

Background: Highly conserved PII signaling proteins monitor carbon/nitrogen balance and energy status.
Results: Crystals of homotrimeric Synechococcus elongatus PII with ADP reveal the structural basis of anti-cooperative ADP binding. Interaction with PipX receptor changes the ATP/ADP preference of PII.
Conclusion: PII signaling is modulated by conformational states imposed by the receptor protein.
Significance: Molecular information is processed through protein structure plasticity.
Keywords: Adenosine, ADP, Cooperativity, Cyanobacteria, Nitrogen Metabolism, Protein-Protein Interactions, 2-Oxoglutarate, Nitrogen Control factor NtcA
Abstract
PII signaling proteins comprise one of the most versatile signaling devices in nature and have a highly conserved structure. In cyanobacteria, PipX and N-acetyl-l-glutamate kinase are receptors of PII signaling, and these interactions are modulated by ADP, ATP, and 2-oxoglutarate. These effector molecules bind interdependently to three anti-cooperative binding sites on the trimeric PII protein and thereby affect its structure. Here we used the PII protein from Synechococcus elongatus PCC 7942 to reveal the structural basis of anti-cooperative ADP binding. Furthermore, we clarified the mutual influence of PII-receptor interaction and sensing of the ATP/ADP ratio. The crystal structures of two forms of trimeric PII, one with one ADP bound and the other with all three ADP-binding sites occupied, revealed significant differences in the ADP binding mode: at one site (S1) ADP is tightly bound through side-chain and main-chain interactions, whereas at the other two sites (S2 and S3) the ADP molecules are only bound by main-chain interactions. In the presence of the PII-receptor PipX, the affinity of ADP to the first binding site S1 strongly increases, whereas the affinity for ATP decreases due to PipX favoring the S1 conformation of PII-ADP. In consequence, the PII-PipX interaction is highly sensitive to subtle fluctuations in the ATP/ADP ratio. By contrast, the PII-N-acetyl-l-glutamate kinase interaction, which is negatively affected by ADP, is insensitive to these fluctuations. Modulation of the metabolite-sensing properties of PII by its receptors allows PII to differentially perceive signals in a target-specific manner and to perform multitasking signal transduction.
Introduction
The PII signal transduction system responds to nitrogen, carbon, and energy of the cell and is highly conserved and widespread in all three domains of life (1–5). PII proteins receive metabolic information by binding ATP, ADP, and 2-oxoglutarate (2-OG)4 and integrate the signal through conformational changes and covalent modifications (1, 3). Depending on the signal, PII proteins control regulatory and metabolic enzymes, transport proteins, and transcription factors involved in central nitrogen metabolism (5).
PII proteins are homotrimers of 12–13-kDa subunits with a cylindrical body and three flexible T-loops that extend into the solvent and take part in binding of effector molecules and protein-protein interactions (3, 5). Each subunit also has two smaller loops, the B- and C-loops, that face each other in the intersubunit clefts and participate in effector binding. The PII trimer thus contains three effector nucleotide-binding sites, one in each intersubunit cleft (6–8), where the adenylyl nucleotides ATP and ADP compete for the same sites. In the PII protein from the cyanobacterium Synechococcus elongatus PCC 7942, the binding of ATP and ADP to the three sites is anti-cooperative, with ATP having a higher affinity than ADP (9). When Mg2+-ATP is ligated to a binding site of bacterial PII, the site can also bind 2-OG, whereas ADP binding does not support the binding of 2-OG (10, 11). The 2-OG binding site is located at the base of the T-loop in direct vicinity of the β- and γ-phosphate of ATP, which ligates 2-OG through a chelated Mg2+ ion (11, 12). Consequently, binding of ATP and 2-OG is synergistic (13). Because ADP competes with Mg2+-ATP in binding to PII, it negatively affects the binding of 2-OG and thereby interferes with the ability of PII to sense the signal of nitrogen starvation (10, 14, 15). The three 2-OG binding sites exhibit negative cooperativity relative to each other, which is mediated through intersubunit signaling inside the trimer (9, 11).
In living cells ATP and ADP are simultaneously present, and they compete for the three nucleotide-binding sites of the PII trimer. The total concentration of these effectors in bacterial cells is in the millimolar range (16–18), well above the KD for the PII binding sites (9); therefore, all three binding sites of PII are presumably always occupied by either ATP or ADP. Considering that 2-OG is only able to bind to the sites occupied by Mg2+-ATP, PII proteins could theoretically adopt 20 different ligand binding states, but considering that in vivo all sites are filled with either ATP or ADP, only 10 states would be possible. If the ligand binding properties are taken into account, only 5–7 states are probable in vivo. At high ATP concentrations, all PII sites are filled with Mg-ATP (PII-ATP3) and up to three 2-OG molecules can be bound, thereby measuring this effector with the highest sensitivity. As the ADP concentration increases, the PII-ATP2-ADP1 population forms. Only when the ADP concentration exceeds that of ATP would the PII-ATP1-ADP2 population accumulate, as the binding affinity of ADP is lower than that of ATP. In general, the mixed populations (PII-ATP2-ADP1 and PII-ATP1-ADP2) have fewer 2-OG binding sites than PII-ATP3, and calorimetric titration experiments have shown that at an ATP:ADP ratio of 1:1, only one 2-OG is bound (15). The failure to detect two binding sites for 2-OG under these conditions indicates either that the PII-ATP2-ADP1 species can bind only one 2-OG molecule or, more unlikely, that in the presence of equimolar ADP and ATP, the PII-ATP1-ADP2 species is preferentially formed.
PII proteins from oxygenic phototrophs (cyanobacteria and plants) control arginine biosynthesis through the arginine feedback-inhibited enzyme N-acetyl-l-glutamate kinase (NAGK) (19, 20). PII binding to NAGK enhances the activity of the enzyme and protects it from feedback inhibition by arginine (21). PII in the ATP-ligated state avidly binds to NAGK, whereas in the presence of ADP, complex formation is impaired, and as a consequence, NAGK activity is not enhanced (15). Likewise, the effector 2-OG strongly inhibits PII-NAGK complex formation. Thus, the inhibitory effect of 2-OG is not antagonized by ADP and is, therefore, robust against different ATP/ADP ratios (15, 21). In addition to the arginine pathway control, cyanobacterial PII proteins indirectly control gene expression by sequestering PipX, which is a co-activator of the global nitrogen control transcription factor NtcA (22–24). PII binds PipX in the absence of 2-OG, with ADP strongly enhancing the PII-PipX interaction and antagonizing the inhibitory 2-OG effect even at low ADP concentrations (15, 24).
Binding of ATP and ADP is a general property of PII proteins in living cells (5). In Escherichia coli, which has two PII paralogues, GlnB and GlnK, ADP acts as an antagonist of 2-OG in the GlnB-mediated regulation of the NRI/NRII two-component system and of the adenylyltransferase-glutamine synthetase monocycle in vitro. ADP also inhibits uridylylation of GlnB by the bifunctional enzyme uridylyltransferase/uridylyl-removing enzyme (UTase) (10, 25). In the presence of ADP, the PII paralogue GlnK forms a complex and thereby blocks the ammonium transporter AmtB, a reaction that is antagonized by Mg2+/ATP/2-OG (14). Recently, Radchenko et al. (26) showed that in the absence of 2-OG, GlnK displays intrinsic ATPase activity, slowly converting ATP-GlnK to ADP-GlnK, which then tightly binds AmtB. Sensing of the energy charge levels has also been suggested for the PII proteins from Rhodospirillum rubrum (27, 28). ADP stabilizes the GlnJ-AmtB1 complex (GlnJ is a GlnK paralogue) even in the presence of complex-dissociating ATP-2-OG levels (29). The affinities toward ATP and ADP and the anti-cooperativity between the binding sites are key features of PII proteins that determine their ability to function as energy sensors. In this respect it is important to note that the fine-tuned ligand binding properties of PII proteins from different sources are variable (30). One of the best-studied PII proteins is that of S. elongatus. Many structure-function details related to 2-OG signaling have been elucidated (3, 11, 24, 31) (an overview of the various S. elongatus PII structures is presented in Table 1). However, the structural implications of ADP binding and the functional consequences for the integrated NAGK-PII·PipX interaction network remain elusive.
TABLE 1.
