CD22 ligands. J. Biol. Chem. 269, 10637–10643 [PubMed] [Google Scholar]
- 20. Kitazume S., Imamaki R., Ogawa K., Komi Y., Futakawa S., Kojima S., Hashimoto Y., Marth J. D, Paulson J. C., Taniguchi N. (2010) α2,6-sialic acid on platelet endothelial cell adhesion molecule (PECAM) regulates its homophilic interactions and downstream antiapoptotic signaling. J. Biol. Chem. 285, 6515–6521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Skelton T. P., Zeng C., Nocks A., Stamenkovic I. (1998) Glycosylation provides both stimulatory and inhibitory effects on cell surface and soluble CD44 binding to hyaluronan. J. Cell Biol. 140, 431–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Duchesne L., Tissot B., Rudd T. R., Dell A., Fernig D. G. (2006) N-Glycosylation of fibroblast growth factor receptor 1 regulates ligand and heparin sulfate co-receptor binding. J. Biol. Chem. 281, 27178–27189 [DOI] [PubMed] [Google Scholar]
- 23. Chiodelli P., Urbinati C., Mitola S., Tanghetti E., Rusnati M. (2012) (2012) Sialic acid associated with αvβ3 integrin mediates HIV-1 Tat protein interaction and endothelial cell proangiogenic activation. J. Biol. Chem. 287, 20456–20466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Varki A. (2008) Sialic acids in human health and disease. Trends Mol. Med. 14, 351–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Schauer R. (2009) Sialic acids as regulators of cellular interactions. Curr. Opin. Struct. Biol. 19, 507–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Harduin-Lepers A., Mollicone R., Delannoy P., Oriol R. (2005) The animal sialyltransferases and sialyltransferase-related genes: a phylogenetic approach. Glycobiology 15, 805–817 [DOI] [PubMed] [Google Scholar]
- 27. Wang P. H. (2005) Altered glycosylation in cancer: sialic acids and sialyltransferases. J. Cancer Mol. 1, 73–81 [Google Scholar]
- 28. Monti E., Preti A., Venerando B., Borsani G. (2002) Recent development in mammalian sialidase molecular biology. Neurochem. Res. 27, 649–663 [DOI] [PubMed] [Google Scholar]
- 29. Monti E., Bonten E., D'Azzo A., Bresciani R., Venerando B., Borsani G., Schauer R., Tettamanti G. (2010) (2010) Sialidases in vertebrates: a family of enzymes tailored for several cell functions. Adv. Carbohydr. Chem. Biochem. 64, 403–479 [DOI] [PubMed] [Google Scholar]
- 30. Miyagi T., Yamaguchi K. (2012) Mammalian sialidases: physiological and pathological roles in cellular functions. Glycobiology 22, 880–896 [DOI] [PubMed] [Google Scholar]
- 31. Giacopuzzi E., Bresciani R., Schauer R., Monti E., Borsani G. (2012) New insights on the sialidase protein family revealed by a phylogenetic analysis in metazoa. PLoS One 7, e44193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cross A. S., Hyun S. W., Miranda-Ribera A., Feng C., Liu A., Nguyen C., Zhang L., Luzina I. G., Atamas S. P., Twaddell W. S., Guang W., Lillehoj E. P., Puché A. C., Huang W., Wang L. X., Passaniti A., Goldblum S. E. (2012) NEU1 and NEU3 sialidase activity expressed in human lung microvascular endothelia. NEU1 restrains endothelial cell migration whereas NEU3 does not. J. Biol. Chem. 287, 15966–15980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bonten E. J., d'Azzo A. (2000) Lysosomal neuraminidase. Catalytic activation in insect cells is controlled by the protective protein/cathepsin A. J. Biol. Chem. 275, 37657–37663 [DOI] [PubMed] [Google Scholar]
- 34. Pshezhetsky A. V., Ashmarina M. (2001) Lysosomal multienzyme complex: biochemistry, genetics, and molecular pathophysiology. Prog. Nucleic Acid Res. Mol. Biol. 69, 81–114 [DOI] [PubMed] [Google Scholar]
- 35. van der Spoel A., Bonten E., d'Azzo A. (1998) (1998) Transport of human lysosomal neuraminidase to mature lysosomes requires protective protein/cathepsin A. EMBO J. 17, 1588–1597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bonten E. J., Campos Y., Zaitsev V., Nourse A., Waddell B., Lewis W., Taylor G., d'Azzo A. (2009) Heterodimerization of the sialidase NEU1 with the chaperone protective protein/cathepsin A prevents its premature oligomerization. J. Biol. Chem. 284, 28430–28441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lillehoj E. P., Hyun S. W., Feng C., Zhang L., Liu A., Guang W., Nguyen C., Luzina I. G., Atamas S. P., Passaniti A., Twaddell W. S., Puché A. C., Wang L.-X., Cross A. S., Goldblum S. E. (2012) NEU1 sialidase expressed in human airway epithelia regulates epidermal growth factor receptor and MUC1 signaling. J. Biol. Chem. 287, 8214–8231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Amith S. R., Jayanth P., Franchuk S., Finlay T., Seyrantepe V., Beyaert R., Pshezhetsky A. V., Szewczuk M. R. (2010) Neu1 desialylation of sialyl α-2,3-linked β-galactosyl residues of TOLL-like receptor 4 is essential for receptor activation and cellular signaling. Cell. Signal. 22, 314–324 [DOI] [PubMed] [Google Scholar]
