

Background: Peroxisomal proliferator activator receptor γ coactivator (PGC)-1α induces both mitochondrial biogenesis and angiogenesis in skeletal muscle.
Results: Hypoxic induction of an alternative spliced form of PGC-1α induces only angiogenesis in skeletal muscle, and not mitochondrial biogenesis.
Conclusion: Alternative splicing of PGC-1α explains how PGC-1α achieves specific induction of angiogenesis during hypoxia.
Significance: The findings suggest novel ways to induce angiogenesis in muscle without simultaneously inducing mitochondrial biogenesis.
Keywords: Angiogenesis, Hypoxia, Hypoxia-inducible Factor (HIF), Metabolism, Mitochondria, RNA Splicing, PGC-1, PGC-1alpha
Abstract
The transcriptional coactivator peroxisome proliferator-activator receptor γ coactivator (PGC)-1α is required for full hypoxic induction of vascular endothelial growth factor (VEGF) in skeletal muscle cells. Under normoxic conditions, PGC-1α also strongly induces mitochondrial biogenesis, but PGC-1α does not activate this program under hypoxic conditions. How this specificity is achieved is not known. We show here that hypoxia specifically induces alternatively spliced species encoding for truncated forms of PGC-1α: NT-PGC-1α and PGC-1α4. NT-PGC-1α strongly induces VEGF expression, whereas having little effect on mitochondrial genes. Conditioned medium from cells expressing NT-PGC-1α robustly induces endothelial migration and tube formation, hallmarks of angiogenesis. Transgenic expression of PGC-1α4 in skeletal muscle in mice induces angiogenesis in vivo. Finally, knockdown of these PGC-1α isoforms and hypoxia-inducible factor-1α (HIF-1α) abrogates the induction of VEGF in response to hypoxia. NT-PGC-1α and/or PGC-1α4 thus confer angiogenic specificity to the PGC-1α-mediated hypoxic response in skeletal muscle cells.
Introduction
Skeletal muscle is uniquely adaptable to extracellular physiological cues. For example, endurance exercise triggers mitochondrial biogenesis and neovascularization, ultimately improving fatigue resistance; nerve stimulation controls fiber type composition and maintains pro-growth signals; and ischemia triggers complex pathways, including a robust response to hypoxia. The many molecular pathways that regulate these processes are only beginning to be understood.
The transcription coactivator PGC-1α4 is induced by exercise, nerve stimulation, and hypoxia in skeletal muscle (1). It is regarded as a crucial regulator of oxidative metabolism in muscle (2). PGC-1α binds to and augments the activity of several transcription factors at the promoters of nuclear-encoded genes. Co-activation of nuclear respiratory factor (NRF)-1 and NRF-2 induces the expression of nuclear-encoded mitochondrial genes, including almost all genes involved in oxidative phosphorylation. At the same time, PGC-1α indirectly regulates mitochondrial DNA (mtDNA) replication and transcription via increased expression of mitochondrial transcription factor A (TFAM) and other nuclear-encoded factors. PGC-1α thus coordinates metabolic gene expression in both the nuclear and the mitochondrial genomes. Transgenic expression of PGC-1α in muscle induces functional mitochondrial biogenesis, leading to decreased muscle fatigue and increased running endurance of the mice (3, 4).
We recently showed that PGC-1α also dramatically induces angiogenesis, thereby coordinating oxygen/fuel delivery (via vessels) with their consumption (in mitochondria) (5, 6). PGC-1α expression is induced by hypoxia in muscle cells and is required for full hypoxic induction of angiogenic genes such as VEGF. PGC-1α induces VEGF by co-activating the transcription factor estrogen-related receptor α (ERRα) on a novel enhancer located in the first intron of the VEGF gene. Transgenic expression of PGC-1α in muscle induces dramatic neovascularization, and these same mice are protected from muscle ischemic injury (5). Conversely, deletion of PGC-1α in skeletal muscle prevents exercise-mediated angiogenesis (7).