Overview of structural data used in this work
AU, asymmetric unit.
| Structure PDB entry | Protein(s) and organism | Number of subunits in the AU | Detailed description of AU | Detailed co-factors description | Nomenclature of the PII trimer in this paper | Nomenclature of individual effector sites in this paper | Resolution (Å) | Reference |
|---|---|---|---|---|---|---|---|---|
| 4C3L | PII from | One monomer | None | PIINCI | 1.6 | This work | ||
| S. elongatus | ||||||||
| 4C3M | PII from | One trimer | None | PIINCII | 2.2 | This work | ||
| S. elongatus | ||||||||
| 4C3K | PII from | Two trimers | PIIADP | 3.1 | This work | |||
| S. elongatus | ||||||||
| Trimer I | One ADP | PIIADP1 | PIIADP1-S1 | |||||
| Trimer II | Three ADP | PIIADP3 | PIIADP3-S1 | |||||
| PIIADP3-S2 | ||||||||
| PIIADP3-S3 | ||||||||
| 2XZW | PII from S. elongatus | Three trimers | Trimer I | Three ATP | PIIOG1 | PIIOG1-S1 | 1.95 | (11) |
| One Mg2+ | PIIOG1-S2 | |||||||
| One 2-OG | PIIOG1-S3 | |||||||
| Trimer II | Three ATP | PIIOG2 | PIIOG2-S1 | |||||
| Two Mg2+ | PIIOG2-S2 | |||||||
| Two 2-OG | PIIOG2-S3 | |||||||
| Trimer III | Three ATP | PIIOG3 | PIIOG3-S1 | |||||
| Three Mg2+ | PIIOG3-S2 | |||||||
| Three 2-OG | PIIOG3-S3 | |||||||
| 2XUL | PII from S. elongatus | Two identical trimers | Three ATP | PIIOGex | 2.2 | (11) | ||
| Three Mg2+ | ||||||||
| Three 2-OG | ||||||||
| 2XBP | PII from S. elongatus (I86N mutant) | One monomer | One ATP | 1.2 | (9) | |||
| One Mg2+ | ||||||||
| 4AFF | PII from S. elongatus (I86N mutant) | One monomer | One ATP | 1.05 | (31) | |||
| One Mg2+ | ||||||||
| One citrate | ||||||||
| 1QY7 | PII from S. elongatus (S49A mutant) | One trimer | None | 2.0 | (6) | |||
| 2J9D | GlnK1 from M. jannashii | Four trimers | Trimer I Trimer II Trimer III Trimer IV |
One ADP One AMP One ADP Two ADP |
2.1 | (42) | ||
| 1V9O | PII from T. thermophilus | One trimer | Three ADP | (8) | ||||
| 3TA1 | PII from A, fulgidus | One trimer | Three ADP | 1.9 | (30) | |||
| 3NCR | PII from A. fulgidus | One trimer | Three ADP | 1.44 | (41) | |||
| 2XG8 | PII/PipX from S. elongatus | One trimeric complex | None | 3.2 | (24) | |||
| 3N5B | PII/PipX from Nostoc sp. | One monomeric complex | ADP | 1.9 | (48) | |||
| 2V5H | PII/NAGK from S. elongatus | One NAGK hexamer two PII trimers | N-Acetyl-l-glutamate (bound to NAGK) | 2.75 | (46) |
In the present study we investigated the structural transitions of effector-free and ADP-loaded PII subunits from S. elongatus and propose a mechanism of ADP sensing by PII. We also analyzed the role of energy charge sensing (energy charge measured as the ATP/ADP ratio) for the PipX/NtcA-PII·NAGK interaction network. Based on our data and previously published data, we conclude that the PII signal transduction system does not work as an on/off switch governed by the intrinsic ATPase activity of PI, but rather as a multitasking signal integration device with flexible signal output dependent on its receptor protein.
EXPERIMENTAL PROCEDURES
Overexpression and Purification Of Recombinant PII, NAGK, PipX, and NtcA
The glnB and pipX genes from S. elongatus, cloned into the Strep-tag fusion vector pASK-IBA3 (IBA, Göttingen, Germany), were overexpressed in E. coli RB9060 (32), and the PII and PipX proteins, each with a C-terminal fused Strep-tag peptide, were purified using affinity chromatography as described previously (20, 22). The genes encoding NAGK and NtcA from S. elongatus, cloned in pET15b vector, were overexpressed in E. coli strain BL21(DE3) (33), and the His6-tagged recombinant proteins were purified as reported previously (20, 22).
Surface Plasmon Resonance (SPR) Analysis
SPR experiments were performed using a Biacore X biosensor system (Biacore AB, Uppsala, Sweden) at 25 °C in HBS-Mg buffer containing 10 mm HEPES, 150 mm NaCl, 1 mr MgCl2, and 0.005% Nonidet P-40, pH 7.5, as described previously (21). To immobilize purified His6-NAGK on the Ni2+-loaded NTA sensor chip of flow cell 2 (FC2), 50 μl of a 30 nm His6-NAGK solution (of the hexamer) was injected until a binding signal of ∼3000 resonance units (RU) was reached, which corresponds to a surface concentration change of 3 ng/mm2. To measure the effect of PipX on the association of the PII·NAGK complex, a solution of 100 nm PII was injected over the His6-NAGK surface immobilized on the sensor chip in the presence or absence of 100 nm PipX and with various ratios of ATP:ADP (with 1 mm MgCl2). To determine the influence of PII on the NtcA-PipX interaction, 125 nm His6-NtcA (of the dimer) in a volume of 60 μl was injected onto the Ni2+-loaded NTA sensor chip to receive a binding signal of ∼1500 RU. PipX (500 nm) was injected in the presence of various effector concentrations, 1 mm MgCl2, and 500 nm PII. To load fresh proteins on the NTA sensor chip, bound proteins were first removed by injection of 25 μl of 0.4 m EDTA, pH 7.5; the chip could then be loaded again with 5 mm NiSO4 solution and the His6-tagged protein.
Isothermal Titration Calorimetry for Determination of Binding Constants
Isothermal titration calorimetry experiments were performed on a VP-ITC microcalorimeter (MicroCal, LCC) in buffer containing 10 mm HEPES, pH 7.4, 50 mm KCl, 50 mm NaCl, and 1 mm MgCl2 at 20 °C. Isotherms of ADP and ATP binding to PII (25 μm trimer concentration) were determined in the presence of 75 μm PipX. For one measurement, 5 μl of 1 mm 2-OG was injected 35 times (2.1–146.8 μm) into the measuring cell containing PII protein (cell volume 1.4285 ml) with stirring at 350 rpm. The binding isotherms were calculated from the recorded data and fitted to the three-site binding model using the MicroCal ORIGIN software (Northampton, MA) as indicated.
Crystallization of Recombinant S. elongatus PII Protein
PII protein was crystallized using the sitting-drop technique by mixing 400 nl of the protein solution with equal amounts of the reservoir solution with the honeybee robot (Genomic Solutions Ltd). Drops were incubated at 20 °C, and images were recorded by the RockImager system (Formulatrix, Waltham, MA). For the PII-ADP form, the crystallization buffer was composed of 10 mm Tris, pH 7.4, 0.5 mm EDTA, 100 mm NaCl, 1% glycerol, and 2 mm ADP; crystals formed with the precipitant polyethylene glycol (PEG) 4000 at 20% in 100 mm Tris. Crystals in the absence of ADP were achieved under similar conditions with 18% PEG6000 and 100 mm Hepes, pH 6.5. Using glycerol or PEG 400 as the cryoprotectant, crystals were flash-frozen in liquid nitrogen. Diffraction data were collected at the Swiss Light Source (Villigen, Switzerland). Diffraction images were recorded with a MarCCD camera 225 (MAR Research, Norderstedt, Germany), and images were processed using XDS/XSCALE software (34). The crystal structure was solved by molecular replacement using the program Molrep (35) for which the high resolution structure 4AFF was used as a starting model. The structure was rebuilt and refined using the programs Coot and Refmac (36, 37). The quality of the structure was analyzed by the Procheck program (38). Figures were generated using PyMOL. The resolution of the wild type structures can be considered as high (1.6 Å) and medium resolutions (2.2 Å), and hence structural details and changes at this resolution can be clearly monitored. The structure in complex with ADP is at 3.1 Å, and the exact location of residues and co-factors becomes more critical. Details of the structure (such as side chains or termini) were introduced only on the basis of a clear electron density distribution for all atoms. Therefore, we believe that we discuss details that were clearly verified. At this lower resolution the structure is not being discussed on the basis of exact bond lengths and intra-residue distances as these may become imprecise while we focus on relative changes such as residue presence or absence and residual geometries.
RESULTS
PII Structures Carry Flexible Modules to Adapt to Effector and Protein Interaction
Crystal structures of PII enzymes have shown a variety of conformational states, with transitions typically affecting flexible modules surrounding the structurally invariant β-sheet core. These changes are expressed in particular in the T-loop element and in the C terminus in response to protein-protein interactions or effector interactions, or they are artificially stabilized through crystallographic contacts. More subtle changes, detectable in the vicinity of the ligand-binding sites, may be involved in allosteric regulation of neighboring effector binding sites after e.g. partial ligand occupation in trimeric structures.
In the PII protein from S. elongatus, the conformational states resulting from metabolic signal sensing and signal transduction of the protein have been thoroughly studied. However, snapshots of the protein alone and in complex with ADP have been lacking. Hence, we crystallized the S. elongatus PII protein in the absence and presence of ADP. Two crystal structures of the effector-free protein were resolved, one at 1.6 Å and one at 2.2 Å resolution, with one monomer or one trimer (PIINC1 and PIINC2) in the asymmetric unit, respectively. We subsequently crystallized the ADP-enriched protein and determined the structure at 3.1 Å with two trimers in the asymmetric unit. All structures were solved by molecular replacement, and the statistics of data collection and refinement are given in Table 2. We report these structures along with a discussion of their conformational transitions from the cofactor-free PII protein (PIINC1) to the PII protein with a single occupied ADP site (PIIADP1). Interestingly, the structure of PII in complex with three ADP molecules (PIIADP3) was simultaneously trapped, and the differences in the binding mode of the protein with the first, second, and third ADP molecule were dissected. We then placed this subset of structures into a series of previously reported structures (Table 1) to allow a detailed comparison of PIINC2, PIIADP1/PIIADP3, PIIOG1–3 (three PII structures occupied by three ATP and one, two, or three Mg/2-OG effectors), PIIOGex (structure equivalent to PIIOG-3), and PII·PipX structural states, which reflect the whole range of subtle transitions in effector binding pockets as well as significant conformational changes in the β-core periphery.
TABLE 2.
