

Background: Post-translational regulation of CTP:phosphoethanolamine cytidylyltransferase (Pcyt2) is largely unexplored.
Results: Pcyt2 isoforms are characteristically phosphorylated within a newly identified central linker segment not present in other cytidylyltransferases.
Conclusion: Pcyt2 phosphorylation influences cell growth and is regulated by conventional PKCs and serum deficiency.
Significance: The isoform-specific phosphorylation reveals a post-translational regulation of Pcyt2 that is distinct from other cytidylyltransferases.
Keywords: Lipid Synthesis, Phospholipid, Phospholipid Metabolism, Phosphorylation, Protein Kinase C (PKC), CTP:Phosphoethanolamine Cytidylyltransferase, Pcyt2, Ethanolamine Phospholipids, Protein Kinase C
Abstract
CTP:phosphoethanolamine cytidylyltransferase (Pcyt2) is the main regulatory enzyme for de novo biosynthesis of phosphatidylethanolamine by the CDP-ethanolamine pathway. There are two isoforms of Pcyt2, -α and -β; however, very little is known about their specific roles in this important metabolic pathway. We previously demonstrated increased phosphatidylethanolamine biosynthesis subsequent to elevated activity and phosphorylation of Pcyt2α and -β in MCF-7 breast cancer cells grown under conditions of serum deficiency. Mass spectroscopy analyses of Pcyt2 provided evidence for isoform-specific as well as shared phosphorylations. Pcyt2β was specifically phosphorylated at the end of the first cytidylyltransferase domain. Pcyt2α was phosphorylated within the α-specific motif that is spliced out in Pcyt2β and on two PKC consensus serine residues, Ser-215 and Ser-223. Single and double mutations of PKC consensus sites reduced Pcyt2α phosphorylation, activity, and phosphatidylethanolamine synthesis by 50–90%. The phosphorylation and activity of endogenous Pcyt2 were dramatically increased with phorbol esters and reduced by specific PKC inhibitors. In vitro translated Pcyt2α was phosphorylated by PKCα, PKCβI, and PKCβII. Pcyt2α Ser-215 was also directly phosphorylated with PKCα. Mapping of the Pcyt2α- and -β-phosphorylated sites to the solved structure of a human Pcyt2β showed that they clustered within and flanking the central linker region that connects the two catalytic domains and is a novel regulatory segment not present in other cytidylyltransferases. This study is the first to demonstrate differences in phosphorylation between Pcyt2 isoforms and to uncover the role of the PKC-regulated phosphorylation.
Introduction
Phosphatidylethanolamine (PE)3 and its alkenylacyl derivatives (PE plasmalogens) are essential membrane phospholipids implicated in the regulation of various cellular processes, including cell proliferation, signaling, autophagy, and apoptosis (1–5). PE phospholipids are synthesized de novo from ethanolamine and diacylglycerol/alkylacylglycerols through the CDP-ethanolamine-Kennedy pathway and by decarboxylation of phosphatidylserine in the mitochondria. The de novo Kennedy pathway is quantitatively the most important route for the biosynthesis of PE in mammalian cells (6). In this pathway, ethanolamine is first phosphorylated by ethanolamine kinase to phosphoethanolamine (P-Etn), which is then converted to CDP-ethanolamine (CDP-Etn) by CTP:phosphoethanolamine cytidylyltransferase (Pcyt2; also known as ET (7, 8) or ECT (9)). In the final step, CDP-ethanolamine:1,2-diacylglycerol ethanolamine phosphotransferase transfers P-Etn from CDP-Etn to DAG/alkylacylglycerol to produce PE/plasmalogens. We have recently reviewed the role of Pcyt2 in phospholipid homeostasis (10).
Recent studies using Pcyt2-deficient mouse models established a direct link between PE synthesis and triglyceride metabolism (11–13). Heterozygous Pcyt2 deletion (Pcyt2−/+) reduced the metabolic flux through the CDP-ethanolamine pathway and generated an excess of DAG. The unused DAG was redirected toward triglyceride synthesis, which also stimulated fatty acid synthesis and resulted in excessive accumulation of fat in multiple tissues. Consequently, Pcyt2+/− mice developed metabolic syndrome, characterized by adult-onset obesity, liver steatosis, hypertriglyceridemia, and insulin resistance (11). Liver-conditional knock-out mice (l-Pcyt2−/−), however, relied on mitochondrial phosphatidylserine decarboxylation to form liver PE. They also developed severe fatty liver phenotypes; however, they did not develop insulin resistance (13).
Complete Pcyt2 knock-out (Pcyt2−/−) mice die early during embryogenesis (2), showing that Pcyt2 and the CDP-ethanolamine pathway are essential for animal development. Pcyt2 is activated during muscle cell differentiation (14), and the importance of Pcyt2 for growth and development has also been demonstrated in several genome-wide studies (15–19). Pcyt2 is coexpressed with genes important for ES self-renewal and differentiation (15), and preimplantation embryos in culture conditions overexpress Pcyt2 (16). Pcyt2 is a target gene of histone deacetylase activity and was down-regulated by histone deacetylase and microRNA transcriptional repression during oligodendrocyte differentiation (17). Pcyt2 was up-regulated by NGF-initiated but not by IL6-initiated PC12 cell differentiation (18), and it is a candidate gene in the regulation of mouse brain and retinal development (19).
The importance of PE and Pcyt2 activity for tumor growth has also been well documented (1, 20–23). Breast cancer cells have reduced Pcyt2 activity and PE/plasmalogen levels relative to noncancerous cells (24), and a limited Pcyt2 activity has also been reported in lymphomatous liver (20) and ras-transformed 3T3 fibroblasts (21). Genome models of cancer metabolism predict Pcyt2 as a synthetic lethal gene in pairing with PS synthase-2 and an important drug target (22). Transcription factor TFAP2C plays an important role in breast cancer biology, and Pcyt2 is induced by TFAP2C in estrogen-responsive breast cancer cells MCF-7 (23). Pcyt2 is regulated by the early growth response protein 1 (EGR1) and NFκB (1). Pcyt2 is, however, still less expressed in MCF-7 cells compared with the nontumorigenic MCF-10A cells because of the reduced activity of EGR1 and increased activity of NFκB (24).
Unlike other members of the cytidylyltransferase family, Pcyt2 monomers contain two cytidylyltransferase catalytic folds, which we refer to as N-cat (N-terminal domain) and C-cat (C-terminal domain; Fig. 1). Mammals express two functional Pcyt2 proteins, the longer Pcyt2α and the shorter Pcyt2β, produced by alternative splicing of exon 7 (25). Exon 7 encodes an 18-amino acid peptide in Pcyt2α that is contained within the ∼75-residue linker region that joins the two catalytic domains and may contribute to distinct kinetic properties of the two isoforms (26). The longer isoform, Pcyt2α, is more active at higher substrate concentrations (P-Etn and CTP), whereas Pcyt2β is more active at lower substrate concentrations (26). The crystal structure of human Pcyt2β has been solved by x-ray diffraction (Protein Data Bank code 3elb).
FIGURE 1.
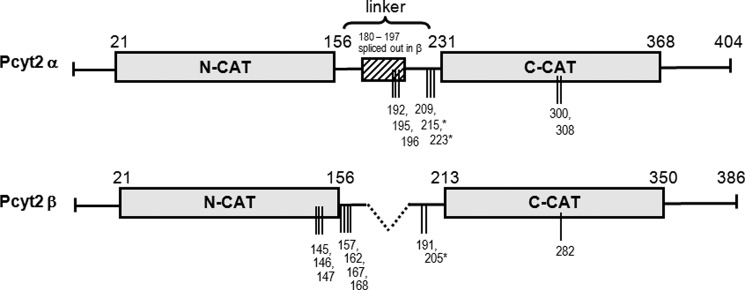
Domain structure of Pcyt2α and -β and phosphorylation sites identified in this study. Pcyt2β splicing results in the deletion of residues 180–197 from the central linker segment. The layout of the catalytic domains (N-cat and C-cat) is scaled proportionately, as is the linker segment. Phosphorylation sites identified by mass spectroscopy are represented by vertical lines, and * indicates phosphorylations at PKC consensus sites. Pcyt2α is longer and has 404 residues. Pcyt2β is the product of internal splicing of central 18-mer α-specific motif and it has 386 residues.