These findings, however, raised a perplexing question. Hypoxia leads to the induction of VEGF and other hypoxic genes, but does not lead to the induction of mitochondrial genes. If PGC-1α is induced by hypoxia, and if PGC-1α is a potent activator of mitochondrial genes, then why are these genes not induced during hypoxia? We thus hypothesized that a specific form of PGC-1α protein must confer this specificity during hypoxia. Zhang et al. (8) recently reported the existence in liver and brown fat of an alternatively spliced biologically active truncated isoform of PGC-1α, termed NT-PGC-1α. Alternative splicing of the exon 6/7 boundary leads to a missense splicing event, the encoding of a premature stop codon, and a protein product less than one-half the size of full-length PGC-1α. Ruas et al. (9) demonstrated that transcription of this splice variant is likely initiated from an alternative promoter, and named this alternative messenger mRNA PGC-1α4. Neither study reported on the effects of these isoforms on angiogenic programs. PGC-1α4 and NT-PGC-1α differ in their N-terminal 12 and 16 amino acids, respectively. The remaining 250 amino acids are identical between both proteins and retain the sequences necessary for binding to ERRα (10), but not those required for binding to NRF-1 and NRF-2 (11). We thus reasoned that NT-PGC-1α and PGC-1α4 would likely induce VEGF and angiogenic genes more robustly than mitochondrial genes. We further hypothesized that alternative splicing at the exon 6/7 boundary of PGC-1α may be specifically induced by hypoxia, thus conferring specificity to the PGC-1α hypoxic response.
EXPERIMENTAL PROCEDURES
Mice and Cells
All animal experiments were performed according to procedures approved by the Beth Israel Deaconess Medical Center Institutional Animal Care and Use Committee (IACUC). HIF-1α floxed mice were from The Jackson Laboratory (007568), and both the PGC-1α4 transgenic mice, and the PGC-1α KO mice were from Dr. Spiegelman. Isolation and culture of primary skeletal myocytes were performed on entire hindlimb muscle after collagenase/dispase digestion, as described previously (15). Primary myocytes were differentiated in DMEM (5% horse serum).
Generation of ad-NT-PGC-1α
A blunt-end NT-PGC-1α product was generated by PCR from FL-PGC-1α using forward primer consisting of first 32 bp of PGC-1α and reverse primer consisting of 23 bp of homology to the 5′ end of exon 6 plus an additional 13 bp coding for the additional Leu-Phe-Leu-STOP sequence of NT-PGC1α (forward, CACCGGATCCCCGGATCACCACCATGGCTTGG; reverse, GCCTCGAGTTATAAAAACAAATTTGGTGACTGTGG), and cloned into pAD/CMV/V5-DEST (Gateway).
Real-time PCR
Total RNA was isolated from cultured cells using Turbocapture (Qiagen). Samples for real-time PCR analyses were reverse-transcribed (Applied Biosystems), and quantitative real-time PCRs were performed on the cDNAs in the presence of fluorescent dye (SYBR green; Bio-Rad). Relative expression levels were determined using the comparative cycle threshold method. The quantitative real-time PCR (qPCR) data were normalized against control primers for hprt, tbp, and 36B4.
Endothelial Cell Migration Assay
Differentiated myotubes in 24-well plates were infected with adenovirus expressing GFP, FL, or NT-PGC-1α for 34 h. BSA or soluble Flt1 (100 ng/ml; R&D Systems) was added to the medium for 12 h. Then, 5 × 104 cells of HUVECs were plated on the upper compartment of Transwell inserts (8.0-μm pore size) prewarmed with EBM-2 medium for 16 h at 37 °C. HUVEC migration to the lower compartment of Transwell inserts was measured after 12 h. Migrated HUVECs were fixed with 4% paraformaldehyde in PBS for 20 min at room temperature, and cells that remained in the upper compartment were removed with cotton swabs. Cells were blocked with 5% BSA in PBS-Tween 20 (PBST; 0.2% Tween 20) and stained with phalloidin-FITC in PBST for 4 h to visualize filamentous actin. Transwell inserts were washed three times in PBST and mounted onto slides with DAPI mounting medium.
Tube Formation Assay
HUVEC cells were starved for 16 h in 0.5% FBS, EBM-2 medium. A 96-well plate was then coated with 50 μl of Matrigel/well. 1.5 × 104 HUVECs were seeded per well in 100 μl of starvation medium. 1 h after seeding, the medium was removed and replaced with 100 μl of either NT-PGC-1α or GFP conditioned medium. 6 h later, the HUVECs were examined under a light microscope at 4× magnification. Photographs were taken, and total tube length per field was quantified using ImageJ.