Data collection and refinement statistics
| PIINC1 | PIINC2 | PIIADP | |
|---|---|---|---|
| Data collection | |||
| Space group | P321 | P321 | P61 |
| Cell dimensions | |||
| a, b, c (Å) | 65.69, 65.69, 49.20 | 73.42, 73.42, 95.63 | 122.46, 122.46, 80.47 |
| α, β, γ (°) | 90, 90, 120 | 90, 90, 120 | 90, 90, 120 |
| Resolution (Å) | 33-1.6 (1.7-1.6) | 95.6-2.15 (2.28-2.15) | 48.7-3.1 (3.29-3.1) |
| Rsym or Rmerge | 0.03 (0.34) | 0.07 (0.57) | 0.13 (0.76) |
| I/σI | 32.3 (4.9) | 20.2 (2.4) | 8.8 (1.8) |
| Completeness (%) | 99.9 (99.4) | 100.0 (99.8) | 87.8 (90.1) |
| Redundancy | 8.1 (7.8) | 13.7 (13.6) | 3.2 (3.5) |
| Refinement | |||
| Resolution (Å) | 33-1.6 (1.7-1.6) | 63-2.15 (2.28-2.15) | 45-3.1 (3.41-3.10) |
| No. reflections | 16,524 | 16,722 | 11,089 |
| Rwork/Rfree | 0.21/0.23 | 0.23/0.27 (0.31/0.37) | 0.21/0.27 (0.30/0.36) |
| No. atoms (all) | |||
| Protein | 695 | 2.057 | 4.525 |
| Water | 56 | 48 | |
| B-Factors | |||
| Protein | 27.8 | 15.1 | 81 |
| Water | 35.5 | 31.8 | |
| r.m.s.d. | |||
| Bond lengths (Å) | 0.008 | 0.004 | 0.005 |
| Bond angles (°) | 1.28 | 0.78 | 1.05 |
| Ramachandran statistics | |||
| Residues in favored region, no. (%) | 253 (95.8) | 245 (97.0) | 546 (96.8) |
| Residues in allowed region, no. (%) | 261 (100.0) | 258 (100.0) | 564 (100.0) |
| Residues in outlier region, no. (%) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| PDB entry | 4C3I | 4C3M | 4C3K |
Structural Variability in PII in the Absence of Cofactors
PII structures devoid of cofactors were crystallized in two different space groups and superimposed to estimate deviations due to the influence of pH, chemical composition of the crystallization buffer, or crystal contacts (Fig. 1A). Interestingly, when we compared the structures with each other and with the previously reported structure of the PIIS49A variant, we found a significant deviation for PIINC1/PIINC2, resulting in an r.m.s.d. of 1.6 Å for the monomer with 76 of 91 Cα atoms superimposed, 1.4 Å for PIINC1/1QY7, and 0.9 Å for PIINC2/1QY7. The significant structural deviation between PIINC1 and PIINC2 likely results from the unique crystal packing obtained in the high resolution structure PIINC1, which shows crystal contacts exclusively mediated through anti-parallel strand pairing of the surface-exposed C-terminal β5-strand (Fig. 1, A and B). This structural arrangement known as β-augmentation restricts the back-folding of the C terminus in PIINC1 and thereby prevents the formation of the terminal β6-strand (Fig. 1B). As a consequence, helix α2 is not further stabilized through interactions with the β5/β6-strands and is released to move outward from the β-sheet core (Fig. 1B). Whereas in the mutant structure PIIS49A the T-loop is extended in a 90° angle relative to the trimeric molecular axis and is stabilized by crystal contacts, in PIINC1 and PIINC2, the T-loops are entirely disordered (Figs. 1A and 2). Residues at the basis of the T-loop located between β2 and β3 strands (Gly-35/Phe-36 and Phe-55/Leu-56) are stabilized through backbone interactions with each other and by backbone interaction with residues Thr-28 and Met-29 of the β2* strand of the neighboring subunit (Fig. 2A). In the following analysis and comparisons, we selected the PIINC2 structure because its overall geometry is the least affected by crystallographic packing artifacts. The three subunits of this structure are also identical, with an r.m.s.d. of 0.2 Å, and the comparison is not distorted after superposition of individual subunits.
FIGURE 1.

Comparison of PII structures from S. elongatus in the absence of cofactors. All structures are shown as schematics prepared using PyMOL. A, superposition of the two wild-type structures PIINC1 (green) and PIINC2 (red) with the previously published mutant structure (1QY7; blue). The structures are shown in two perspectives (top and side views), which are related to each other through a rotation around the x axis by 90°. The structures show significant deviations (yellow arrows) in the T-loop conformations and the C-terminal extensions (CT), which strongly depend on the crystallographic environment of the protein trimer. B, crystallographic packing of the PIINC1 structure, which shows that the C-terminal β-strand β6 is not formed due to crystallographic contacts (encircled by dotted lines) of trimers via their β5 strands.
FIGURE 2.
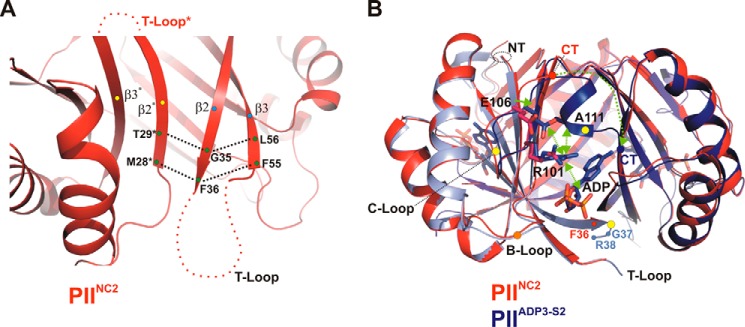
Transitions between structures PIINO-COF and PIIADP3. A, organization of the T-loop structure in the ligand-free PII trimer leads to an entirely flexible T-loop conformation. The PIINO-COF structure is shown in red as a schematic with a close-up view of the β-sheet core (β2, β3, β2*, and β3*). The H-bond interactions between terminal residues are marked by dotted lines. The terminal residues of the secondary structures are labeled. B, side view focusing on the S2 site of the trimeric superimposed PIINO-COF (red) and PIIADP3 (blue) complex structures. Residues that change their side-chain conformations are depicted, and changes are traced by green arrows. Secondary structure elements that significantly change their conformations upon ADP binding are marked with yellow dots (C terminus (CT), T-loop, and B-loop).
Structural Changes upon ADP Binding and Allosteric Modulation
A comparison of the trimeric structure of PIINC2 and of PIIADP1 revealed a small r.m.s.d. of 0.7 Å for 269 Cα atoms, which is in the same range as that determined for the PIIADP1/PIIADP3 trimers, i.e. 0.6 Å for 280 Cα atoms superimposed. After superimposing the trimers, we analyzed changes of mobile elements in these structures (T-loop and C terminus) and in the vicinity of the effector-binding sites S1-S3. The small changes surrounding the binding sites are in particular indicative of the anti-cooperative behavior of PII proteins (see data below). Per our definition, the individual sites in the PIIADP1 state are denoted PIIADP1-S1/S2/S3, with site S1 occupied by the single ADP and sites S2 and S3 ligand free. The three occupied sites of the PIIADP3 complex are termed accordingly, i.e. PIIADP3-S1/S2/S3.
First, to decipher the structural transitions in response to ADP binding, we visualized changes using fully occupied PIIADP3 relative to the ligand-free PIINC2 structure (Fig. 2B). ADP binding in sites S1 and S2 causes stabilization and translocation of the C terminus toward the ADP-binding sites (Figs. 2B and 3, A and C) through a cascade of small changes in the side and main chains. ADP binding induces the realignment of Arg-101 toward the β-phosphate. This shift is further transferred to Glu-106 located on the C-loop, which forms a salt bridge with Arg-101. Both side chains contact the backbone of Ala-111, whereby a small helix motif is moved into the subunit interface (Fig. 2B). ADP binding further leads to the stabilization of terminal residues of the T-loop (Gly-37–Arg-38–Gln-39), which form pure backbone interactions with the phosphate moiety, mainly mediated by Arg-38 (Figs. 2B and 3B).
FIGURE 3.

Transition between structures PIIADP1 and PIIADP3 and differential binding mode of ADP. A, superposition of the PIIADP1 (orange and yellow) and PIIADP3 (dark blue and magenta) structures shown as schematics with side views of the S1 effector binding sites. Secondary structure elements are marked with α1-α2 and β1-β4 (with * for the second molecule). The ADP cofactors are represented by stick structures and color-coded according to the protein backbone (orange and blue). B, close-up of the S1 site of PIIADP3 highlighting residues (labeled and represented by stick structures) involved in ADP binding. C, comparison of the structures of the S2 binding site of PIIADP1 and PIIADP3. The color codes are the same as for A, but additional subunits are light orange (PIIADP1) and light blue (PIIADP3). Significant structural changes in B-loop, T-loop, and C termini induced by ADP binding are enclosed in dotted rectangles. D, close-up of the S2 site of PIIADP3. ADP is bound only via main-chain interactions to residues of the B-loop and T-loop.