There is no information available regarding the post-translational regulation of Pcyt2. We have recently reported an unexpected stimulation of Pcyt2 activity and phosphorylation in MCF-7 cells when grown under conditions of serum deprivation (1). In this study, we identified the isoform-specific phosphorylation sites and mapped them to the inter-domain linker and flanking regions. Mainly nonphosphorylated in the presence of serum, Pcyt2α became phosphorylated in the linker segment, including the α-specific motif, and in the C-cat domain in the absence of serum. Pcyt2β was uniquely phosphorylated in the N-cat and proximal linker region and shared most of the phosphorylation with Pcyt2α within the C-cat domain. We show that the Pcyt2α isoform is regulated by protein kinase C (PKC) phosphorylation at two conserved sites within the C-terminal domain. Mutation analysis and phosphorylation studies establish that conventional PKCs are activators of Pcyt2 and contribute to the elevated Pcyt2 activity and PE synthesis required for stimulated cancer cell growth. To our knowledge, this is the first study to identify the phosphorylation sites and types of regulation of Pcyt2 by phosphorylation that is fundamentally different from other cytidylyltransferases.
EXPERIMENTAL PROCEDURES
Cell Culture and Transfections
MCF-7 human breast cancer cells were transfected with plasmids containing His6-tagged Pcyt2α and Pcyt2β as described (1). The Pcyt2 expression plasmids, pcDNA3.1ETα and pcDNA3.1ETβ, were produced from the full-length clones BC003473 for Pcyt2α and BU557590 for Pcyt2β and made in-frame with the V5/His6 tag peptide sequence at the C terminus. Approximately 60% confluent MCF-7 cells grown in a 100-mm dish were transfected with 24 μg of the pcDNA3.1ETα or pcDNA3.1ETβ using Lipofectamine (Invitrogen), and the cells were grown 24 h in serum-deficient minimum Eagle's medium or in the same media supplemented with 10% fetal bovine serum.
Pcyt2α and Pcyt2β Analysis with MALDI-Qq-TOF Mass Spectroscopy
Purification of His-tagged Pcyt2α and -β proteins overexpressed in MCF-7 cells was performed by nickel column chromatography as described (1). Purified His-tagged Pcyt2α and -β proteins were digested overnight at 37 °C both with trypsin and AspN in 50 mm ammonium bicarbonate, pH 8.2. The phosphopeptides from digested samples were enriched through specific interactions of phosphate groups with immobilized SwellGel gallium(III) disc, according to the phosphopeptide isolation kit (Pierce, catalog no. 89853). In brief, after adjusting the pH to 2–3, digested samples were added to SwellGel discs and bound phosphopeptides, eluted with 50 μl of 100 mm ammonium bicarbonate (acidified to pH <2.5 by acetic acid), and further purified using zip-tips (Millipore). Pcyt2α and -β phosphopeptide analysis was conducted using LCQ DECA XP ion-trap (Thermo Finnigan, San Jose, CA) and MALDI-Qq-TOF mass spectrometers (MDS SCIEX, Toronto, Canada) as described (26). The enriched phosphopeptide mixtures were separated on a Vydac reversed-phase column (Grace Vydac, Hesperia, CA) using 5% dilution of a mixture of 99.9% acetonitrile + 0.1% acetic acid, at the flow-rate of 10 μl/min for 5 min. The eluted peptides were then detected by MALDI-Qq-TOF mass spectrometer equipped with a low flow metal needle assembly and operating in a data-dependent mode (27). Peptide sequencing data were analyzed by Sequest software (Lisle, IL). Raw spectra were compared against the theoretical tandem mass spectra in the Sequest database to which the Pcyt2α and -β protein sequences were added on our initiative because they were not present in the database prior to this analysis (27).
Cloning and Expression of Pcyt2 Phosphorylation Mutants
To prepare the S215A, S223A, and S215A,S223A mutants, we used the QuikChange II site-directed mutagenesis kit (Stratagene). We designed a specific pair of primers to introduce an S215A mutation into V5-His6-tagged Pcyt2α (S215A forward, 5′-gtgtcccagtttctacagacagccca-gaagatca-3′; S215A reverse, 5′-tgatcttctgggctgtctgtag-aaactgggacac-3′) and a specific pair of primers to generate the S223A mutant (S223A forward, 5′-atcatccagtttgctgctgggaaggagcccc-3′; S223A reverse, 5′-ggggctccttcccagcagcaaactggatgat-3′). Subsequently, the S215A plasmid was used as a template to introduce an additional mutation to obtain the double mutant (S215A,S223A), using S223A-forward- and S223A-reverse-specific primers. PCR amplification was carried out using Pfu Ultra polymerase (Stratagene). The integrity of mutants was verified by sequencing and expression.
Effect of Pcyt2α Mutations on Enzyme Activity and Cell Growth
The enzyme activity of the cells transfected with plasmids containing V5 and His-tagged versions of the wild-type Pcyt2α or αS215A, αS223A, and αSer215A,S223A Pcyt2α mutants was assayed as described (1). We used 50 μg of the cell lysate in 100 μl of the reaction mixture of 10 mm Tris-HCl, pH 7.8, 5 mm MgCl2, 5 mm dithiothreitol, 2 mm CTP, 0.01 μCi of [14C]P-Etn, 1 mm unlabeled P-Etn. The [14C]CDP-Etn product formed after 15 min was separated from the [14C]P-Etn substrate on silica gel G plates by methanol, 0.5% NaCl, ammonia (50:50:5 v/v/v), visualized by 0.1% ninhydrin in butanol, and analyzed by scintillation counting. Protein content was determined using the bicinchoninic acid protein assay (Pierce).
Cell proliferation of MCF-7 cells transfected with Pcyt2α or αS215A, αS223A, and αS215A,S223A Pcyt2α mutants was monitored using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay. Cells (4 × 104) were plated in 96-well plates; 24 h after transfection in serum-free medium, cells were incubated with 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (0.05 mg/ml) for 3 h at 37 °C. Formed crystals of formazan were dissolved using 50 μl of DMSO, and the A570 nm was measured using a microplate reader.
Furthermore, the induction of cell survival by autophagy was examined. Content and lipidation of the autophagy-specific marker LC3 was measured by Western blotting as described previously (5).
Treatments with Phorbol Esters and PKC Inhibitors
Serum-starved MCF-7 cells were treated with 25 nm phorbol 12-myristate 13-acetate (PMA, Sigma) for 0, 0.5, 1, and 2 h. At the end of treatment periods, cells were homogenized in a lysis buffer and centrifuged to clear the lysates, and supernatants were subjected to immunoprecipitation using Pcyt2(α+β) antibody (1). The precipitated proteins were separated on SDS-PAGE, transferred onto PVDF membrane, and probed with Ser(P) and Pcyt2(α+β) antibodies. The cell lysates were also used for determination of total Pcyt2 activity as described above. In a second set of experiments, serum-starved cells were treated for 2 h with 25 nm PMA alone or together with the PKC inhibitors, Gö6976 (50 nm) or myristoylated EGF-R fragment 651–658, (50 nm) (both from Calbiochem). The Pcyt2 activity and the analyses using anti-Pcyt2(α+β) and anti-Ser(P) were performed as above.
Radiolabeling with [14C]Ethanolamine
MCF-7 cells (106 cells/60-mm dish) were grown as described under “Cell Culture and Transfections.” Twenty four hours after transfection with plasmids containing His-tagged Pcyt2α, S215A, S223A, or S215A,S223A mutants, the cells were labeled for 1 h with [14C]ethanolamine (0.1 μCi/dish) in the presence of 50 μm unlabeled ethanolamine. Radiolabeled lipid- and water-soluble fractions were separated by the method of Bligh and Dyer (1). Total ethanolamine incorporation into PE was determined by evaporation of the entire chloroform phase under nitrogen, followed by the separation of PE from lyso-PE in a TLC system of chloroform/methanol/ammonia (65:35:5). Spots corresponding to PE were collected and subjected to liquid scintillation analysis.