Histological Analysis
CD31 staining quantification of capillaries was performed computationally, using ImageJ, on three random fields chosen from transverse sections from the tibialis anterior muscle. All quantifications were performed blindly.
Oxygen Consumption Rate
Primary satellite cells were plated at 2 × 105 cells/well and then differentiated into myotubes. Oxygen consumption rates (pmol/min) were assessed using a XF Flux Analyzer (Seahorse Biosciences), at baseline and after the addition of the uncoupler 2,4-dinitrophenol, to determine maximum respiration rate.
Statistics
Data are presented as means ± S.E. Statistical analysis was performed with Student's t test. p values of less than 0.05 were considered statistically significant.
RESULTS
Alternative Splicing of PGC-1α Is Preferentially Induced by Hypoxia in Skeletal Muscle Cells
Oligonucleotides were generated to amplify specifically exon 6/7 alternative splice forms of PGC-1α, including both NT-PGC-1α and PGC-1α4, by qPCR (Fig. 1A). qPCR of plasmids encoding each specific isoform demonstrated the specificity of the qPCR primers (Fig. 1B). To evaluate expression of these spliced forms in skeletal muscle, differentiated primary murine skeletal myotubes were used. We have shown previously that hypoxia induces PGC-1α in these cells (5), but those results did not evaluate which isoform of PGC-1α was induced. Primary myoblasts were thus made to differentiate in cell culture, and were then treated with 0.5% oxygen for 16 h, versus normoxic control. Activation of a hypoxic program was confirmed by measuring expression of VEGF, Glut1, and PDK1, genes well known to be induced by hypoxia (Fig. 1C). As shown in Fig. 1D, hypoxia induced the expression of alternatively spliced PGC-1α 8-fold. The expression of FL-PGC-1α, on the other hand, was unaltered by hypoxic treatment. Hypoxia thus preferentially induces alternatively spliced PGC-1α expression in skeletal muscle cells.
FIGURE 1.

Alternative splicing of PGC-1α is preferentially induced by hypoxia in skeletal muscle cells. A, schematic of FL-PGC-1α, NT-PGC-1α, and PGC-1α4. Locations of specific primers for qPCR are indicated. Alternatively spliced (Alt) primers identify the exon 6/7 alternative splicing event in both NT-PGC-1α and PGC-1α4. B, specific qPCR amplification of FL-PGC-1α and Alt-PGC-1α from FL-PGC-1α and NT-PGC-1α, respectively. C, induction of VEGFA, PDK1, and GLUT1 in primary myotubes treated with 0.5% oxygen for 16 h. D, specific induction of NT-PGC-1α expression by hypoxia in myotubes treated as in C. *, p < 0.05 by Student's t test.
NT-PGC-1α Only Weakly Induces Mitochondrial Genes in Skeletal Muscle Cells
To evaluate the function of NT-PGC-1α in muscle cells, an adenovirus that expresses NT-PGC-1α was generated. This virus was then used to infect primary myotubes. To be sure that any effects are independent of full-length PGC-1α present in myotubes, primary myoblasts were isolated from PGC-1α−/− mice and made to differentiate in culture. Infection of these primary myotubes lacking endogenous PGC-1α led to robust expression of NT-PGC-1α, as measured by Western blotting (Fig. 2A). The effects of infection with adeno-NT-PGC-1α were next compared with those of adeno-FL-PGC-1α, versus GFP-only control. Multiplicities of infection of each adenovirus were chosen that achieved equivalent, and moderate, induction of FL and NT-PGC-1α (Fig. 2B). As shown in Fig. 2C, FL-PGC-1α led to robust induction of various nuclear genes encoding important components of the mitochondrion, including cytochromes (cycs), electron transport chain complex IV (cox5b), ATP synthase (Atp5o), the TCA cycle (cs), and mitochondrial replication and transcription (TFAM). In sharp contrast, NT-PGC-1α only marginally induced these same genes (Fig. 2C). Measurements by Western blotting of mitochondrial complex III and V proteins revealed similarly poor induction by NT-PGC-1α (Fig. 2D). NT-PGC-1α thus only weakly induces mitochondrial genes in skeletal myotubes in culture. Finally, to directly and sensitively test the effects of NT-PGC-1α on mitochondrial function, NT-PGC-1α was more strongly overexpressed in primary myotubes, and cellular respiration was measured, using a Seahorse extracellular flux analyzer. As shown in Fig. 2E, despite the marked 5-fold greater overexpression of NT-PGC-1α versus FL-PGC-1α in this experiment, oxygen consumption rate was only moderately induced by NT-PGC-1α, roughly only half as much as that induced by FL-PGC-1α.