Next we compared the single and triple occupied ADP states to identify small changes that may reflect the anti-cooperative behavior of PII from S. elongatus (Fig. 3). Analysis of the PIIADP1-S1 and PIIADP3-S1 binding sites indicates only minor changes in backbone conformations, possibly introduced through the different diphosphate torsion angle geometries (Fig. 3A). The general binding mode of ADP in both S1 sites is based on main-chain H-bond interactions formed with Gly-87 and Asp-88 and a weaker additional contribution with Gly-89 (∼3.5 Å apart from ADP) located in the B-loop. The backbone of Arg-38 from the T-loop further stabilizes ADP from the “bottom” side (Fig. 3B). Side chains of Lys-90 and of C-loop residues Arg-101 and Arg-103 contribute to the further alignment and stabilization of the phosphate moiety of ADP from the top side (Figs. 3B and 4B, left). All residues that contribute side chains and glycines that allow backbone flexibility to adapt to the various effector molecules are highly conserved in the canonical PII proteins (31). Interestingly, the coordination of ADP in the PIIADP3-S2 and PIIADP3-S3 sites is quite different from that in PIIADP3-S1; in agreement with this difference, the superposition of the three PIIADP3 subunits shows an significantly increased structural heterogeneity at the C-terminal extensions and at the ADP-binding site. Specifically, in the S2 and S3 sites, the side-chain interactions of Lys-90, Arg-101, and Arg-103 with ADP are lacking, and the phosphate moiety of ADP is only stabilized by main-chain interactions with Gly-87, Gly-37, and Arg-38 (Figs. 3D and 4B). Of particular interest is the structural rearrangement of the side chain of Lys-58, which plays a key role in various PII conformations (12). In the S1 site, Lys-58 interacts with the side chain of Gln-39, whereas in the S2 and S3 sites it approaches the ADP-ribose moiety instead (Fig. 3D). Furthermore, in the S1 site, the backbone of B-loop residues Ile-86 and Gly-87 interact with the side chains of Gln-39 and Lys-58, respectively. These interactions result in a trapezoidal arrangement between the B-loop backbone (Ile-86, Gly-87), Gln-39 from the T-loop basis, and Lys-58 from the core of the PII body (Fig. 5). This arrangement, which shifts the basal part of the T-loop closer to the B-loop in the S1 site, does not exist in the S2 and S3 sites.
FIGURE 4.

Comparison of PIIADP3 and PIIOG1 structures. A, superposition of the two trimer structures PIIADP3 (dark blue) and PIIOG1 (magenta) showing the side view of the three binding sites. B, comparison of the structures of the ADP and ATP binding modes in PIIADP3-S1, PIIADP3-S2, and PIIOG1-S2. Interacting residues are represented by stick structures and are labeled.
FIGURE 5.
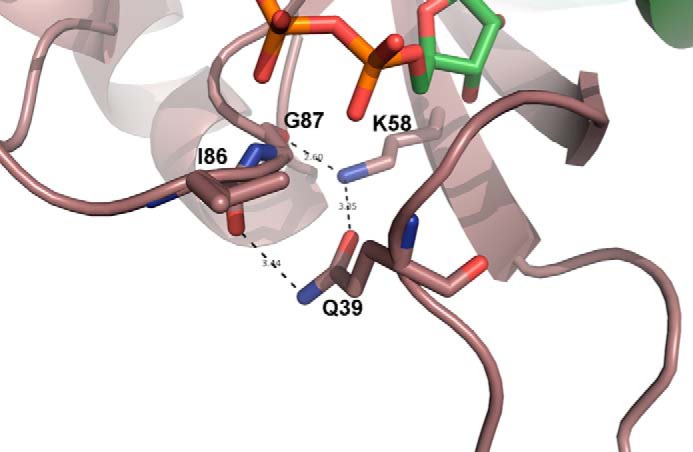
Close-up of the S1 site of PIIADP3. Residues Lys-58, Gln-39, Gly-87, and Ile-86 form a hydrogen-bonding network that drags the basal part of the T-loop (around Gln-39) close to the B loop (Ile-86/Gly-87).
With regard to the transition of the S2 site from the PIIADP1 state to the PIIADP3 state (see Fig. 3C), the most obvious movement is observed at the C terminus. A short C-terminal α-helix (αCT) of PIIADP3-S2 is formed and folds back toward the ADP-binding site. This movement is similar to that observed in the transition of the S1 site from the co-factor-free state to the ADP-filled state (see above). By contrast, this transition does not occur in the S3 site (Fig. 4A). The S2 site of the PIIADP1 state displays several subtle transitions near the ADP-binding site. In the PIIADP1 state, the N-terminal end of the β2-sheet is slightly shifted toward the empty binding pocket (Fig. 3C, flexed arrow). Furthermore, subtle changes in the B-loop and C-loop are visible. Interestingly, the side chain of the key residue Lys-58 is displaced in the S2 site of PIIADP1 as compared with the S1 site of the PIIADP1 or PIINC2 state, where it contacts the backbone of Gly-87. These subtle changes in S2 are apparently caused by the binding of ADP to the S1 site in PII and could affect the binding mode of the second ADP molecule described above.
Structural Differences between ADP and ATP Binding to PII from S. elongatus
To compare ADP binding of PIIADP3 with the most closely related PII/ATP structure from S. elongatus, we decided to use the PIIOG1 trimer complex with three ATP and one Mg2+/2-OG (note that the PDB entry 2XZW contains three independent trimers: PIIOG1 with one active site occupied by ATP/Mg2+/2-OG and PIIOG2 and PIIOG3 in the asymmetric unit). The superposition of this structure onto PIIADP3 resulted in an r.m.s.d. of 0.7 Å for 292 Cα atoms. Overall, the changes in the C terminus and in the B-loop are minute (Fig. 4A). However, the binding modes of ADP and ATP are significantly different (Fig. 4B). Binding of ATP is more similar to binding of ADP to the S1 site (involving side-chain interactions of Lys-90 and Arg-101/-103) than to binding of ADP to the weakly coordinated S2 and S3 sites of PIIADP3 (see above). In contrast to the binding of ADP to the S1 site, ATP interacts with side chains of Arg-38 and Gln-39 of the basal part of the T-loop. As a consequence, the trapezoidal arrangement between the B-loop backbone, Lys-58, and Gln-39 in PIIADP3-S1 is broken. Overall, the larger number of interactions increases the affinity of ATP toward all three binding sites but leads to only a minor stabilization of the distal part of the T-loop (Fig. 4A).
Influence of Interaction Partner PipX on PII Effector Binding
To understand the structural basis for the positive effect of ADP on the interaction of PII with the PII-target protein PipX described previously (15), we compared the PIIADP3 structure with the structures of the PII·PipX complex from S. elongatus and the PII·PipX-ADP complex from Anabaena sp. strain PCC 7120 (Table 1). The PII·PipX complexes from these two cyanobacteria show very similar vertically extended T-loop conformations (Fig. 6, A and B) that clasp the PipX partner protein. In our comparison of these complexes with PIIADP3, the PII conformation in the PII·PipX-ADP complex from Anabaena sp. PCC 7120 is almost identical to the structure of the S1 site of PIIADP3 but differs from that of the S2 and S3 sites (Fig. 7). Notably, the hydrogen-bonding network organized by residues Lys-58, Gln-39, Gly-87, and Ile-86 that drags the basal part of the T-loop close to the B loop is conserved, which causes a similar folding of the T-loop basis (see also Fig. 5). We questioned whether this particular T-loop conformation, which appears to be stabilized by PipX binding, influences the interaction between PII and its effectors. Using isothermal titration calorimetry, we determined the binding affinities of ADP and ATP to PII in the presence of equimolar amounts of PipX and PII (monomers). The data obtained were calculated assuming a model with three binding sites and indicated that PipX influences both affinity and anti-cooperativity of the binding reaction (Table 3). In the presence of PipX, binding site S1 has increased affinity for ADP (1.6-fold) but a 6-fold decreased affinity for ATP, which results in an inversion of the adenylate nucleotide preference, with a 3.5-fold higher affinity for ADP than for ATP. For binding site S2, PipX strongly decreases the affinity for both ATP and ADP binding (8- and 13-fold increase in KD, respectively), whereas binding site S3 is almost unaffected by PipX. In conclusion, only the first binding site in the PII·PipX complex becomes very sensitive to ADP. This behavior fits very well with the asymmetric structure of the PII-ADP complex; PipX binding stabilizes the S1 site conformation of PIIADP3, whereas the conformation of the S2 and S3 sites is displaced by PipX. By this mechanism, PipX enhances the sensitivity of PII toward ADP by favoring the PipX-PII-ADP1ATP2 population over PipX-PII-ATP3. Because coordination of ADP prevents the binding of 2-OG, the above results explain our previous observation that ADP can strongly antagonize the inhibitory effect of 2-OG on PII·PipX interaction (15).
FIGURE 6.

A, comparison of PII/PipX structures from Anabaena sp. PCC 7120 crystallized in the presence of ADP effector molecules (PDB entry 3N5B) and from S. elongatus (PDB entry 2XG8) in the absence of effector molecules. Orange, PII from Anabaena sp.; red, PII from S. elongatus; light green, PipX from Anabaena sp.; dark green, PipX from S. elongatus; ball-and-stick structure, ADP effector molecule in Anabaena sp. structure. B, close-up of A highlighting the T-loop conformations of the two structures. ADP binding of the structure has an influence on the T-loop conformation, whereby the anchoring residues are bent toward the ligand molecule. In the effector-free structure, no additional constrictions are imposed on the conformation.
FIGURE 7.