In Vitro Transcription and Translation of Pcyt2
To obtain a nonphosphorylated Pcyt2 protein, an untagged Pcyt2α cDNA was prepared and translated in vitro using the PURExpress in vitro protein synthesis kit (New England Biolabs). This system produces a protein product devoid of post-translational modifications. The untagged Pcyt2α cDNA template was generated by PCR using the pcDNA.ETα plasmid. We amplified the entire open reading frame and the required flanking sequences, including the ribosomal binding site, but deleted the V5 and His tags using Pcyt2-specific primers (forward primer, 5′-taagaaggagatataccaatgatccggaacgggcacgg-3′-3′; reverse primer, 5′-tattcattagtcaatctcccctccaggcggcgcc-3′). In the second round of PCR, we added the T7 promoter at the 5′-end of Pcyt2 using the universal primer (5′-gaaattaatacgactcactatagggagaccacaacggtttccctctagaaataattttgtttaactttaagaaggagatatacca-3′) and the Pcyt2 reverse primer. Pcyt2 cDNA was purified using a gel extraction kit (Sigma) and 250 ng of the cDNA incubated with mixture I (PURExpress) at 37 °C for 1 h to generate Pcyt2α protein. The mixture was concentrated (60 min at 1,500 × g at 4 °C) using a Microcon YM-100 spin concentrator (Millipore Corp.) and mixed with 0.25 volumes of nickel-nitrilotriacetic acid-agarose beads (Qiagen) (mixture II) and incubated at 4 °C for 45 min to remove the translation assay components. Mixture II was then applied to a micro-spin column (Bio-Rad), and the unbound Pcyt2α protein was released by centrifugation (2 min at 1500 × g at 4 °C). Pcyt2α proteins from multiple reactions were combined and pooled samples applied in the in vitro PKC phosphorylation assays below.
In Vitro Pcyt2 Phosphorylation with PKCα, PKCβI, and PKCβII
Pcyt2α free from tags and post-translational modifications was subjected to phosphorylation with [γ-32P]ATP and either PKCα, PKCβI, or PKCβII (Enzo Life Sciences) (28). The reaction was carried out at 30 °C for 10 min in 100 μl of mixture containing 20 mm HEPES, pH 7.4, 10 mm MgCl2, 0.1 mm CaCl2, 100 μm ATP, 100 μg/ml phosphatidylserine, 20 μg/ml diacylglycerol, 0.03% Triton X-100, 1 μl of [γ-32P]ATP, and different aliquots (20 or 40 μl) of Pcyt2α protein. Reactions were initiated by adding 0.4 μg of PKC and terminated after 10 min by adding 20 μl of 1% BSA and 1 ml of 10% TCA. After 5 min of incubation on ice, the aliquots (40 μl) from each reaction were transferred to a nitrocellulose membrane on an aspirator funnel and washed three times with 2 ml of 5% TCA. Membranes were dried under vacuum and the γ-32P was incorporated into Pcyt2α protein determined by counting. The assays were performed in triplicate, and the background radioactivity for the reactions without Pcyt2α or PKC served as controls (28).
Statistical Analysis
All measurements were expressed as means ± S.D. from at least three independent experiments. GraphPad Prism 3.0 software (San Diego) was used to perform one- or two-way analysis of variance and Student's paired t test. Differences were considered statistically significant at p ≤ 0.05. Densitometry analysis was performed by Scion Image software (Scion Inc).
RESULTS
Pcyt2α and Pcyt2β Catalytic Domains and Phosphorylation Sites
Fig. 1 provides a simplified representation of the Pcyt2α and -β structures showing the position of the two catalytic (CT) domains. The two splice isoforms differ only in the length of the linker connecting the two CT domains as follows: 74 residues for the α isoform and 56 residues for Pcyt2β. Putative phosphorylation sites were deduced by the Scan-Prosite proteomic server (ExPASy-Swiss Institute of Bioinformatics). Details on how the specific phosphorylations shown in Fig. 1 were characterized are described below.
Pcyt2α and Pcyt2β Phosphorylation Is Distinct and Serum-dependent
In response to serum deficiency, Pcyt2 is activated by phosphorylation, leading to elevated synthesis of PE by the CDP-ethanolamine–Kennedy pathway (1). Contributions of separate isoforms to the increased Pcyt2 activity have not been investigated, and the mechanism behind the increased enzyme phosphorylation in serum-deficient MCF-7 cells remains unknown. In addition, because the predicted phosphorylation sites are conserved, the contribution of each isoform to total activity was difficult to establish. Therefore, we transfected MCF-7 cells separately with His-tagged Pcyt2α and Pcyt2β (1), cultured the cells under different serum conditions, and purified overexpressed proteins using a nickel-agarose column. The purified Pcyt2 proteins were digested with trypsin, and the phosphopeptides were enriched by column chromatography (27) and investigated by mass spectroscopy (MS). The final phosphorylation results obtained from multiple transfections and purifications are summarized in Table 1 and Fig. 1. The initial analysis of the MS data revealed several general features of Pcyt2 phosphorylations as follows. (i) Maximal phosphorylation of both isoforms was observed in the absence of serum as predicted by previous data (1). (ii) The general pattern of phosphorylation was very distinctive for Pcyt2α and -β. (iii) Phosphorylation was mostly at serine residues as indicated previously (1). (iv) Additional phosphorylations were identified at multiple threonine residues, specifically in Pcyt2β (Thr-146/Thr-147, and Thr-157), and one phosphorylation at a tyrosine residue in Pcyt2α (Tyr-308).
TABLE 1.
Pcyt2α and Pcyt2β phosphopeptides
His-tagged Pcyt2α and -β proteins expressed in MCF-7 cells in the presence (+) or the absence of serum (−) were purified and analyzed by Ion-Trap/MALDI-Qq-TOF MS. Shown are the specific positions of the phosphorylated amino acid residues in Pcyt2α and -β. The PKC-targeted sites are underlined and boldface. The asterisk indicates that a consecutive y or b ion spanning the potential site was absent, suggesting a lower confidence in determination.
| Peptides containing phosphorylation sites | Position | Phosphorylated | Pcyt2 | Serum |
|---|---|---|---|---|
| Residue TQGVpSpTpTDLVGRMLLVpTK | 141–158 | Ser-145; Thr-146, -147, -157 | β | − |
| AHHpSSQEMpSpSEYR | 159–171 | Ser-162, -167, -168 | β | − |
| pSQFLQTSQKIIQFASGKEPQPGE ‘ | 191–213 | *Ser-191 | β | + |
| pSQCPGGQSPWTGVpSQFLQTSQKIIQFA | 196–222 | *Ser-196, Ser-209 | α | − |
| PTPAGDTLSpSEVpSSQCPGGQSPWT | 183–206 | Ser-192, -195 | α | − |
| GVSQFLQTpSQKIIQFASGKE | 207–226 | Ser-215 | α | − |
| KIIQFApSGKEPQ | 199–210 | Ser-205 | β | +/− |
| KIIQFApSGKEPQ | 217–228 | Ser-223 | α | − |
| CRYVpSEVVIGAPYSV | 278–292 | Ser-282 | β | − |
| CRYVpSEVIGAPYSV | 296–310 | Ser-300 | α | +/− |
| RYVpSEVVIGAPpY | 297–308 | Ser-300, Tyr-308 | α | − |
Pcyt2β Is Uniquely Phosphorylated within the First Catalytic Domain
Strikingly distinct phosphorylation was established at the N-terminal catalytic domain (Fig. 1). Pcyt2α was not phosphorylated in the N-Cat domain under any experimental conditions. Pcyt2β, however, was phosphorylated at multiple residues within the N-Cat domain as follows: 26–152 and in the linker segment immediately following the N-Cat domain. All Pcyt2β N-terminal phosphorylations were identified under serum-deficient conditions (Table 1), when the enzyme activity was elevated (1). Pcyt2β was specifically phosphorylated at βSer-145 in the CKII consensus motif (141TQGVpSpTpTDLVGR152) and at three threonine residues βThr-146, -147 and -157 (Fig. 2 and Table 1). Most importantly, part of the 141–152 peptide belongs to the cytidylyltransferase signature motif 140RTQGVSTT147, which interacts with the enzyme substrate CTP. Three additional Pcyt2β-specific phosphorylations were detected at βSer-162 and βSer-167/βSer-168 (Table 1). Specific kinase(s) responsible for this phosphorylation were not predicted by the ScanProSite. There are, however, two records for the same Pcyt2 phosphorylated peptide in the MS database PhosphoSitePlus (βSer-162 and βSer-163).