FIGURE 2.

NT-PGC-1α only weakly induces mitochondrial genes in skeletal muscle cells. A, Western blot showing protein expression of NT-PGC-1α 48 h after infection of PGC-1α−/− myotubes with adenovirus expressing NT-PGC-1α. B, adenoviral multiplicities of infection were chosen to achieve matched overexpression of NT-PGC-1α and FL-PGC-1α expression. C and D, the expression of the indicated mitochondrial genes (C) and proteins (D) is induced in primary murine PGC-1α−/− myotubes by adenovirus encoding FL-PGC-1α, but only weakly by NT-PGC-1α. E, oxygen consumption rate (OCR) in primary myotubes infected with adenovirus expressing full-length PGC-1α, NT-PGC-1α, and GFP control. Left, gene expression; right, basal and maximal oxygen consumption rate. *, p < 0.05 by Student's t test; §, p < 0.05 versus PGC-1α.
NT-PGC-1α Strongly Induces VEGF and a Pro-angiogenic Phenotype in Muscle Cells
Infection of PGC-1α−/− myotubes with FL-PGC-1α led to a 3–4-fold induction of the canonical pro-angiogenic peptide VEGF (Fig. 3A), as we have shown previously (5). We have shown that the induction of VEGF occurs via coactivation of the nuclear receptor ERRα (5), and indeed, PGC-1α induces the expression of ERRα itself as well (Fig. 3A). Infection with ad-NT-PGC-1α achieved more than double the induction of VEGF that was seen with FL-PGC-1α (Fig. 3A). This marked induction of VEGF coincided with only minimal induction of mitochondrial genes (Fig. 2C). The ratio of induction of VEGF to cycs by NT-PGC-1α is thus more than 5-fold higher than by FL-PGC-1α (Fig. 3B). NT-PGC-1α directly coactivates ERRα in GAL-luciferase assays Fig. 3C), consistent with its ability to induce VEGF (5) and its known ability to bind peroxisome proliferator-activated receptor α (PPARα) (8).
FIGURE 3.

NT-PGC-1α strongly induces VEGF and a pro-angiogenic phenotype in muscle cells. A and B, infection of primary murine PGC-1α−/− myotubes with virus encoding NT-PGC-1α strongly induces VEGF, measured by qPCR and expressed as absolute induction (A) or ratio of induction of VEGF to that of cycs (B). C, NT-PGC-1α coactivates a GAL4-ERRα fusion protein to activate expression of a luciferase reporter construct. D, NT-PGC-1α expression in myotubes plated in the inferior chamber of a modified Boyden chamber (see diagram at right) robustly induces migration of adjacent endothelial cells in the top chamber, and this process is inhibited by the addition of sFlt1, a potent inhibitor of VEGF. Numbers of endothelial cells per high power field that have migrated toward the myotubes are indicated in the y axis. E, conditioned medium (CM) from primary murine myotubes infected with adenovirus expressing NT-PGC-1α promotes formation of spontaneous tube-like structures by endothelial cells, as compared with control myotubes infected with GFP-encoding adenovirus. *, p < 0.05 by Student's t test.
Endothelial migration and tube formation are hallmarks of angiogenesis. As shown in Fig. 3D, conditioned medium from skeletal muscle cells that overexpress FL-PGC-1α led to a 3-fold induction in the migration of endothelial cells in a Transwell assay, as we have shown before (5). Overexpression of NT-PGC-1α more than quintupled that induction, achieving 15-fold induction of endothelial migration over baseline (Fig. 3D). The addition of soluble fms-like tyrosine kinase 1 (sFlt1), a potent inhibitor of VEGF, completely abrogated the increase in endothelial migration (Fig. 3D), indicating that NT-PGC-1α induces endothelial migration largely via the secretion of VEGF. Lastly, conditioned medium from myotubes expressing NT-PGC-1α strongly stimulated the formation of tubes by endothelial cells (Fig. 3E). Together, these data indicate that, as compared with FL-PGC-1α, NT-PGC-1α strongly favors the activation of a pro-angiogenic program over a pro-mitochondrial program in muscle cells.