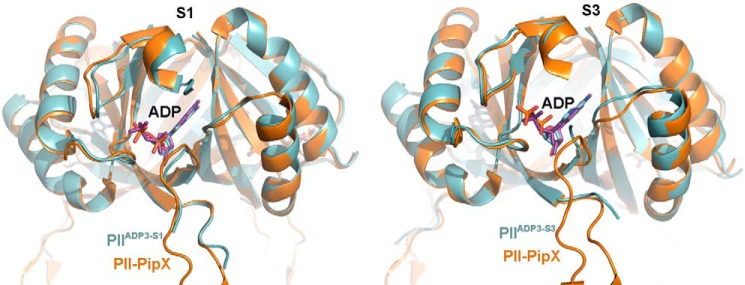
PipX binding favors the S1 state of PIIADP3. Superposition of PIIADP3 (blue) with Anabaena sp. PII in complex with PipX and ADP is shown. Ochre, protein backbone; purple, ADP molecule. PipX molecule is hidden for clarity. Left, superposition of the S1 site of PIIADP3; right, superposition of the S3 site of PIIADP3.
TABLE 3.
Influence of PipX on PII affinity towards ATP or ADP
Values correspond to the mean of three experiments ± S.E. The raw data were fitted using a three-site binding model for a PII trimer. For comparison, the original data for ATP and ADP binding in the absence of PipX are given in parentheses.
| KD1 | KDD2 | KD3 | |
|---|---|---|---|
| μm | μm | μm | |
| ATP | |||
| + PipX | 23.5 ± 8.1 | 101.5 ± 2.1 | 42.6 ± 25.5 |
| − PipX | (4.0 ± 0.1) | (12.5 ± 0.9) | (47.4 ± 21.9) |
| ADP | |||
| + PipX | 6.7 ± 2.4 | 255.7 ± 96.1 | 123.5 ± 32.5 |
| − PipX | (10.6 ± 3.2) | (19.3 ± 2.3) | (133.4 ± 5.2) |
Control of the PII·PipX-NtcA Interaction Network by the ATP/ADP Ratio
PipX is a co-activator of transcription factor NtcA (23). When loaded with 2-OG, the dimeric NtcA protein binds two PipX monomers (24). The interaction of PII with PipX serves to sequester PipX and to prevent NtcA activation. From the biochemical properties it is evident that 2-OG levels control the distribution of PipX between NtcA and PII (the PII·PipX-NtcA interaction network), with higher 2-OG levels shifting the equilibrium toward NtcA. However, the modulation of 2-OG-controlled PII·PipX interaction by ADP raises the question of whether PipX-NtcA interaction in the presence of PII may respond to the cellular energy state (measured as ATP/ADP ratio). To resolve this issue, we investigated the PII·PipX-NtcA interaction network using an SPR spectroscopy-based assay in which the NtcA protein was fixed on the surface of a sensor chip and challenged with a mixture of PipX and PII in the presence of ATP and ADP at various ratios and with a fixed concentration of 2-OG. In the presence of 2-OG, PipX binds to NtcA unless prevented by PII. Therefore, an increase in RU, which measures the binding of PipX to NtcA, indicates the distribution of PipX between PII and NtcA. In control experiments in the absence of PII, the adenylyl nucleotide regime did not influence the NtcA-PipX interaction (Fig. 8A, gray lines). In the presence of PII and 2 mm Mg2+-ATP + 4 mm 2-OG, the NtcA-PipX interaction was not affected because PII in its ATP-2-OG conformation is not suitable for PipX binding (24). In the presence of 2 mm ADP and 4 mm 2-OG, however, PII has a higher affinity for PipX and suppresses the NtcA-PipX complex formation as a competitive factor. In the presence of ATP and ADP at a 1:1 ratio and 4 mm 2-OG, the association of PipX to immobilized NtcA was reduced to one-third that in the absence of PII.
FIGURE 8.
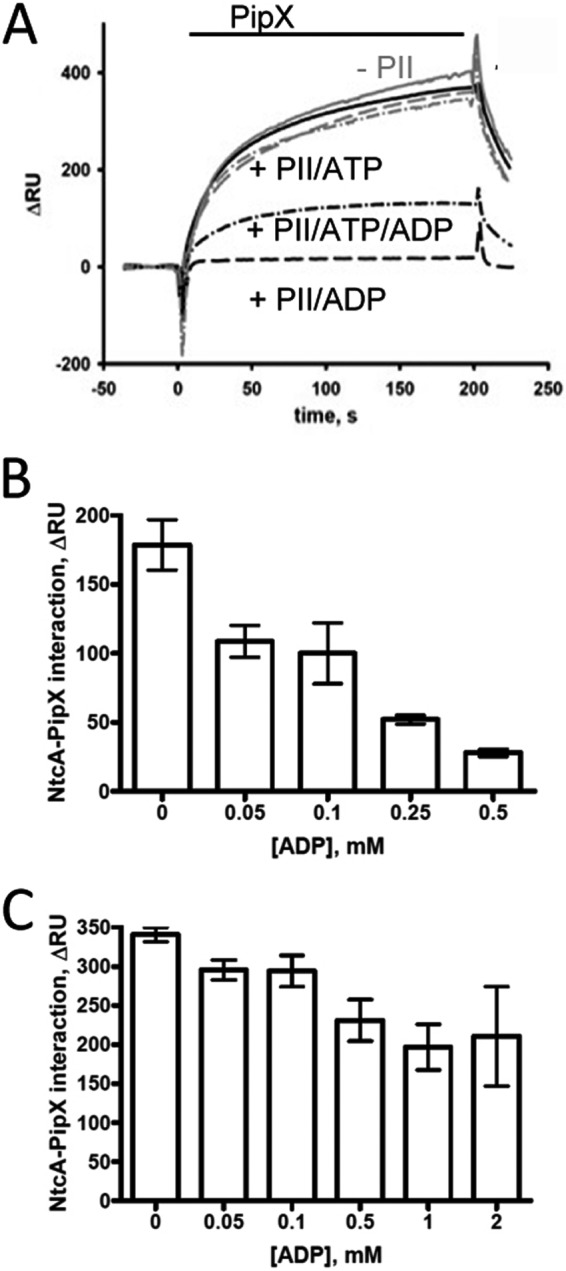
SPR analysis of PipX binding to NtcA in the presence of competitor PII. NtcA was bound to FC2 of a Ni2+-loaded NTA sensor chip (see “Experimental Procedures”), and FC1 was used as a control. The response difference (ΔRU) between FC2 and FC1 is shown. A, PipX was preincubated with PII in the presence of 4 mm 2-OG and with various concentrations of ATP and ADP (solid line, 2 mm ATP; dashed line, 2 mm ADP; dot-dashed line, 2 mm ATP plus 2 mm ADP) and then injected onto immobilized NtcA. The PipX binding experiments in the presence of PII are shown in black, the control binding experiment without PII is in gray. B and C, PipX was preincubated with PII in the presence of 1 mm ATP together with various amounts of ADP in the presence of either 1 mm (B) or 4 mm (C) 2-OG and then injected onto immobilized NtcA. The response signal at t = 197 s after the start of the injection phase was taken as a measure of protein binding; error bars indicate standard deviations from three independent measurements.
We then investigated the effect of subtle changes in the energy state (ATP/ADP ratio) on the PII·PipX-NtcA interaction network at high (4 mm) and intermediate (1 mm) 2-OG levels using the SPR assay described above. The response signal at the end of the PipX injection phase (t = 197 s) was taken as the value of protein binding (Fig. 8). In the presence of 1 mm 2-OG, the NtcA-PipX-PII network was extremely sensitive to the ATP/ADP ratio (Fig. 8A). Even small amounts of ADP (at an ADP:ATP ratio of 0.05:1) allowed PII to reduce the binding of PipX to NtcA by 40% (setting the binding of PipX to NtcA in the presence of 2-OG and ATP to 100%). When the 2-OG concentration was increased to 4 mm, the effect of ADP (in presence of 1 mm ATP) was greatly diminished. For example, 0.5 mm ADP decreased NtcA-PipX interaction only by 35% (Fig. 8B) compared with >80% reduction in the presence of 1 mm 2-OG.
Competition between NAGK and PipX for PII
In addition to interacting with PipX, PII is further involved in the activation of NAGK in S. elongatus. In vitro the interaction of NAGK with PII is antagonized by both ATP/Mg2+/2-OG and ADP. High ADP/ATP ratios, which are probably non-physiological, are required to noticeably impair NAGK activation by PII (39). Because PipX and NAGK compete for the 2-OG-free PII populations, we investigated the possibility that PipX may successfully compete with NAGK for PII in an energy state (ATP/ADP ratio)-dependent manner. To determine which receptor (PipX or NAGK) has higher affinity toward PII, we devised an SPR assay similar to that used for the analysis of PII·PipX-NtcA described above. In this case, NAGK was immobilized on a sensor chip and challenged with PII·PipX mixtures (each 100 nm monomers) in the presence of different effectors. PII interacted strongly with NAGK in the presence of 1 mm ATP regardless of the presence of PipX (Fig. 9A). It should be noted that in the presence of PipX, the association of the PII·NAGK complex was slightly but reproducibly slower than in the absence of PipX (Fig. 9A, dotted and solid lines), which indicated that NAGK actively recruited PII from its complex with PipX, which in turn slows down complex formation. In the presence of 1 mm ADP, however, PipX completely prevented the residual binding of PII to NAGK. At ATP and ADP ratios of 1:1 and 1:2, PipX only weakly affected PII·NAGK interaction (Fig. 9B). Under both conditions, the binding of PII to NAGK was decreased by the presence of PipX only by ∼30%, which indicated that PII has higher affinity to NAGK than to PipX even at a very low energy charge.