FIGURE 2.

Identification of Pcyt2β-specific phosphorylation. Cells overexpressing V5/His-tagged Pcyt2β were grown in the presence and absence of serum for 24 h. Pcyt2β was isolated, and phospho-enriched peptides were analyzed by MALDI-Qq-TOF tandem mass spectroscopy. Panels are fragmentation patterns for the phosphopeptide species indicated at the top. A and B are spectra for two Pcyt2β peptides with specific phosphorylation at βSer-145, βThr-146 and -147, and βThr-157. The identified phosphorylations are located in Pcyt2β N-cat domain (Fig. 1) and were not retrieved from analyses on Pcyt2α. The “y1”-ion series corresponds to the C-terminal peptide fragmentation, and “b1”-ion series corresponds to the N-terminal peptide fragmentation. Amino acids corresponding to m/z are shown at the bottom of each spectrum. The specific ions (m/z) of the phosphorylated residues are indicated. Spectra are representatives of at least three independent experiments. The panels show results from samples obtained from cells without serum. The phosphopeptides are not detected in the presence of serum.
Pcyt2α Is Uniquely Phosphorylated at the α-Specific Motif
Generally, Pcyt2α was not phosphorylated in the presence of serum, with the exception of αSer-300 located in the C-cat domain, which was phosphorylated under both growth conditions (296CRYVpSEVVIGAPYS309; Table 1). The corresponding serine in Pcyt2β, Ser-282, was phosphorylated only in the absence of serum. Independent evidence for the phosphorylation of the αSer-300/βSer-282 exists in the PhosphoSitePlus database (Ser(P)-282) and likely represents a constitutive phosphorylation site. In the absence of serum, Pcyt2α was specifically phosphorylated at two serine residues αSer-192 and αSer-195 within the α-specific motif (192SpSEVpSSQCPG201 in Fig. 3A). The ScanProSite analyses did not identify a kinase that could be responsible for this phosphorylation.
FIGURE 3.
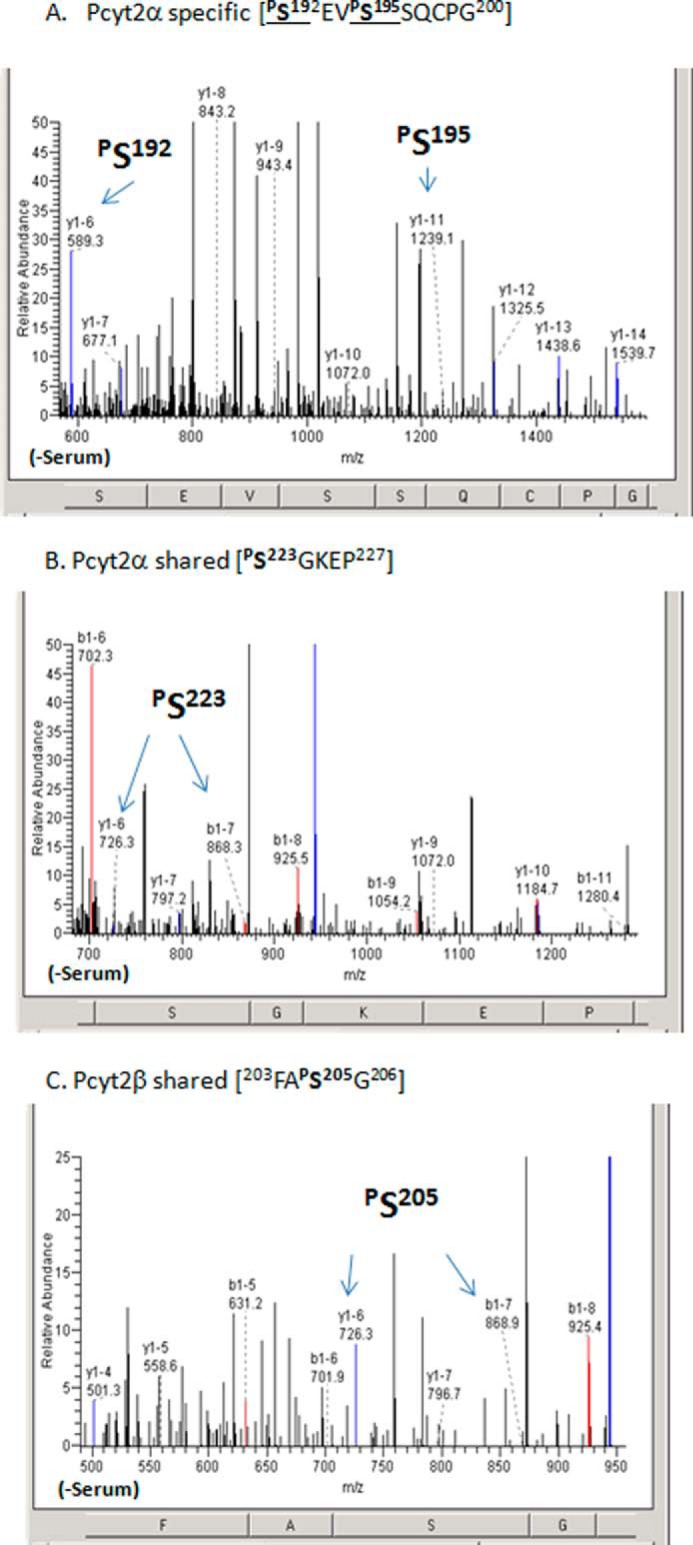
Identification of shared and Pcyt2α-specific phosphorylation. MS fragmentation analysis of phosphorylated peptides for Pcyt2α-specific phosphorylation at the linker peptide at αSer-192 and αSer-195 (A) and Pcyt2α and Pcyt2β shared phosphorylation within the linker segment and immediately upstream of the C-cat domain (C and B), which is also part of the PKC consensus site (Table 1). The specific ions (m/z) of the phosphorylated residues are indicated. Spectra are representative of at least three transfections and protein purifications.
Phosphorylation of Pcyt2α and -β within the Second Catalytic Domain
Unlike the N-cat domain, the C-cat domain was phosphorylated in both Pcyt2 isoforms with some specific variations. Phosphorylations at αSer-215 and αSer-223, which are part of PKC consensus sites 215SQK and 223SGK, were identified only under serum-deficient conditions when total phosphorylation and activity were enhanced (1). Pcyt2β was not phosphorylated at the corresponding βSer-197, but it was phosphorylated at the second PKC site βSer-205 under both growth conditions. Representative spectra identifying those identical phosphorylations (αSer-223 and βSer-205) are in Fig. 3, B and C. An additional common phosphorylation was detected at αSer-209/βSer-191 (Table 1), however, the specific kinase(s) for this site could not be identified.
From the composite of MS data shown in Table 1, we can conclude that most phosphorylations were associated with serum-deficient conditions when the enzyme was more active. Pcyt2 isoforms were phosphorylated at specific and mutual sites and therefore could experience both distinct and identical types of regulation. The most noticeable phosphorylation differences were present within the N-cat domain, which was exclusively phosphorylated in Pcyt2β. Pcyt2α, however, was phosphorylated at the α-specific motif spliced out in Pcyt2β. Both isoforms were phosphorylated within the second catalytic domain and shared the phosphorylation at one PKC site (αSer-223/βSer-205).