- 39. Duca L., Blanchevoye C., Cantarelli B., Ghoneim C., Dedieu S., Delacoux F., Hornebeck W., Hinek A., Martiny L., Debelle L. (2007) The elastin receptor complex transduces signals through the catalytic activity of its Neu-1 subunit. J. Biol. Chem. 282, 12484–12491 [DOI] [PubMed] [Google Scholar]
- 40. Lukong K. E., Seyrantepe V., Landry K., Trudel S., Ahmad A., Gahl W. A., Lefrancois S., Morales C. R., Pshezhetsky A. V. (2001) Intracellular distribution of lysosomal sialidase is controlled by the internalization signal in its cytoplasmic tail. J. Biol. Chem. 276, 46172–46181 [DOI] [PubMed] [Google Scholar]
- 41. Nan X., Carubelli I., Stamatos N. M. (2007) Sialidase expression in activated human T lymphocytes influences production of IFN-γ. J. Leukoc. Biol. 81, 284–296 [DOI] [PubMed] [Google Scholar]
- 42. Liang F., Seyrantepe V., Landry K., Ahmad R., Ahmad A., Stamatos N. M., Pshezhetsky A. V. (2006) Monocyte differentiation up-regulates the expression of the lysosomal sialidase, Neu1, and triggers its targeting to the plasma membrane via major histocompatibility complex class II-positive compartments. J. Biol. Chem. 281, 27526–27538 [DOI] [PubMed] [Google Scholar]
- 43. Hinek A., Pshezhetsky A.V., von Itzstein M., Starcher B. (2006) Lysosomal sialidase (neuraminidase-1) is targeted to the cell surface in a multiprotein complex that facilitates elastic fiber assembly. J. Biol. Chem. 281, 3698–3710 [DOI] [PubMed] [Google Scholar]
- 44. D'Avila F., Tringali C., Papini N., Anastasia L., Croci G., Massaccesi L., Monti E., Tettamanti G., Venerando B. (2013) Identification of lysosomal sialidase NEU1 and plasma membrane sialidase NEU3 in human erythrocytes. J. Cell. Biochem. 114, 204–211 [DOI] [PubMed] [Google Scholar]
- 45. Newman P. J. (1997) Perspectives series: cell adhesion in vascular biology. J. Clin. Invest. 99, 3–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ilan N., Madri J. A. (2003) PECAM-1: old friend, new partners. Curr. Opin. Cell Biol. 15, 515–524 [DOI] [PubMed] [Google Scholar]
- 47. Newman P. J., Newman D. K. (2003) Signal transduction pathways mediated by PECAM-1: new roles for an old molecule in platelet and vascular cell biology. Arterioscler. Thromb. Vasc. Biol. 23, 953–964 [DOI] [PubMed] [Google Scholar]
- 48. Gong N., Chatterjee S. (2003) Platelet endothelial cell adhesion molecule in cell signaling and thrombosis. Mol. Cell. Biochem. 253, 151–158 [DOI] [PubMed] [Google Scholar]
- 49. Biswas P., Canosa S., Schoenfeld D., Schoenfeld J., Li P., Cheas L. C., Zhang J., Cordova A., Sumpio B., Madri J. A. (2006) PECAM-1 affects GSK-3β-mediated β-catenin phosphorylation and degradation. Am. J. Pathol. 169, 314–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lukong K. E., Elsliger M. A., Chang Y., Richard C., Thomas G., Carey W., Tylki-Szymanska A., Czartoryska B., Buchholz T., Criado G. R., Palmeri S., Pshezhetsky A. V. (2000) Characterization of the sialidase molecular defects in sialidosis patients suggests the structural organization of the lysosomal multienzyme complex. Hum. Mol. Genet. 9, 1075–1085 [DOI] [PubMed] [Google Scholar]
- 51. Hyun S. W., Anglin I. E., Liu A., Yang S., Sorkin J. D., Lillehoj E., Tonks N. K., Passaniti A., Goldblum S. E. (2011) Diverse injurious stimuli reduce protein tyrosine phosphatase-μ expression and enhance epidermal growth factor receptor signaling in human airway epithelia. Exp. Lung Res. 37, 327–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Cao G., O'Brien C. D., Zhou Z., Sanders S. M., Greenbaum J. N., Makrigiannakis A., DeLisser H. M. (2002) Involvement of human PECAM-1 in angiogenesis and in vitro endothelial cell migration. Am. J. Physiol. Cell Physiol. 282, C1181–C1190 [DOI] [PubMed] [Google Scholar]