FIGURE 9.
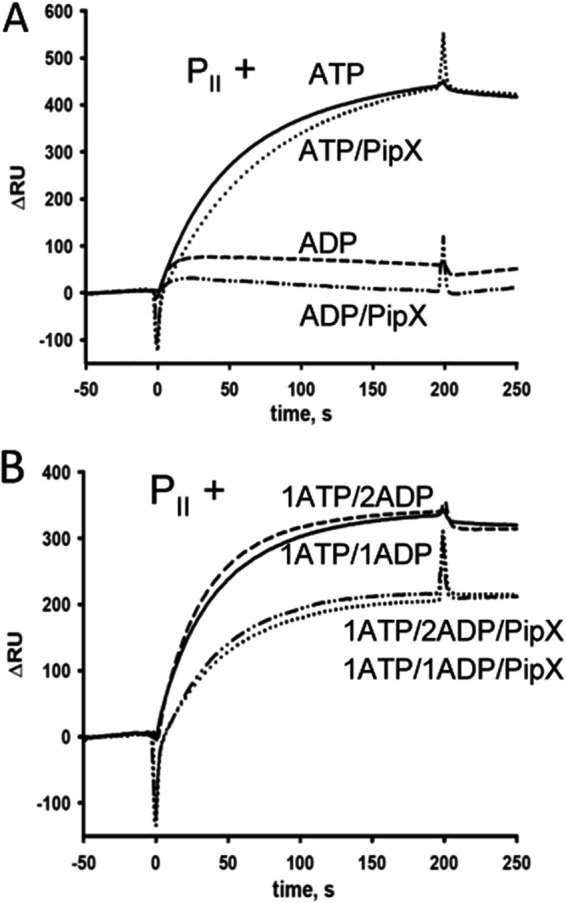
PipX effect on the PII-NAGK interaction. For SPR measurements NAGK was bound to FC2 of a Ni2+-loaded NTA sensor chip (see “Experimental Procedures”), and FC1 was used as a control. The response difference (ΔRU) between FC2 and FC1 is shown. A, association of PII-NAGK complex influenced by PipX and effectors; 100 nm PII was preincubated with 1 mm ATP (solid line), 1 mm ATP + 100 nm PipX (dotted line), 1 mm ADP (dashed line), or 1 mm ADP + 100 nm PipX (dot-dot-dashed line) and afterward injected onto immobilized NAGK. B, association of PII-NAGK complex influenced by PipX and ATP and ADP at different ratios: 1 mm ATP + 1 mm ADP without PipX (solid line), 1 mm ATP + 1 mm ADP + 100 nm PipX (dotted line), 1 mm ATP + 2 mm ADP (dashed line), and 1 mm ATP + 2 mm ADP + 100 nm PipX (dot-dot-dashed line).
DISCUSSION
Structures of PII signaling proteins, with their archetypical motifs including a conserved quaternary structure, T-, B-, and C-loops, and effector binding, have been studied in detail. However, most of the structural studies interpret the complex as being symmetric, with ligand-binding sites treated equivalently with respect to the stereochemistry. Moreover, the T-loop as prototype of a highly sophisticated signaling device has been observed in many different, mostly elongated conformations, which are always artificially stabilized by crystal contacts. By contrast, most of the PII structures from the cyanobacterium S. elongatus are not influenced by any crystallographic artifacts, and hence this system may provide the appropriate structural information to understand the function of PII. Our work presented here and previous studies have led to a repertoire of structures of PII from S. elongatus: PII without cofactors, PIIADP1, PIIADP3, PIIOG1, PIIOG2, PIIOG3, PII in complex with NAGK or PipX, and the mutant structures PII(S49A) and PII(I86N) as well as PII(I86N) in complex with ATP/Mg2+ and citrate in place of 2-OG. Hence, only PII in complex with ATP and mixed ADP/ATP structures are lacking for describing the structural basis of PII physiology. A summary of the various cyanobacterial structures together with other PII structures discussed in the following is shown in Table 1. A comprehensive table summarizing all PII structures resolved up to 2013 is given in Huergo et al. (40).
Structural Basis of Anti-cooperativity
A number of functional studies have shown that binding of effector molecules may occur cooperatively and/or anti-cooperatively. For PII from S. elongatus, anti-cooperativity for all three sites has been reported (9, 11). By contrast, the first and second 2-OG sites of the archaeal Archaeoglobus fulgidus GlnK3 protein show strong anti-cooperativity (−40-fold), but the second and third binding sites show cooperativity (+10-fold) (30). A similar anti-cooperative/cooperative coupling occurs with ATP binding to A. fulgidus GlnK2 (first/second sites: −10-fold; second/third sites: +4-fold) (41). Although these differences are obvious, they require explanation on a structural basis. Yet structural asymmetry is often not analyzed in detail even though previously published structures from different organisms indicate such an asymmetric behavior of PII proteins. As found for PII from S. elongatus, some binding sites in the same trimeric molecule of some structures were occupied, and others were unoccupied. For example, in the structure of GlnK1 from the archaeon Methanocaldococcus jannaschii crystallized in the presence of ADP with four trimers in the asymmetric unit, one or two ADP molecules occupied the binding sites (see Table 1) (42). Interestingly, all these partially occupied trimers occurred in the same asymmetric unit. Similarly, the ATP-Mg·2-OG complex of S. elongatus PII could be crystallized with three PII trimers in the asymmetric unit containing one, two, or three bound 2-OG molecules. The increase in KD value from site S1 (5.1 ± 4.0 μm) to site S2 (11.1 ± 1.8 μm) to site S3 (106.7 ± 14.8 μm) were in line with changes in the structures. The absence of 2-OG in the S2 and S3 binding sites was accompanied by the absence of Mg2+. Moreover, binding of the first 2-OG/Mg2+ molecules introduced a structural transition in the geometry of the neighboring ATP molecule of the S2 site. In site S3, the ATP was clamped differently than in sites S1 and S2, and structural changes in the vicinity of S3 were visible (e.g. C terminus does not fold back to the ligand). In our presented studies, we wanted to analyze structural changes in PII after binding to ADP to trace the changes induced by the effector molecule. We obtained a crystal with two PII molecules in the asymmetric unit, one of which contained only one ADP molecule whereas the second was fully occupied. In the latter structure the second and third ADP molecules are less tightly coordinated, and the binding pocket is not shielded by a structured T-loop basis. These observations are in line with the binding constants of ADP, showing only one high affinity site. A similar effect is seen in the GlnK1 structure of M. jannaschii occupied by ADP (42), where one of four trimers is occupied by two ADP molecules. The first ADP molecule is tightly coordinated (involving the C-loop contacts similar to the S1 state of S. elongatus PIIADP3), and the binding site is in a closed conformation with a distance between T- and B-loop (measured between Gln-39 and Pro-86) of 5 Å. By contrast, the second site shows fewer coordinations and is in an open conformation (distance Gln-39–Pro-86 of 7.8 Å). Asymmetry in ADP binding occurs also in the TT1020 protein structure, a PII homolog from Thermus thermophilus. Superposition of PIIADP3 with TT1020 reveals that the corresponding S1 sites yield the lowest r.m.s.d. values for the overall structure geometry. All these structures are in support of an asymmetric arrangement of PII proteins, which is obviously not unique to the PII protein from S. elongatus. A recent dynamic correlation analysis of 2-OG binding to S. elongatus PII (43) calculated the binding pocket sizes and revealed that ligation of 2-OG to site S1 alters the binding pocket size in sites S2 and S3. In accord with this result, the binding of ADP to site S1 of S. elongatus PII leads to subtle structural changes in the other two sites, which results in their reduced affinity toward ligands. Although it seems plausible that the transmission of intersubunit signals occurs along the central β2/β3 core of PII, which directly links the effector binding sites with each other (see Figs. 1A and 2A), a detailed atomic description of structural transitions is limited by the resolution of the structure of 3.1 Å.
T-loop Conformations
The only conformation of a fully resolved T-loop unaffected by crystal contact has been trapped in the structure of the S. elongatus PII-I86N variant in the presence of ATP/Mg2+; the structure shows the T-loop stabilized through the formation of an engineered additional intramolecular H-bond (9, 31). None of the structures of wild-type S. elongatus PII occupied by effector molecules (ADP, ATP, ATP/Mg2+/2-OG) shows the T-loop in an ordered conformation. In a number of PII proteins crystallized so far, an artificial T-loop structure is strongly stabilized by crystal contacts, and individual changes due to effector binding are, therefore, difficult to detect (e.g. PDB entries 3TA1, 3NCR, 2J9D; see Table 1). Yildiz et al. (42) argue that upon binding of ADP, the entire T-loop becomes stabilized. In the four trimers of the asymmetric unit, all five T-loops associated with AMP or ADP were strongly stabilized by crystallographic contacts and showed significantly different orientations. Moreover, in one binding site not occupied by any effector molecule, the T-loop was stabilized only through crystallographic contacts. This finding coincides with a structure of the S49A variant of PII from S. elongatus, where T-loops are also stabilized in the absence of effector molecules by simple crystallographic contacts (6). A plausible explanation for this variability assumes the T-loop as a very flexible “device” in solution that can adapt very easily to protein complexes of different size and follow different structural constraints given by proteins such as PipX, NAGK, or AmtB.