Phorbol Esters and PKC Inhibitors Regulate Pcyt2 Activity and Phosphorylation
We next determined whether the phosphorylation and activity of endogenous Pcyt2 could be modified by a PKC activator, PMA, and PKC-specific inhibitors, Gö6976 and EGF-R fragment 651–658. Serum-deficient MCF-7 cells were treated with PMA for 0–2 h followed by immunoprecipitation with Pcyt2(α+β) antibody and probing with Ser(P)- and Pcyt2(α+β) antibodies (Fig. 4, A and C) or used for determination of Pcyt2 activity (Fig. 4, B and D). Serine phosphorylation of Pcyt2(α+β) was increased by PMA addition, and both PKC inhibitors reduced the PMA-induced phosphorylation (Fig. 4, A and C). At the same time, Pcyt2 activity was elevated by PMA treatments, and Gö6976 or EGF-R attenuated the PMA-induced Pcyt2 activity (Fig. 4, B and D). These results established that endogenous Pcyt2 proteins were subjects of activation by phosphorylation that is regulated by PKC.
FIGURE 4.

In vivo and in vitro regulation of Pcyt2 with PKC. Phosphorylation (A) and catalytic activity (B) of endogenous Pcyt2 were analyzed after a short term treatment (0.5, 1, and 2 h) of serum-starved MCF-7 cells with the PKC activator PMA. PMA (25 nm)-induced Pcyt2 phosphorylation (C) and catalytic activity (D) were measured after 0.5 h of treatment with specific PKC inhibitors Gö6976 (50 nm) and EGF-R (50 nm); Pcyt2 immunoprecipitation and activity assays were performed as described under “Experimental Procedures.” Endogenous Pcyt2 was immunoprecipitated with Pcyt2total antibody, and phosphorylated Pcyt2 was identified by Ser(P) antibody. Pcyt2 enzyme activity was determined under the same conditions using [14C]phosphoethanolamine as a substrate. Pcyt2 activity is in picomoles/mg/protein ± S.D. at p < 0.05 (*), p <0.01 (**), and p <0.001 (***). Results are from three independent experiments. E, in vitro transcribed-translated Pcyt2α (produced untagged and devoid of modifications by phosphorylation) was phosphorylated with [32P]ATP and PKCα, PKC βI, or PKCβII. The incorporation of [32P]ATP was measured in triplicate (dpm ± S.D.) at two Pcyt2α concentrations (0.1 μg (hatched bars) and 0.2 μg (open bars). The 32P activity incorporated into Pcyt2α was compared with assays without Pcyt2 or PKC added. F and G, overexpressed V5/His-tagged Pcyt2α was purified from serum-treated MCF-7 cells (conditions established to produce minimal phosphorylation) and was subjected to the PKCα assays as above, however, using unlabeled ATP. The phosphorylated peptides were separated and characterized by MALDI-Qq-TOF tandem mass spectroscopy. Shown are the two m/z fragmentations for PKCα-treated Pcyt2α. Both patterns identify Pcyt2α phosphorylation at Ser-215.
Pcyt2α Is Phosphorylated with PKCα, PKC βI, and PKC βII
We next tested whether different PKC isozymes could directly phosphorylate Pcyt2α. To avoid the interference from other types of phosphorylation, we produced an untagged Pcyt2α protein by an in vitro transcription-translation system that generates a protein product devoid of post-translational modifications, including phosphorylation. The translated Pcyt2α protein was pooled from multiple reactions and used at two different concentrations in PKC phosphorylation assays with [32P]ATP and either PKCα, PKCβI, or PKCβII (Fig. 4E). All three PKC isoforms increased the incorporation of 32P into nascent Pcyt2α protein, and the incorporation was proportional to the concentration of Pcyt2α. Kinase-catalyzed phosphorylation was ranked in the order PKCα > PKCβII > PKCβI. These results established that Pcyt2α could be directly phosphorylated by all three conventional PKCs but is the best substrate for PKCα.
To further demonstrate that Pcyt2α and Pcyt2β are substrates for PKCα, the isoforms were overexpressed in serum-treated cells (conditions established to produce minimal Pcyt2 phosphorylation), purified, and subjected to phosphorylation with PKCα using unlabeled ATP. The PKCα-phosphorylated proteins were than characterized by MS. As indicated in Fig. 4, F and G, the PKCα-specific phosphorylation was detected on one of the predicted sites, αSer-215. Neither αSer-223 nor the same sites in Pcyt2β were phosphorylated by PKCα under the same conditions (data not shown). This site could be the target for PKCβ.
Mutations of Both PKC Sites Reduced Pcyt2α Phosphorylation and Activity
To probe whether the phosphorylation at the PKC-directed sites has a regulatory function, we created single (αS215A and αS223A) and double (αS215A,S223A) Pcyt2α mutants and performed transfections under serum-deficient conditions (conditions established to produce maximal phosphorylation). The overexpressed proteins were immunoprecipitated with Pcyt2 tag V5 antibody and probed with anti-Ser(P). As shown in Fig. 5A, the single mutations αS215A and αS223A similarly reduced Ser phosphorylation, with the double mutant αS215A,S223A showing an almost complete loss of phosphorylation. The activities of αS215A, αS223A, and αS215A,S223A mutants were reduced to ∼50, 8, and 36% of the wild-type Pcyt2α, respectively (Fig. 5B). In addition, as shown for the double mutant in Fig. 5C, the hypophosphorylation of the PKC double-site mutant could not be further modified with PMA or EGF-R (Fig. 5C). When the effect of various PMA concentrations on endogenous and overexpressed Pcyt2α was examined (Fig. 5D), PMA strongly stimulated endogenous activity with maximal 7-fold activation at 100 nm PMA in serum-deficient conditions. The activity of the overexpressed Pcyt2α was ∼4-fold above the endogenous level and showed no further activation with PMA (Fig. 5D). This is not surprising because the overexpressed Pcyt2α was already highly phosphorylated under serum-deficient conditions, including both PKC sites, as established by multiple MS analyses described earlier.
FIGURE 5.

Characterization of PKC-directed phosphorylation and functional analysis of PKC site mutants. MCF7 cells were transiently transfected with WT-Pcyt2α or Pcyt2α mutants (S215A, S223A, S215A,S223A) and cultured in serum-free medium for 24 h. A, effects on phosphorylation. V5/His-tagged Pcyt2α and mutated proteins were immunoprecipitated (IP) with V5 antibody, and the phosphorylated Pcyt2α proteins were detected by immunoblotting using anti-Ser(P) (top). Blots were probed with anti-V5 to assess the level of the expressed proteins (bottom). B, in vitro activity of Pcyt2α and Pcyt2α mutants was performed using lysates from cells transfected with the indicated Pcyt2α construct, as described under “Experimental Procedures.” Student's t test was performed to compare the activity of each mutant against the wild-type enzyme. Results are means ± S.D. of three independent experiments; p < 0.001 (***). C, MCF7 cells were transiently transfected with V5/His-tagged WT or Pcyt2α PKC site double mutant (S215A,S223A). Cells were cultured in serum-deficient medium for 24 h and subsequently treated for 2 h with an activator (25 nm PMA) and an inhibitor (50 nm EGF-R) of PKCs as indicated. Subsequent immunoprecipitation was performed using anti-V5 antibody. Phosphorylation level was detected by immunoblotting with Ser(P) (pSer) antibody (top). Anti-V5 antibody (bottom) was used to confirm protein expression. D, MCF7 cells were transiently transfected with empty vector (endogenous) or V5/His-tagged-Pcyt2α (overexpressed) as indicated. Cells were cultured in serum-deficient medium for 24 h and subsequently treated for 2 h with 25–1000 nm PMA. Subsequently, in vitro activity assays were performed using cell lysates, as described under “Experimental Procedures.” Results are means ± S.D. of two independent experiments: p < 0.05 (*), p < 0.01 (**). Student's t test was performed to compare the activity of the indicated variables in each treatment category as indicated. E, transfected cells were radiolabeled with [14C]ethanolamine (0.1 μCi/106cells; 1 h) in the presence of 50 μm unlabeled ethanolamine, and PE synthesis was measured, normalized to the total protein level, adjusted for expression efficiency (E, bottom), and represented relative to that of WT Pcyt2α. Data are means ± S.D. of three individual experiments; p < 0.05 (*) and p < 0.01 (**). F, MCF-7 cells (4 × 104 per well) were transfected with the appropriate plasmid (empty vector (mock), Pcyt2α, S215A, S223A, or S215A,S223A) using Lipofectamine in the absence of serum. Viability test was performed at the indicated time points (0–72 h) to quantify changes in cell proliferation. Data shown are means ± S.D. of three independent experiments. G, immunoblotting of the autophagy marker LC3I/II in cells grown with and without serum showing decreased LC3 lipidation (LC3II amounts) in the absence of serum. H, transfections with Pcyt2α WT did not modify the MCF7 cell autophagy marker LC3 under the same conditions, assessed with an LC3-specific antibody (Novus Biologicals).