- 53. Pierce A. D., Anglin I. E., Vitolo M. I., Mochin M. T., Underwood K. F., Goldblum S. E., Kommineni S., Passaniti A. (2012) Glucose-activated RUNX2 phosphorylation promotes endothelial cell proliferation and an angiogenic phenotype. J. Cell. Biochem. 113, 282–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Gratzinger D., Canosa S., Engelhardt B., Madri J.A. (2003) Platelet endothelial cell adhesion molecule-1 modulates endothelial cell motility through the small G-protein Rho. FASEB J. 17, 1458–1469 [DOI] [PubMed] [Google Scholar]
- 55. DeLisser H. M., Christofidou-Solomidou M., Strieter R. M., Burdick M. D., Robinson C. S., Wexler R. S., Kerr J. S., Garlanda C., Merwin J. R., Madri J. A., Albelda S. M. (1997) Involvement of endothelial PECAM-1/CD31 in angiogenesis. Am. J. Pathol. 151, 671–677 [PMC free article] [PubMed] [Google Scholar]
- 56. Zhou Z., Christofidou-Solomidou M., Garlanda C., DeLisser H.M. (1999) Antibody against murine PECAM-1 inhibits tumor angiogenesis in mice. Angiogenesis 3, 181–188 [DOI] [PubMed] [Google Scholar]
- 57. Solowiej A., Biswas P., Graesser D., Madri J. (2003) Lack of platelet endothelial cell adhesion molecule-1 attenuates foreign body inflammation because of decreased angiogenesis. Am. J. Pathol. 162, 953–962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Takashima S., Tsuji S., Tsujimoto M. (2002) Characterization of the second type of human β-galactoside α2,6-sialyltransferase (ST6Gal II), which sialylates Galβ 1,4GlcNAc structures on oligosaccharides preferentially. Genomic analysis of human sialyltransferase genes. J. Biol. Chem. 277, 45719–45728 [DOI] [PubMed] [Google Scholar]
- 59. Newman P. J., Berndt M. C., Gorski J., White G. C., 2nd, Lyman S., Paddock C., Muller W. A. (1990) PECAM-1 (CD31) cloning and relation to adhesion molecules of the immunoglobulin gene superfamily. Science 247, 1219–1222 [DOI] [PubMed] [Google Scholar]
- 60. Simmons D. L., Walker C., Power C., Pigott R. (1990) Molecular cloning of CD31, a putative intercellular adhesion molecule closely related to carcinoembryonic antigen. J. Exp. Med. 171, 2147–2152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Newton J. P., Hunter A. P., Simmons D. L., Buckley C. D., Harvey D. J. (1999) CD31 (PECAM-1) exists as a dimer and is heavily N-glycosylated. Biochem. Biophys. Res. Commun. 261, 283–291 [DOI] [PubMed] [Google Scholar]
- 62. Sun Q. H., DeLisser H. M., Zukowski M. M., Paddock C., Albelda S. M., Newman P. J. (1996) Individually distinct Ig homology domains in PECAM-1 regulate homophilic binding and modulate receptor affinity. J. Biol. Chem. 271, 11090–11098 [DOI] [PubMed] [Google Scholar]
- 63. Leavesley D. I., Oliver J. M., Swart B. W., Berndt M. C., Haylock D. N., Simmons P. J. (1994) Signals from platelet/endothelial cell adhesion molecule enhance the adhesive activity of the very late antigen-4 integrin of human CD34+ hemopoietic progenitor cells. J. Immunol. 153, 4673–4683 [PubMed] [Google Scholar]
- 64. Tanaka Y., Albelda S. M., Horgan K. J., van Seventer G. A., Shimizu Y., Newman W., Hallam J., Newman P. J., Buck C.A., Shaw S. (1992) CD31 expressed on distinctive T cell subsets is a preferential amplifier of β1 integrin-mediated adhesion. J. Exp. Med. 176, 245–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Berman M. E., Muller W. A. (1995) Ligation of platelet/endothelial cell adhesion molecule 1 (PECAM-1/CD31) on monocytes and neutrophils increases binding capacity of leukocyte CR3 (CD11b/CD18). J. Immunol. 154, 299–307 [PubMed] [Google Scholar]
- 66. Chiba R., Nakagawa N., Kurasawa K., Tanaka Y., Saito Y., Iwamoto I. (1999) Ligation of CD31 (PECAM-1) on endothelial cells increases adhesive function of αvβ3 integrin and enhances β1 integrin-mediated adhesion of eosinophils to endothelial cells. Blood 94, 1319–1329 [PubMed] [Google Scholar]
- 67. Berman M. E., Xie Y., Muller W. A. (1996) Roles of platelet/endothelial cell adhesion molecule-1 (PECAM-1, CD31) in natural killer cell transendothelial migration and β2 integrin activation. J. Immunol. 156, 1515–1524 [PubMed] [Google Scholar]
- 68. Piali L., Albelda S. M., Baldwin H. S., Hammel P., Gisler R. H., Imhof B. A. (1993) Murine platelet endothelial cell adhesion molecule (PECAM-1)/CD31 modulates β2 integrins on lymphokine-activated killer cells. Eur. J. Immunol. 23, 2464–2471 [DOI] [PubMed] [Google Scholar]