PGC-1α4 Induces Angiogenesis in Vivo
Ruas et al. (9) recently generated transgenic mice that express PGC-1α4 in skeletal muscle, using the MEF2C enhancer/myogenin promoter. We thus used these transgenic mice to test whether an alternatively spliced, truncated form of PGC-1α induces VEGF and angiogenesis in vivo. Tibialis anterior muscles were isolated from 12-week-old animals, and total RNA was prepared, and gene expression measured by qPCR. As shown in Fig. 4A, transgenic expression of PGC-1α4 induced total PGC-1α expression ∼5-fold. Despite this induction, the expression of mitochondrial genes was only marginally induced (Fig. 4B). On the other hand, VEGF expression was significantly induced in these animals (Fig. 4C). PGC-1α4 thus favors the activation of a pro-angiogenic program over a pro-mitochondrial program in intact muscle. Thin frozen sections from the tibialis anterior muscle were next stained with antibodies against CD31, an endothelial specific marker that identifies capillaries. As shown in Fig. 4B, capillary density, expressed as number of capillaries per high powered field, or as capillaries per myofiber, was doubled in transgenic animals. PGC-1α4 thus induces angiogenesis in vivo.
FIGURE 4.

PGC-1α4 induces angiogenesis in vivo. A, induction of total and PGC-1α4, but not other PGC-1 family members, as determined by qPCR, in muscle of transgenic animals overexpressing PGC-1α4 specifically in skeletal muscle. B and C, PGC-1α4 transgenic animals induce oxidative phosphorylation (OXPHOS) genes poorly in skeletal muscle (B), whereas significantly inducing angiogenic genes (C), as determined by qPCR. D–F, induction of capillary density in tibialis anterior muscle from transgenic animals, as determined by staining for the endothelial-specific marker CD31. D–F, representative images (D) and quantification of capillaries/fiber (E) and capillaries/high power field (HPF) (F). *, p < 0.05 by Student's t test.
Alternatively Spliced PGC-1α Mediates Hypoxic Induction of VEGF in Muscle Cells
The transcription factor HIF-1α mediates a large part of the transcriptional response to hypoxia in many, if not most, cells (12). To separate effects mediated by PGC-1α from those mediated by HIF-1α, we generated cells that lack HIF-1 activity. Primary myoblasts were isolated from animals carrying floxed alleles of HIF-1α. The myoblasts were then infected with adenovirus encoding for the Cre recombinase, leading to inactivation of the HIF-1α locus. The cells were then stably infected with lentivirus expressing shRNA targeted against HIF-1β (ARNT (aryl hydrocarbon receptor nuclear translocator)), the obligate heterodimer of both HIF-1 and HIF-2 transcription factors. Finally, the cells were made to differentiate into myotubes. As shown in Fig. 5A, HIF-1α and HIF-1β expression was largely absent in these cells. Despite strongly reduced HIF-1 expression, however, exposing the cells to 0.2% oxygen still led to significant induction of VEGF (Fig. 5B). Hypoxic induction of VEGF can thus occur HIF-independently in these cells.
FIGURE 5.

Alternatively spliced PGC-1α mediates hypoxic induction of VEGF in muscle cells. A, efficient reduction of expression of HIF-1α and HIF-1β by combined shHIF-1β expression and adenovirally mediated Cre expression in primary murine HIF-1α lox/lox myotubes. B, hypoxia induces VEGF and hypoxic genes in primary murine HIF-1α lox/lox myotubes pretreated with shHIF-1β and adeno-Cre despite severely reduced HIF-1 (as shown in A). C, hypoxia induces exon 6/7 alternatively spliced (Alt) PGC-1α despite severely reduced HIF-1, as in B. D, shAlt-PGC-1α abrogates the remaining hypoxia response in myotubes with reduced HIF-1 activity treated as in B. *, p < 0.05 by Student's t test.