The effect of Pcyt2α mutations on PE synthesis by the CDP-ethanolamine–Kennedy pathway is shown in Fig. 5E. Transfected cells were radiolabeled with [14C]ethanolamine, and its incorporation into newly synthesized PE was determined. Data showed that the reduction in PE synthesis (Fig. 5E) was mirrored by the reduction in Pcyt2 activity (Fig. 5B). Therefore, we concluded that the mutation of αSer-215 and αSer-223 significantly impacted the enzyme phosphorylation and activity, showing that both sites are important for regulation by serum deficiency. Interestingly, the double mutant showed a maximal reduction in phosphorylation that, however, did not further suppress the enzyme activity and PE synthesis, suggesting that the two PKC sites were not synergistically regulated and are likely independent.
Pcyt2α Mutants Do Not Support Cell Growth
Regulation of Pcyt2 activity by PKCα, also an important marker of breast cancer cell proliferation (29), suggests that the stimulation of Pcyt2α and PE synthesis under serum deficiency (1) is important in supporting the cancer cell growth and survival. To test if the Pcyt2α mutants could influence the MCF-7 cell growth, the cells were transfected with wild-type or the phosphorylation site mutants described above, and cell growth was monitored under serum-deficient conditions. We have previously shown that the endogenous Pcyt2 was elevated and supported the MCF-7 cell proliferation under serum-deficient conditions (1). The overexpressed Pcyt2α increased the growth further by additional stimulation of membrane PE synthesis (1). Overexpressed mutated proteins did not support the cell growth and were toxic at longer times (Fig. 5F). The observed effect on cell proliferation was concomitant with the decrease in PE synthesis under the same conditions (Fig. 5E).
Under conditions of serum/nutrient deprivation, cell growth is typically supported by the process of autophagy (30, 31). Hence, we further tested contribution of autophagy to the observed changes in PE synthesis and cell proliferation. We have previously demonstrated that Pcyt2 could increase the supply of PE for LC3-I lipidation during induction of autophagy (5, 31), which could stimulate cell growth (Fig. 5F). The main event in the autophagy is a covalent attachment of PE to the autophagy-specific marker LC3-I (lipidation) to form the PE-bound LC3-II. Therefore, we tested if the overexpressed Pcyt2α increased the MCF7 cell growth (Fig. 5F) by stimulating the LC3-I lipidation. We monitored the content of LC3-I (PE-free form) and LC3-II (PE-bound form) by Western blotting (Fig. 5, G and H). However, in mock-transfected cells, the amount of LC3-I and LC3-II was reduced in the absence of serum. Although the ratio of LC3-II to LC3-I in the presence of serum was ∼2.60, the same ratio was unchanged in serum-deprived cells (∼1.09) (Fig. 5G), showing that the autophagy was generally impaired and therefore did not contribute to the observed changes in cell growth in serum deficiency (Fig. 6B). Hence, it is not surprising that the overexpression of Pcyt2α did not increase the LC3 lipidation either after 24 h (LC3II/LC3I ratio in mock versus Pcyt2α was 1.14:1.06) or 48 h (LC3II/LC3I ratio in mock versus Pcyt2α was 0.98:1.01) (Fig. 5H). We concluded that the increased Pcyt2 activity observed in serum-deficient cells (1) stimulated the cell growth by increasing the membrane PE formation (1) without additional effects on cell renewal by autophagy.
FIGURE 6.

Structural elements and new phosphorylation domains. A, sequence alignment of murine Pcyt2α (UniProt Q922E4) and human Pcyt2β (UniProt Q99447), is displayed. The structural elements for the human β isoform were obtained from Protein Data Bank code 3elb. N- and C-domains contain identical Rossman folds of six helices (αA to αE) and five β-sheets (β1 to β5); (h are α-helix residues and s are β-strand residues). The area unresolved in the Pcyt2β crystal is inside the boxed sequence, indicated by a dashed line, and includes the α-specific peptide (gray box). The identified phosphorylated residues are shown for mPcyt2α in red and for mPcyt2β in blue. B, phosphorylation sites mapped onto the solved structure of Pcyt2β. The image of human Pcyt2β (Protein Data Bank code 3elb) was prepared using PyMOL. The N-terminal catalytic domain is blue, and the C-terminal catalytic domain is in gray. A CMP ligand in stick representation occupies the active site of the C-cat domain. The linker region connecting the two domains is highlighted red. The side chains of the phosphorylated residues are shown as spheres (carbon, green; oxygen, red); the numbering is for Pcyt2β (Pcy2α numbering in parentheses). The broken line denotes the portion of the linker that is unresolved in 3elb and is not to scale. The unresolved segment contains residues 157–186 in Pcyt2β and residues 157–203 in Pcyt2α; these include identified phosphorylation sites at residues 157, 162, 167, and 168 in Pcyt2β and residues 192 and 195 in Pcyt2α.
Identification of a Centrally Located Pcyt2 Phosphorylation Region
To determine where the common and isoform-specific phosphorylation sites are located in the Pcyt2 tertiary structure, we mapped them onto the solved crystal structure of the human Pcyt2β, available in the Protein Data Bank under code 3elb. The human (NP_002852) and murine (EDL34775.1) Pcyt2β sequences are 92% identical, and all of the phosphorylation sites we have identified are conserved between the two species. A comparison of the sequences of murine Pcyt2α and human Pcyt2β is shown in Fig. 6A along with the phosphorylation sites we have identified by MS and the secondary structure motifs obtained from the solved structure (3elb). As shown in Fig. 6B, Pcyt2 is composed of two nearly identical α/β Rossman folds, each composed of five β-strands (β1–β5) and six α-helices. Other cytidylyltransferases, including CTP:phosphocholine cytidylyltransferase (Pcyt1/CCT) or CTP:phosphoglycerol cytidylyltransferase (GCT), contain one α/β Rossman fold, and one catalytic domain. Two monomers of CCT or GCT associate to form a dimer that is analogous to the Pcyt2 monomer with its two catalytic domains (9, 32, 33). Fig. 6A also highlights the linker segment that connects the two catalytic folds. In the solved structure of human Pcyt2β, 30 residues of this 57-residue segment were unresolved, as indicated by the broken line in Fig. 6B. This unresolved region includes the α-specific 18-mer motif. From residues 187 to 212, the Pcyt2β structure is ordered, including a short helix (αF) that makes contact with the C-cat domain. Our phospho-site mapping indicated that all the phosphorylated sites cluster within the linker area or at sites proximal to it in the tertiary structure (residues βSer-145, βThr-146, and βThr-147 in N-cat and residues βSer-197, βSer-205, and βSer-282 in C-cat (Fig. 6B).