PII Protein-Effector Distribution in Cells
As a consequence of the extensive (anti-)cooperative properties of PII proteins, distinct PII populations will form in cells depending on the effector concentrations, with each of them having its own signaling properties. Moreover, modulation of signal perception by receptor interaction allows the PII-receptor system to respond with different sensitivity toward the different metabolites. For example, regulation of NAGK by PII is highly sensitive to 2-OG but only weakly responds to fine-tuned changes in ADP levels, whereas the PII·PipX-NtcA interaction network responds strongly to the ADP/ATP ratio. Of the 10 theoretical states of PII with completely occupied ATP/ADP sites, only 5 states are physiologically meaningful: the 3 PII-ATP3 states with 1–3 bound 2-OG molecules and the 2 PII-ATP2-ADP1 states with 0 or 1 2-OG molecule bound. The 2-OG-free PII-ATP3 state is unlikely to be formed as the Kd of the first site (5 μm) is much lower than physiological 2-OG concentrations. The theoretically expected species PII-ATP2-ADP1 with two molecules of 2-OG is probably not formed, as concluded from previous isothermal titration calorimetry analysis (15). This implies that ADP, bound to one site of the PII trimer, not only prevents 2-OG from binding to its own site but also has a negative impact on 2-OG binding to one neighboring subunit. The PII-ATP1-ADP2 species is probably irrelevant in S. elongatus cells, as its formation requires an excess of ADP over ATP, which is unlikely in living cells. The distribution of the various PII states is further affected by the interaction with receptor proteins, as shown by the effect of PipX on ATP and ADP affinities for PII. The fully ATP-ligated form of PII will probably prevail in the absence of PipX, and this form clearly shows the highest sensitivity toward 2-OG and regulates NAGK. However, in complex with PipX, one binding site of PII has a preference for ADP. Binding of ADP tunes down the antagonistic effect of 2-OG on PipX-PII interaction, and therefore, the PII·PipX-NtcA interaction network responds to the energy status: at very low 2-OG levels, which represents nitrogen excess (44), PipX cannot bind to NtcA and, hence, will always be bound to PII, irrespective of the ADP/ATP ratio. In the presence of intermediate concentrations of 2-OG (in vitro at 1 mm), PipX could bind to the NtcA·2-OG complex. As long as the ADP levels are low enough to enforce the PII-ATP3-2-OG3 state, PII is unable to compete for PipX binding, and PipX is in fact bound to NtcA. However, a slight increase in ADP levels (0.05 mm) already affects the network; at a 0.05:1 ADP:ATP ratio, NtcA-PipX interaction is reduced by 40%. PipX increases the ADP sensitivity of PII and thereby displaces 2-OG, allowing formation of the PII·PipX complex. At even higher ADP levels (0.5 mm), most PipX (>80%) is bound to PII and removed from NtcA. In essence, these data demonstrate that the PipX distribution between PII and NtcA at intermediate (physiological) 2-OG levels is highly sensitive to the ADP/ATP ratio. At very high 2-OG levels (4 mm), the interaction between PipX and NtcA is so stable that ADP has only a marginal influence on the distribution of PipX. In physiological context, these results suggest that a main function of PII in the NtcA-PipX-PII network is to switch off PipX-activated NtcA gene expression when the energy supply of the cell ceases transiently under conditions of intermediate cellular 2-OG levels.
Recent Advances in PII Biology or Is PII a Slow ATPase?
In the GlnK-AmtB complex, the T-loop blocks the pore for ammonium transport (5). As in the PII·PipX interaction in cyanobacteria, the GlnK-AmtB complex in E. coli is formed in the presence of ADP and dissociates in the presence of ATP-Mg/2-OG. Although the regulated processes are completely different, the PII response to metabolites and target regulation appeared to undergo the same conserved mechanism. A recent paper challenged this view; Radchenko et al. (26) showed that in the absence of 2-OG, GlnK displays intrinsic ATPase activity, slowly converting ATP-GlnK to ADP-GlnK, which then binds AmtB. The authors suggested that it is the 2-OG-regulated switch from the ATP-PII to the ADP-PII state that promotes association with AmtB. Although this mechanism makes sense for PII complexes stabilized by the ADP state, it contradicts the formation of PII-receptor complexes stabilized by the ATP state of PII, such as the PII·NAGK association. PII binds in its ATP state in the absence of 2-OG to its target NAGK. The 2-OG-regulated ATPase activity of PII would prevent PII·NAGK interaction due to formation of the ADP state of PII. However, the experimental evidence of PII·NAGK interaction contradicts this assumption (15, 39, 45, 46). Therefore, even if the intrinsic ATPase activity of GlnK is relevant for AmtB interaction, this mechanism cannot explain the diversity of PII signal transduction in the whole PII family. Numerous examples in other organisms have demonstrated that the PII regulatory network is as complex as that in cyanobacteria. The combined effect of the metabolites ADP, ATP, and 2-OG on PII signaling has been studied in E. coli and R. rubrum, and the sensing mechanism undergoes the same principle as in cyanobacteria (10, 25, 27, 29, 47). However, the aspect of competition between different PII targets in these organisms has not yet been addressed and further complicates the system.
In summary, the array of PII signal-input states is not only organized by (anti-)cooperative intersubunit and effector molecule interactions but is further modulated by the receptor protein to which PII is signaling. This enables the PII proteins to simultaneously regulate multiple targets with different sensitivity in response to its metabolic effectors ATP, ADP, and 2-oxoglutarate. The asymmetry behavior of PII proteins and its modulation by target proteins appears to be a key requisite for its function as a multitasking information processor.
Acknowledgments
We thank Jan Lüddecke for critically reading the manuscript, Karen A. Brune for linguistic revision of the manuscript, Reinhard Albrecht and Vasuki Ranjani-Chellamuthu for help with crystallization and crystal handling, and the entire beamline staff of PXII at the Swiss Light Source for beamline maintenance and continuous support.
This work was supported by the Max Planck Society, the University of Tübingen, Germany, and German Science Foundation Grant FO195/9-1 (to K. F.).
The atomic coordinates and structure factors (codes 4C3M, 4C3K, and 4C3L) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- 2-OG
- 2-oxoglutarate
- NAGK
- N-acetyl-l-glutamate kinase
- SPR
- surface plasmon resonance
- RU
- resonance unit
- NTA
- nitrilotriacetic acid
- r.m.s.d.
- root mean square deviation
- FC2 and FC3
- flow cell 2 and 3, respectively
- PDB
- Protein data bank.
REFERENCES
- 1. Ninfa A. J., Jiang P. (2005) PII signal transduction proteins. Sensors of α-ketoglutarate that regulate nitrogen metabolism. Curr. Opin. Microbiol. 8, 168–173 [DOI] [PubMed] [Google Scholar]
- 2. Leigh J. A., Dodsworth J. A. (2007) Nitrogen regulation in bacteria and archaea. Annu. Rev. Microbiol. 61, 349–377 [DOI] [PubMed] [Google Scholar]
- 3. Forchhammer K. (2008) PII signal transducers. Novel functional and structural insights. Trends Microbiol. 16, 65–72 [DOI] [PubMed] [Google Scholar]
- 4. Sant'Anna F. H., Trentini D. B., de Souto Weber S., Cecagno R., da Silva S. C., Schrank I. S. (2009) The PII superfamily revised. A novel group and evolutionary insights. J. Mol. Evol. 68, 322–336 [DOI] [PubMed] [Google Scholar]
- 5. Huergo L. F., Pedrosa F. O., Muller-Santos M., Chubatsu L. S., Monteiro R. A., Merrick M., Souza E. M. (2012) PII signal transduction proteins. Pivotal players in post-translational control of nitrogenase activity. Microbiology 158, 176–190 [DOI] [PubMed] [Google Scholar]
- 6. Xu Y., Carr P. D., Clancy P., Garcia-Dominguez M., Forchhammer K., Florencio F., Vasudevan S. G., Tandeau de Marsac N., Ollis D. L. (2003) The structures of the PII proteins from the cyanobacteria Synechococcus sp. PCC 7942 and Synechocystis sp. PCC 6803. Acta Crystallogr. D 59, 2183–2190 [DOI] [PubMed] [Google Scholar]
- 7. Xu Y., Cheah E., Carr P. D., van Heeswijk W. C., Westerhoff H. V., Vasudevan S. G., Ollis D. L. (1998) GlnK, a PII-homologue. Structure reveals ATP binding site and indicates how the T-loops may be involved in molecular recognition. J. Mol. Biol. 282, 149–165 [DOI] [PubMed] [Google Scholar]
- 8. Sakai H., Wang H., Takemoto-Hori C., Kaminishi T., Yamaguchi H., Kamewari Y., Terada T., Kuramitsu S., Shirouzu M., Yokoyama S. (2005) Crystal structures of the signal transducing protein GlnK from Thermus thermophilus HB8. J. Struct. Biol. 149, 99–110 [DOI] [PubMed] [Google Scholar]
- 9. Fokina O., Chellamuthu V. R., Zeth K., Forchhammer K. (2010) A novel signal transduction protein PII variant from Synechococcus elongatus PCC 7942 indicates a two-step process for NAGK-PII complex formation. J. Mol. Biol. 399, 410–421 [DOI] [PubMed] [Google Scholar]