We have shown previously that PGC-1α is required for maximal induction of VEGF expression by hypoxia in muscle cells (5). The data presented here suggest that alternative spliced forms of PGC-1α may be responsible for this effect. Consistent with this notion, Alt-PGC-1α was induced by hypoxia in HIF− cells (Fig. 5C) as robustly as in wild type cells (Fig. 1). To test the role of these isoforms directly, lentivirus was generated that encodes for shRNA to target specifically the alternatively spliced PGC-1α transcripts, while leaving the FL-PGC-1α transcript unaffected. The myoblasts were then made to differentiate into myotubes and exposed to 0.2% oxygen, versus normoxia control. As shown in Fig. 5D, sh-Alt-PGC-1α completely abrogated the induction of VEGF in response to hypoxia, whereas sh-scrambled control had no effect. Truncated forms of PGC-1α thus mediate hypoxic induction of VEGF in skeletal muscle cells. The data also indicate that the remnant hypoxic induction was not dependent on low grade HIF activity (either HIF-1 or HIF-2) because it was abrogated by shRNA directed at PGC-1α.
DISCUSSION
We show here that N-truncated forms of PGC-1α, as compared with FL-PGC-1α, preferentially induce an angiogenic program over a mitochondrial program in skeletal muscle cells. PGC-1α induces mitochondrial genes in large part via the coactivation of NRF-1 and NRF-2. The region of PGC-1α that binds to NRF-1 and NRF-2 remains poorly defined, but almost certainly lies outside the peptide sequences retained in NT-PGC-1α and PGC-1α4 (8, 11). On the other hand, these PGC-1α isoforms retain the LXXLL motifs via which PGC-1α interacts with ERRα, and we show here that NT-PGC-1α can coactivate ERRα. We have shown previously that PGC-1α induces VEGF expression via coactivation of ERRα (5). We thus propose that the specificity of NT-PGC-1α and PGC-1α4 for the angiogenic program is achieved via specific binding to ERRα, but not NRFs (Fig. 6).
FIGURE 6.
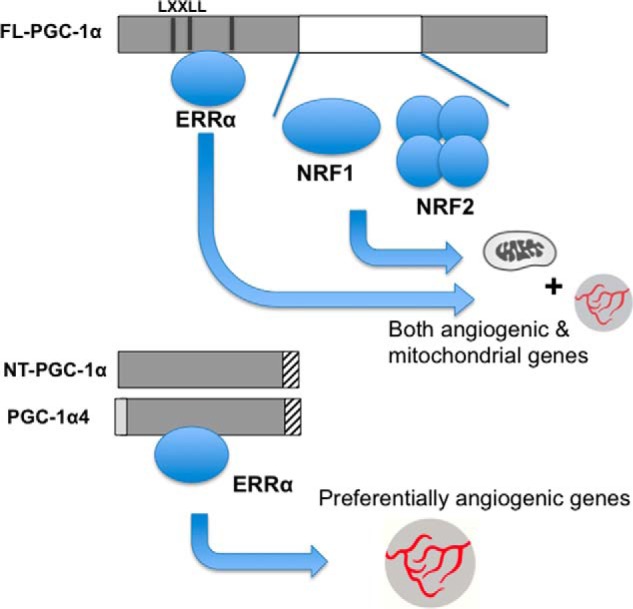
Model of how NT-PGC-1α and PGC-1α4 confer preference for angiogenic genes over mitochondrial genes. FL-PGC-1α binds to ERRα as well as NRFs, allowing for full induction of mitochondrial genes and angiogenic genes. NT-PGC-1α and PGC-1α4, on the other hand, bind only ERRα, which limits their ability to fully induce mitochondrial genes, whereas not affecting ability to induce angiogenic genes such as VEGF.