DISCUSSION
For optimal PE synthesis by the CDP-ethanolamine pathway, Pcyt2 catalytic function must be precisely controlled. However, relatively little is known about the regulation of the activity of Pcyt2 in cells. Studies described herein examine the regulation of Pcyt2 by phosphorylation and for the first time demonstrate distinct phosphorylation properties of two Pcyt2 isoforms. We established the isoform-specific and PKC-mediated phosphorylations that can regulate Pcyt2α activity. We identified a Pcyt2-specific phosphorylation region. Findings presented in this study clearly distinguish the regulation of Pcyt2 from the regulation of structurally and functionally similar CTP-phosphocholine cytidylyltransferase (Pcyt1, CCT) from the CDP-choline branch of the Kennedy pathway (32, 33).
Pcyt2α Is a Physiological Target of PKC
The PKC family of serine/threonine kinases has been strongly implicated in the regulation of PE synthesis and cancer cell growth. DAG and phorbol esters increase the rate of PE synthesis (7), and based on the metabolite pool sizes and the enzyme activity, the regulation of the CDP-ethanolamine pathway was predominantly mediated at the level of Pcyt2 (8, 34, 35). PMA stimulation of PE synthesis is accomplished through stimulation of Pcyt2 activity as well as by increases in the cellular content of the substrates ethanolamine (7) and DAG (8). PMA-resistant cells have an impaired PE synthesis and Pcyt2 activity (34, 35), and phorbol esters target two different PKC isozymes in MCF-7 cells, PKC-βI/II for the stimulation of PE synthesis and PKCα for the stimulation of PE degradation (8). PKCα has been strongly implicated in breast cancer development (29). We provided evidence that part of the cancer cell defense mechanism against unfavorable growth conditions of serum deficiency includes the stimulation of Pcyt2α activity by phosphorylation and increased membrane PE synthesis (1).
Here, we demonstrated that the increased phosphorylation and activity of endogenous Pcyt2 was strongly attributed to the increased activity of PKC. PKC activator PMA increased although PKC inhibitors decreased the enzyme phosphorylation and activity in a time- and dose-dependent manner. Our data established that Pcyt2α is a physiological target of conventional PKCs. The MS analysis identified two PKC consensus site phosphorylations in Pcyt2α at αSer-215 and αSer-223. These residues were specifically phosphorylated under serum-deficient conditions when the enzyme was more active. In vitro analysis showed that Pcyt2α was a better substrate for PKCα than for PKCβI and PKCβII, and the phosphorylation of αSer–215 with PKCα was confirmed by MS. Phosphorylation at αSer-223 appeared to be an independent target of phosphorylation by other PKCs because this site was phosphorylated in serum deficiency. but it was not found to be phosphorylated by PKCα. Importantly, a regulatory role for PKC phosphorylation at the consensus sites was suggested by the inhibition of PE synthesis and/or enzymatic activity when phosphorylation was blocked by mutations at these sites.
How Could Phosphorylation in the Linker Segment Influence Enzyme Activity?
Four phosphorylation sites identified in the linker region were found within the structurally unresolved portion; thus we do not know how their phosphorylation status could impact catalytic activity. The sites in the ordered portion of the linker include the PKC-directed sites αSer-215 and αSer-223 (βSer-205). These sites are located at the start and at the end of the helical segment (αF) of the linker. Phosphorylation could affect the ordering of this helix, which is situated near the interface between the two catalytic folds, making contacts with helices αA and αC of N-cat and helix αC of C-cat. This in turn could impact on cooperativity between active sites (36).
Pcyt2β Phosphorylation Pattern Includes the Signature Motif RTQGVSTT
In contrast to Pcy2α, Pcyt2β was not directly phosphorylated with PKCα, and the consensus βSer-197 (corresponding to αSer-215) and βSer-205 (corresponding to αSer-223) phosphorylation was not regulated by serum deficiency as strongly as in Pcyt2α. The lack of activation of these sites in Pcyt2β possibly contributed to the observed reduced activity of Pcyt2β relative to Pcyt2α established earlier (1, 26). Although the N-terminal domain of Pcyt2α was not found phosphorylated under any experimental conditions, Pcyt2β was specifically phosphorylated at three N-cat sites located within a cytidylyltransferase family signature motif 139RTQGVSTT147. Residues 145–147 are at the start of helix αE. In GCT and CCT crystal structures, this portion of helix αE made several contacts with CTP- or CDP-choline (9, 32). Helix αE has been suggested as a potential target for regulation in other cytidylyltransferases (32, 33). Therefore, Pcyt2β Ser-145, Thr-146, and Tyr-147 phosphorylations could directly modify αE helix contributions to the active site and the progress of the reaction. The Pcyt2β phosphorylation sites in helix αE are potential targets of CK2-mediated phosphorylation. CK2 is, however, constitutively active in different cellular compartments and has been frequently associated with regulation of nuclear proteins. Both Pcyt2 proteins predominantly localize in the cytoplasm, and it is not clear how CK2 would specifically phosphorylate Pcyt2β under serum-deficient conditions. Therefore, a goal of future studies could be to better understand the specific role of the CK2 signaling in αE phosphorylation and Pcyt2β function.
To conclude, our studies provide novel characterization of Pcyt2 phosphorylation. Both isoforms undergo elevated phosphorylation in serum deprivation, suggesting that Pcyt2 regulation is dependent on cell growth and nutritional conditions. The existence of PKC-regulated phosphorylation of Pcyt2α and the specific phosphorylation of important Pcyt2β cytidylyltransferase motifs suggest that different mechanisms of regulation control the activity of individual isoforms under serum-deficient conditions. Our data support the notion that the majority of phosphorylations are restricted to the central linker region and flanking segments. Phosphorylation in the linker area is generally accompanied by stimulation of enzyme activity. As the linker is a distinct structural feature of Pcyt2, it may have evolved as a discrete regulatory region to control the enzyme's catalytic function by phosphorylation.
Acknowledgments
We thank Dr. Rulin Zhang for allowing us to use the Mass Spectrometry facility and for helping us with data interpretation and analysis.
This work was supported by grants from the Canadian Institutes of Health Research (to M. B. and R. C.) and Ontario President Research Excellence Award (to M. B.).
- PE
- phosphatidylethanolamine
- DAG
- diacylglycerol
- P-Etn
- phosphoethanolamine
- PMA
- phorbol 12-myristate 13-acetate
- EGF-R
- EGF receptor
- Pcyt2
- CTP:phosphoethanolamine cytidylyltransferase
- quadrupole
- CCT
- CTP:phosphoethanolamine cytidylyltransferase
- GCT
- CTP:glycerol-3-phosphate cytidylyltransferase.