- 10. Jiang P., Ninfa A. J. (2007) Escherichia coli PII signal transduction protein controlling nitrogen assimilation acts as a sensor of adenylate energy charge in vitro. Biochemistry 46, 12979–12996 [DOI] [PubMed] [Google Scholar]
- 11. Fokina O., Chellamuthu V. R., Forchhammer K., Zeth K. (2010) Mechanism of 2-oxoglutarate signaling by the Synechococcus elongatus PII signal transduction protein. Proc. Natl. Acad. Sci. U.S.A. 107, 19760–19765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Truan D., Huergo L. F., Chubatsu L. S., Merrick M., Li X. D., Winkler F. K. (2010) A new PII protein structure identifies the 2-oxoglutarate binding site. J. Mol. Biol. 400, 531–539 [DOI] [PubMed] [Google Scholar]
- 13. Forchhammer K., Hedler A. (1997) Phosphoprotein PII from cyanobacteria - analysis of functional conservation with the PII signal-transduction protein from Escherichia coli. Eur. J. Biochem. 244, 869–875 [DOI] [PubMed] [Google Scholar]
- 14. Radchenko M. V., Thornton J., Merrick M. (2010) Control of AmtB-GlnK complex formation by intracellular levels of ATP, ADP, and 2-oxoglutarate. J. Biol. Chem. 285, 31037–31045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fokina O., Herrmann C., Forchhammer K. (2011) Signal-transduction protein PII from Synechococcus elongatus PCC 7942 senses low adenylate energy charge in vitro. Biochem. J. 440, 147–156 [DOI] [PubMed] [Google Scholar]
- 16. Chapman A. G., Fall L., Atkinson D. E. (1971) Adenylate energy charge in Escherichia coli during growth and starvation. J. Bacteriol. 108, 1072–1086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chapman A. G., Atkinson D. E. (1977) Adenine nucleotide concentrations and turnover rates. Their correlation with biological activity in bacteria and yeast. Adv. Microb. Physiol. 15, 253–306 [DOI] [PubMed] [Google Scholar]
- 18. Kallas T., Castenholz R. W. (1982) Internal pH and ATP-ADP pools in the cyanobacterium Synechococcus sp. during exposure to growth-inhibiting low pH. J. Bacteriol. 149, 229–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Burillo S., Luque I., Fuentes I., Contreras A. (2004) Interactions between the nitrogen signal transduction protein PII and N-acetyl glutamate kinase in organisms that perform oxygenic photosynthesis. J. Bacteriol. 186, 3346–3354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Heinrich A., Maheswaran M., Ruppert U., Forchhammer K. (2004) The Synechococcus elongatus PII signal transduction protein controls arginine synthesis by complex formation with N-acetyl-l-glutamate kinase. Mol. Microbiol. 52, 1303–1314 [DOI] [PubMed] [Google Scholar]
- 21. Maheswaran M., Urbanke C., Forchhammer K. (2004) Complex formation and catalytic activation by the PII signaling protein of N-acetyl-l-glutamate kinase from Synechococcus elongatus strain PCC 7942. J. Biol. Chem. 279, 55202–55210 [DOI] [PubMed] [Google Scholar]
- 22. Espinosa J., Forchhammer K., Burillo S., Contreras A. (2006) Interaction network in cyanobacterial nitrogen regulation. PipX, a protein that interacts in a 2-oxoglutarate dependent manner with PII and NtcA. Mol. Microbiol. 61, 457–469 [DOI] [PubMed] [Google Scholar]
- 23. Espinosa J., Forchhammer K., Contreras A. (2007) Role of the Synechococcus PCC 7942 nitrogen regulator protein PipX in NtcA-controlled processes. Microbiology 153, 711–718 [DOI] [PubMed] [Google Scholar]
- 24. Llácer J. L., Espinosa J., Castells M. A., Contreras A., Forchhammer K., Rubio V. (2010) Structural basis for the regulation of NtcA-dependent transcription by proteins PipX and PII. Proc. Natl. Acad. Sci. U.S.A. 107, 15397–15402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jiang P., Ninfa A. J. (2009) α-Ketoglutarate controls the ability of the Escherichia coli PII signal transduction protein to regulate the activities of NRII (NtrB) but does not control the binding of PII to NRII. Biochemistry 48, 11514–11521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Radchenko M. V., Thornton J., Merrick M. (2013) PII signal transduction proteins are ATPases whose activity is regulated by 2-oxoglutarate. Proc. Natl. Acad. Sci. U.S.A. 110, 12948–12953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhang Y., Pohlmann E. L., Halbleib C. M., Ludden P. W., Roberts G. P. (2001) Effect of PII and its homolog GlnK on reversible ADP-ribosylation of dinitrogenase reductase by heterologous expression of the Rhodospirillum rubrum dinitrogenase reductase ADP-ribosyl transferase-dinitrogenase reductase-activating glycohydrolase regulatory system in Klebsiella pneumoniae. J. Bacteriol. 183, 1610–1620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhang Y. P., Pohlmann E. L., Ludden P. W., Roberts G. P. (2003) Regulation of nitrogen fixation by multiple PII homologs in the photosynthetic bacterium Rhodospirillum rubrum. Symbiosis 35, 85–100 [Google Scholar]
- 29. Teixeira P. F., Jonsson A., Frank M., Wang H., Nordlund S. (2008) Interaction of the signal transduction protein GlnJ with the cellular targets AmtB1, GlnE and GlnD in Rhodospirillum rubrum. Dependence on manganese, 2-oxoglutarate, and the ADP/ATP ratio. Microbiology 154, 2336–2347 [DOI] [PubMed] [Google Scholar]
- 30. Maier S., Schleberger P., Lü W., Wacker T., Pflüger T., Litz C., Andrade S. L. (2011) Mechanism of disruption of the Amt-GlnK complex by PII-mediated sensing of 2-oxoglutarate. PLoS ONE 6, e26327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zeth K., Fokina O., Forchhammer K. (2012) An engineered PII protein variant that senses a novel ligand. Atomic resolution structure of the complex with citrate. Acta Crystallogr. D Biol. Crystallogr. 68, 901–908 [DOI] [PubMed] [Google Scholar]
- 32. Bueno R., Pahel G., Magasanik B. (1985) Role of glnB and glnD gene products in regulation of the glnALG operon of Escherichia coli. J. Bacteriol. 164, 816–822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Studier F. W., Rosenberg A. H., Dunn J. J., Dubendorff J. W. (1990) Use of T7 RNA polymerase to direct expression of cloned genes. Methods Enzymol. 185, 60–89 [DOI] [PubMed] [Google Scholar]
- 34. Kabsch W. (2010) XDS. Acta Crystallogr. D 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Vagin A., Teplyakov A. (1997) MOLREP. An automated program for molecular replacement. J. Appl. Crystallogr. 30, 1022–1025 [Google Scholar]
- 36. Murshudov G. N., Vagin A. A., Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 37. Emsley P., Cowtan K. (2004) Coot. Model-building tools for molecular graphics. Acta Crystallogr. D 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 38. Laskowski R. A., Macarthur M. W., Moss D. S., Thornton J. M. (1993) Procheck. A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26, 283–291 [Google Scholar]
- 39. Lüddecke J., Forchhammer K. (2013) From PII signaling to metabolite sensing. A novel 2-oxoglutarate sensor that details PII-NAGK complex formation. PLoS ONE 8, e83181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Huergo L. F., Chandra G., Merrick M. (2013) PII signal transduction proteins. Nitrogen regulation and beyond. FEMS Microbiol. Rev. 37, 251–283 [DOI] [PubMed] [Google Scholar]
- 41. Helfmann S., Lü W., Litz C., Andrade S. L. (2010) Cooperative Binding of MgATP and MgADP in the Trimeric PII Protein GlnK2 from Archaeoglobus fulgidus. J. Mol. Biol. 402, 165–177 [DOI] [PubMed] [Google Scholar]
- 42. Yildiz O., Kalthoff C., Raunser S., Kühlbrandt W. (2007) Structure of GlnK1 with bound effectors indicates regulatory mechanism for ammonia uptake. EMBO J. 26, 589–599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ma C. W., Luddecke J., Forchhammer K., Zeng A. P. (2013) Population shift of binding pocket size and dynamic correlation analysis shed new light on the anticooperative mechanism of PII protein. Proteins 10.1002/prot.24477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Muro-Pastor M. I., Reyes J. C., Florencio F. J. (2001) Cyanobacteria perceive nitrogen status by sensing intracellular 2-oxoglutarate levels. J. Biol. Chem. 276, 38320–38328 [DOI] [PubMed] [Google Scholar]
- 45. Beez S., Fokina O., Herrmann C., Forchhammer K. (2009) N-Acetyl-l-glutamate kinase (NAGK) from oxygenic phototrophs. PII signal transduction across domains of life reveals novel insights in NAGK control. J. Mol. Biol. 389, 748–758 [DOI] [PubMed] [Google Scholar]
- 46. Llácer J. L., Contreras A., Forchhammer K., Marco-Marín C., Gil-Ortiz F., Maldonado R., Fita I., Rubio V. (2007) The crystal structure of the complex of PII and acetylglutamate kinase reveals how PII controls the storage of nitrogen as arginine. Proc. Natl. Acad. Sci. U.S.A. 104, 17644–17649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Jiang P., Ninfa A. J. (2009) Sensation and signaling of α-ketoglutarate and adenylylate energy charge by the Escherichia coli PII signal transduction protein require cooperation of the three ligand-binding sites within the PII trimer. Biochemistry 48, 11522–11531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zhao M. X., Jiang Y. L., Xu B. Y., Chen Y., Zhang C. C., Zhou C. Z. (2010) Crystal structure of the cyanobacterial signal transduction protein PII in complex with PipX. J. Mol. Biol. 402, 552–559 [DOI] [PubMed] [Google Scholar]