We also show here that alternatively spliced forms of PGC-1α are induced by hypoxia in muscle cells and that these isoforms mediate hypoxic induction of VEGF. It is important to note that the HIF-1 pathway also contributes to this induction. For this reason, experiments were conducted in the absence of HIF-1 activity (Fig. 4). Hypoxic induction of PGC-1α occurs independently of HIF-1 activity, and the induction of VEGF by PGC-1α also occurs independently of HIF-1. FL-PGC-1α has been proposed by others to induce VEGF indirectly via HIF-1, by inducing mitochondrial respiration, leading to elevated consumption of oxygen, local hypoxia, and HIF-1 activation (13). Our data suggest that this HIF-dependent mechanism is not at play with truncated forms of PGC-1α because mitochondrial genes are minimally induced. Muscle cells thus appear to activate two entirely separate pathways in response to hypoxia.
In general, prolonged hypoxia tends to repress the expression of mitochondrial complexes in most cell types. Our findings explain why hypoxic induction of PGC-1α does not induce mitochondrial genes, but the findings do not provide a mechanism for the active repression of these genes. The mechanisms remain incompletely understood and likely differ between cell types. PGC-1α isoforms are not known to repress transcriptional activity, and it thus seems unlikely that they would be involved in this active suppression of oxidative phosphorylation genes.
NT-PGC-1α largely resides in the cytoplasm (14). A significant amount of NT, however, must also be nuclear because NT-PGC-1α robustly coactivates ERRα and induces VEGF expression. It will thus be of interest to evaluate whether hypoxia alters cellular localization of NT-PGC-1α. Future studies will also investigate how the expression of alternatively spliced PGC-1α is induced by hypoxia. This could occur at a transcriptional level or during post-transcriptional splicing. We show here that the induction occurs independently of HIF-1 activity, possibly suggesting a post-transcriptional event, such as regulation of alternative splicing.
Cyclic AMP and PKA promote nuclear localization of NT-PGC-1α (14). We have shown previously that PGC-1α is required for exercise-induced angiogenesis and that this also occurs in part via cAMP signaling (7). Testing whether truncated forms of PGC-1α are the principal isoform that mediate exercise-induced angiogenesis will be of interest, but will likely require specific modification of the murine genome. Exercise-induced mitochondrial biogenesis, on the other hand, is unlikely to be mediated by NT-PGC-1α or PGC-1α4 because they appear to be weak inducers of mitochondrial genes in muscle cells and because PGC-1α appears to be dispensable for exercise-induced mitochondrial biogenesis (15).
Ruas et al. (9) have recently shown that PGC-1α4 regulates an IGF-dependent pro-growth program. It will in the future be of interest to determine comprehensively which gene loci are occupied by NT-PGC-1α, PGC-1α4, and FL-PGC-1α. It will also be of interest to determine whether the pro-growth program remains active under hypoxic conditions.
In summary, the data presented here highlight unique features of truncated forms of PGC-1α in skeletal muscle cells and explain the paradoxical observation that PGC-1α mediates induction of VEGF, but not mitochondrial genes, in response to hypoxia in these cells.
Acknowledgment
We thank Dr. Bruce Spiegelman for kindly providing PGC-1α4 transgenic mice.
Footnotes
- PGC-1α
- peroxisome proliferator-activator receptor γ coactivator-1α
- NRF
- nuclear respiratory factor
- HIF-1α
- hypoxia-inducible factor-1α
- ERRα
- estrogen-related receptor α
- HUVEC
- human umbilical vein endothelial cells
- qPCR
- quantitative real-time PCR
- FL
- full-length
- ad
- adenovirus
- Alt
- alternatively spliced
- NT
- N-terminal
- TCA
- tricarboxylic acid.