REFERENCES
- 1. Zhu L., Bakovic M. (2012) Breast cancer cells adapt to metabolic stress by increasing ethanolamine phospholipid synthesis and CTP:ethanolaminephosphate cytidylyltransferase-Pcyt2 activity. Biochem. Cell Biol. 90, 188–199 [DOI] [PubMed] [Google Scholar]
- 2. Fullerton M. D., Hakimuddin F., Bakovic M. (2007) Developmental and metabolic effects of disruption of the mouse CTP:phosphoethanolamine cytidylyltransferase gene (Pcyt2). Mol. Cell. Biol. 27, 3327–3336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Emoto K., Kobayashi T., Yamaji A., Aizawa H., Yahara I., Inoue K., Umeda M. (1996) Redistribution of phosphatidylethanolamine at the cleavage furrow of dividing cells during cytokinesis. Proc. Natl. Acad. Sci. U.S.A. 93, 12867–12872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Emoto K., Umeda M. (2000) An essential role for a membrane lipid in cytokinesis. Regulation of contractile ring disassembly by redistribution of phosphatidylethanolamine. J. Cell Biol. 149, 1215–1224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Girardi J. P., Pereira L., Bakovic M. (2011) De novo synthesis of phospholipids is coupled with autophagosome formation. Med. Hypotheses 77, 1083–1087 [DOI] [PubMed] [Google Scholar]
- 6. Gibellini F., Smith T. K. (2010) The Kennedy pathway–De novo synthesis of phosphatidylethanolamine and phosphatidylcholine. IUBMB Life 62, 414–428 [DOI] [PubMed] [Google Scholar]
- 7. Tijburg L. B., Houweling M., Geelen J. H., van Golde L. M. (1987) Stimulation of phosphatidylethanolamine synthesis in isolated rat hepatocytes by phorbol 12-myristate 13-acetate. Biochim. Biophys. Acta 922, 184–190 [DOI] [PubMed] [Google Scholar]
- 8. Bleijerveld O. B., Klein W., Vaandrager A. B., Helms J. B., Houweling M. (2004) Control of the CDP-ethanolamine pathway in mammalian cells: Effect of CTP:phosphoethanolamine cytidylyltransferase overexpression and the amount of intracellular diacylglycerol. Biochem. J. 379, 711–719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Weber C. H., Park Y. S., Sanker S., Kent C., Ludwig M. L. (1999) A prototypical cytidylyltransferase: CTP:glycerol-3-phosphate cytidylyltransferase from Bacillus subtilis. Structure 7, 1113–1124 [DOI] [PubMed] [Google Scholar]
- 10. Pavlovic Z., Bakovic M. (2013) Regulation of phosphatidylethanolamine homeostasis—The critical role of CTP:phosphoethanolamine cytidylyltransferase (Pcyt2). Int. J. Mol. Sci. 14, 2529–2550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Fullerton M. D., Hakimuddin F., Bonen A., Bakovic M. (2009) The development of a metabolic disease phenotype in CTP:phosphoethanolamine cytidylyltransferase-deficient mice. J. Biol. Chem. 284, 25704–25713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fullerton M. D., Bakovic M. (2010) Complementation of the metabolic defect in CTP:phosphoethanolamine cytidylyltransferase (Pcyt2)-deficient primary hepatocytes. Metabolism 59, 1691–1700 [DOI] [PubMed] [Google Scholar]
- 13. Leonardi R., Frank M. W., Jackson P. D., Rock C. O., Jackowski S. (2009) Elimination of the CDP-ethanolamine pathway disrupts hepatic lipid homeostasis. J. Biol. Chem. 284, 27077–27089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhu L., Michel V., Bakovic M. (2009) Regulation of the mouse CTP:phosphoethanolamine cytidylyltransferase gene Pcyt2 during myogenesis. Gene 447, 51–59 [DOI] [PubMed] [Google Scholar]
- 15. Li H., Liu Y., Shin S., Sun Y., Loring J. F., Mattson M. P., Rao M. S., Zhan M. (2006) Transcriptome coexpression map of human embryonic stem cells. BMC Genomics 7, 103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Giritharan G., Li M. W., Di Sebastiano F., De Sebastiano F., Esteban F. J., Horcajadas J. A., Lloyd K. C., Donjacour A., Maltepe E., Rinaudo P. F. (2010) Effect of ICSI on gene expression and development of mouse preimplantation embryos. Hum. Reprod. 25, 3012–3024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Swiss V. A., Nguyen T., Dugas J., Ibrahim A., Barres B., Androulakis I. P., Casaccia P. (2011) Identification of a gene regulatory network necessary for the initiation of oligodendrocyte differentiation. PLoS One 6, e18088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kunz D., Walker G., Bedoucha M., Certa U., März-Weiss P., Dimitriades-Schmutz B., Otten U. (2009) Expression profiling and ingenuity biological function analyses of interleukin-6- versus nerve growth factor-stimulated PC12 cells. BMC Genomics 10, 90-2164-10-90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Serb J. M., Orr M. C., West Greenlee M. H. (2010) Using evolutionary conserved modules in gene networks as a strategy to leverage high throughput gene expression queries. PLoS One 5, 10.1371/journal.pone.0012525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dixon R. M. (1996) Phosphatidylethanolamine synthesis in the normal and lymphomatous mouse liver; a 13C NMR study. Anticancer Res. 16, 1351–1356 [PubMed] [Google Scholar]
- 21. Momchilova A., Markovska T., Pankov R. (1999) Ha-ras-transformation alters the metabolism of phosphatidylethanolamine and phosphatidylcholine in NIH 3T3 fibroblasts. Cell Biol. Int. 23, 603–610 [DOI] [PubMed] [Google Scholar]
- 22. Folger O., Jerby L., Frezza C., Gottlieb E., Ruppin E., Shlomi T. (2011) Predicting selective drug targets in cancer through metabolic networks. Mol. Syst. Biol. 7, 501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Woodfield G. W., Chen Y., Bair T. B., Domann F. E., Weigel R. J. (2010) Identification of primary gene targets of TFAP2C in hormone responsive breast carcinoma cells. Genes Chromosomes Cancer 49, 948–962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhu L., Johnson C., Bakovic M. (2008) Stimulation of the human CTP:phosphoethanolamine cytidylyltransferase gene by early growth response protein 1. J. Lipid Res. 49, 2197–2211 [DOI] [PubMed] [Google Scholar]
- 25. Poloumienko A., Coté A., Quee A. T., Zhu L., Bakovic M. (2004) Genomic organization and differential splicing of the mouse and human Pcyt2 genes. Gene 325, 145–155 [DOI] [PubMed] [Google Scholar]
- 26. Tie A., Bakovic M. (2007) Alternative splicing of CTP:phosphoethanolamine cytidylyltransferase produces two isoforms that differ in catalytic properties. J. Lipid Res. 48, 2172–2181 [DOI] [PubMed] [Google Scholar]
- 27. Zhang R., Barker L., Pinchev D., Marshall J., Rasamoelisolo M., Smith C., Kupchak P., Kireeva I., Ingratta L., Jackowski G. (2004) Mining biomarkers in human sera using proteomic tools. Proteomics 4, 244–256 [DOI] [PubMed] [Google Scholar]
- 28. Hastie C. J., McLauchlan H. J., Cohen P. (2006) Assay of protein kinases using radiolabeled ATP: a protocol. Nat. Protoc. 1, 968–971 [DOI] [PubMed] [Google Scholar]
- 29. Gupta A. K., Galoforo S. S., Berns C. M., Martinez A. A., Corry P. M., Guan K. L., Lee Y. J. (1996) Elevated levels of ERK2 in human breast carcinoma MCF-7 cells transfected with protein kinase Cα. Cell Prolif. 29, 655–663 [DOI] [PubMed] [Google Scholar]
- 30. Singh R., Cuervo A. M. (2012) Lipophagy: connecting autophagy and lipid metabolism. Int. J. Cell Biol. 2012, 282041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pereira L., Girardi J. P., Bakovic M. (2012) Forms, crosstalks, and the role of phospholipid biosynthesis in autophagy. Int. J. Cell Biol. 2012, 931956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lee J., Johnson J., Ding Z., Paetzel M., Cornell R. B. (2009) Crystal structure of a mammalian CTP: phosphocholine cytidylyltransferase catalytic domain reveals novel active site residues within a highly conserved nucleotidyltransferase fold. J. Biol. Chem. 284, 33535–33548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Fong D. H., Yim V. C., D'Elia M. A., Brown E. D., Berghuis A. M. (2006) Crystal structure of CTP:glycerol-3-phosphate cytidylyltransferase from Staphylococcus aureus: Examination of structural basis for kinetic mechanism. Biochim. Biophys. Acta 1764, 63–69 [DOI] [PubMed] [Google Scholar]
- 34. Kiss Z. (1997) Expression of protein kinase C-β promotes the stimulatory effect of phorbol ester on phosphatidylethanolamine synthesis. Arch. Biochem. Biophys. 347, 37–44 [DOI] [PubMed] [Google Scholar]
- 35. Lahn M., Kohler G., Sundell K., Su C., Li S., Paterson B. M., Bumol T. F. (2004) Protein kinase Cα expression in breast and ovarian cancer. Oncology 67, 1–10 [DOI] [PubMed] [Google Scholar]
- 36. Taneva S., Dennis M. K., Ding Z., Smith J. L., Cornell R. B. (2008) Contribution of each membrane binding domain of the CTP:phosphocholine cytidylyltransferase-α dimer to its activation, membrane binding, and membrane cross-bridging. J. Biol. Chem. 283, 28137–28148 [DOI] [PMC free article] [PubMed] [Google Scholar]