REFERENCES
- 1. Shoag J., Arany Z. Regulation of hypoxia-inducible genes by PGC-1α. Arterioscler. Thromb. Vasc. Biol. 30, 662–666 [DOI] [PubMed] [Google Scholar]
- 2. Rowe G.C., Jiang A., Arany Z. (2010) PGC-1 coactivators in cardiac development and disease. Circ. Res. 107, 825–838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Calvo J. A., Daniels T. G., Wang X., Paul A., Lin J., Spiegelman B. M., Stevenson S. C., Rangwala S. M. (2008) Muscle-specific expression of PPARγ coactivator-1α improves exercise performance and increases peak oxygen uptake. J. Appl. Physiol. 104, 1304–1312 [DOI] [PubMed] [Google Scholar]
- 4. Lin J., Wu H., Tarr P. T., Zhang C. Y., Wu Z., Boss O., Michael L. F., Puigserver P., Isotani E., Olson E. N., Lowell B. B., Bassel-Duby R., Spiegelman B. M. (2002) Transcriptional co-activator PGC-1 α drives the formation of slow-twitch muscle fibres. Nature 418, 797–801 [DOI] [PubMed] [Google Scholar]
- 5. Arany Z., Foo S.-Y., Ma Y., Ruas J. L., Bommi-Reddy A., Girnun G., Cooper M., Laznik D., Chinsomboon J., Rangwala S. M., Baek K.-H., Rosenzweig A., Spiegelman B. M. (2008) HIF-independent regulation of VEGF and angiogenesis by the transcriptional coactivator PGC-1α. Nature 451, 1008–1012 [DOI] [PubMed] [Google Scholar]
- 6. Patten I. S., Rana S., Shahul S., Rowe G. C., Jang C., Liu L., Hacker M. R., Rhee J. S., Mitchell J., Mahmood F., Hess P., Farrell C., Koulisis N., Khankin E. V., Burke S. D., Tudorache I., Bauersachs J., del Monte F., Hilfiker-Kleiner D., Karumanchi S. A., Arany Z. (2012) Cardiac angiogenic imbalance leads to peripartum cardiomyopathy. Nature 485, 333–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chinsomboon J., Ruas J., Gupta R. K., Thom R., Shoag J., Rowe G. C., Sawada N., Raghuram S., Arany Z. (2009) The transcriptional coactivator PGC-1α mediates exercise-induced angiogenesis in skeletal muscle. Proc. Natl. Acad. Sci. U.S.A. 106, 21401–21406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhang Y., Huypens P., Adamson A. W., Chang J. S., Henagan T. M., Boudreau A., Lenard N. R., Burk D., Klein J., Perwitz N., Shin J., Fasshauer M., Kralli A., Gettys T. W. (2009) Alternative mRNA splicing produces a novel biologically active short isoform of PGC-1α. J. Biol. Chem. 284, 32813–32826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ruas J. L., White J. P., Rao R. R., Kleiner S., Brannan K. T., Harrison B. C., Greene N. P., Wu J., Estall J. L., Irving B. A., Lanza I. R., Rasbach K. A., Okutsu M., Nair K. S., Yan Z., Leinwand L. A., Spiegelman B. M. (2012) A PGC-1α isoform induced by resistance training regulates skeletal muscle hypertrophy. Cell 151, 1319–1331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Takacs M., Petoukhov M. V., Atkinson R. A., Roblin P., Ogi F. X., Demeler B., Potier N., Chebaro Y., Dejaegere A., Svergun D. I., Moras D., Billas I. M. (2013) The asymmetric binding of PGC-1α to the ERRα and ERRγ nuclear receptor homodimers involves a similar recognition mechanism. PLoS One 8, e67810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wu Z., Puigserver P., Andersson U., Zhang C., Adelmant G., Mootha V., Troy A., Cinti S., Lowell B., Scarpulla R. C., Spiegelman B. M. (1999) Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 98, 115–124 [DOI] [PubMed] [Google Scholar]
- 12. Rowe G. C., El-Khoury R., Patten I. S., Rustin P., Arany Z. (2012) PGC-1alpha is dispensable for exercise-induced mitochondrial biogenesis in skeletal muscle. PloS one 7, e41817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Semenza G. L. (2007) Life with oxygen. Science 318, 62–64 [DOI] [PubMed] [Google Scholar]
- 14. O'Hagan K. A., Cocchiglia S., Zhdanov A. V., Tambuwala M. M., Cummins E. P., Monfared M., Agbor T. A., Garvey J. F., Papkovsky D. B., Taylor C. T., Allan B. B. (2009) PGC-1alpha is coupled to HIF-1alpha-dependent gene expression by increasing mitochondrial oxygen consumption in skeletal muscle cells. Proc Natl Acad Sci U S A 106, 2188–2193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chang J. S., Huypens P., Zhang Y., Black C., Kralli A., Gettys T. W. (2010) Regulation of NT-PGC-1alpha subcellular localization and function by protein kinase A-dependent modulation of nuclear export by CRM1. J. Biol. Chem. 285, 18039–18050 [DOI] [PMC free article] [PubMed] [Google Scholar]