

Background: Sortase enzymes catalyze a transpeptidation reaction that displays bacterial surface proteins.
Results: Structural and computational studies reveal how the sortase B enzyme recognizes its sorting signal substrate.
Conclusion: Sortase enzymes catalyze transpeptidation using a substrate-stabilized oxyanion hole.
Significance: The results of this work could facilitate the rational design of sortase inhibitors.
Keywords: Cell Surface Enzymes, Crystal Structure, Enzyme Mechanisms, Molecular Dynamics, Staphylococcus aureus
Abstract
Sortase cysteine transpeptidases covalently attach proteins to the bacterial cell wall or assemble fiber-like pili that promote bacterial adhesion. Members of this enzyme superfamily are widely distributed in Gram-positive bacteria that frequently utilize multiple sortases to elaborate their peptidoglycan. Sortases catalyze transpeptidation using a conserved active site His-Cys-Arg triad that joins a sorting signal located at the C terminus of their protein substrate to an amino nucleophile located on the cell surface. However, despite extensive study, the catalytic mechanism and molecular basis of substrate recognition remains poorly understood. Here we report the crystal structure of the Staphylococcus aureus sortase B enzyme in a covalent complex with an analog of its NPQTN sorting signal substrate, revealing the structural basis through which it displays the IsdC protein involved in heme-iron scavenging from human hemoglobin. The results of computational modeling, molecular dynamics simulations, and targeted amino acid mutagenesis indicate that the backbone amide of Glu224 and the side chain of Arg233 form an oxyanion hole in sortase B that stabilizes high energy tetrahedral catalytic intermediates. Surprisingly, a highly conserved threonine residue within the bound sorting signal substrate facilitates construction of the oxyanion hole by stabilizing the position of the active site arginine residue via hydrogen bonding. Molecular dynamics simulations and primary sequence conservation suggest that the sorting signal-stabilized oxyanion hole is a universal feature of enzymes within the sortase superfamily.
Introduction
Surface proteins in bacteria play key roles in the infection process by promoting microbial adhesion to host tissues, nutrient acquisition, host cell entry, and the suppression of the immune response. In Gram-positive bacteria, virulence factors are displayed by sortase enzymes, a superfamily of cysteine transpeptidases that join proteins bearing a cell wall sorting signal to the cell wall or to other proteins to construct pili (1–6). Sortases have proven to be useful molecular biology tools to site-specifically attach proteins to a variety of biomolecules and are considered a potential drug target because they display virulence factors. The clinically important pathogen, Staphylococcus aureus, displays surface proteins using two sortase enzymes, sortase A (SrtA)2 and sortase B (SrtB). srtA− strains of S. aureus are significantly attenuated in virulence, whereas srtB− strains establish less persistent infections (7–9). SrtA plays a “housekeeping” role in the cell, covalently mounting a variety of proteins to the cell wall, whereas SrtB anchors the heme transporter IsdC, a key component of the iron-regulated surface determinant system that captures heme-iron from hemoglobin (10–12). The mechanism of catalysis is best understood for SrtA, the archetypal member of the sortase superfamily. Proteins anchored by SrtA possess a C-terminal cell wall sorting signal that consists of an LPXTG motif that is processed by the enzyme, followed by a hydrophobic transmembrane segment and a positively charged C-terminal tail (13). SrtA operates through a ping-pong mechanism that begins when its active site cysteine residue nucleophilically attacks the carbonyl carbon of the threonine in the LPXTG motif. This results in a tetrahedral intermediate that, after cleavage of the threonine-glycine peptide bond, generates a semistable enzyme-substrate thioacyl intermediate (14, 15). The protein is then transferred by SrtA to the cell wall precursor, lipid II, when the amino group in this molecule nucleophilically attacks the thioacyl linkage creating a second tetrahedral intermediate that collapses to form the covalently linked protein-lipid II product (16–18). Transglycosylation and transpeptidation reactions that synthesize the cell wall then incorporate this product into the peptidoglycan. SrtB anchors the IsdC protein to the cell wall through a similar mechanism. However, unlike SrtA, SrtB recognizes a unique NPQTN sorting signal, and it attaches IsdC to un-cross-linked peptidoglycan instead of heavily cross-linked peptidoglycan (19, 20).
Sortase enzymes adopt an α/β sortase fold that contains three proximally positioned active site residues: His130, Cys223, and Arg233 (SrtB numbering). Although the nucleophilic role of the cysteine residue is well established, various catalytic functions have been proposed for the histidine and arginine residues. His130 was originally thought to activate the cysteine by forming a histidine-cysteine ion pair (21), but more recent data suggest that it instead functions as a general acid/base (14, 22). Arg233 has been proposed to either stabilize substrate binding (23–26), function as a general base (27), or directly stabilize the tetrahedral catalytic intermediates (14, 22, 23). Other residues within the active sites of sortases have also been proposed to participate in catalysis, including Asp225 in SrtB, which was postulated to participate in a His130-Cys223-Asp225 catalytic triad (28). The catalytic mechanism has remained poorly understood because the tetrahedral and acyl intermediates of catalysis are too short lived to be characterized by either NMR or x-ray crystallography. Several sortase structures have been determined in the absence of their substrates (28–32) or covalently bound to generic sulfhydryl modifiers (33). However, only a single structure of a sortase enzyme covalently bound to its sorting signal substrate has been reported (the NMR structure of SrtA bound to an LPAT substrate analog) (1, 24). This structure revealed that the active site in this enzyme undergoes substantial changes in its structure and dynamics that facilitate specific recognition of the sorting signal, but it did not provide an atomic level view of the positioning of atoms within the active site because their coordinates were not well defined in the NMR structure because of resonance line broadening. Thus, the structural features utilized by sortases to stabilize key tetrahedral and thioacyl reaction intermediates remain poorly understood.
Here we report the 2.5 Å crystal structure of SrtB covalently bound to an analog of its NPQTN sorting signal. The structure of the complex closely resembles the thioacyl intermediate formed during catalysis, laying the groundwork for MD simulations to investigate the catalytic mechanism. The results of these simulations and in vitro transpeptidation measurements suggest that Arg233 and the backbone amide of Glu224 form an oxyanion hole that stabilizes high energy tetrahedral catalytic intermediates. Interestingly, a highly conserved threonine residue within the sorting signal actively participates in constructing the oxyanion hole by hydrogen bonding to the active site arginine residue. MD simulations of SrtA, as well as primary sequence conservation, suggest that all sortases will use a similar substrate-stabilized mechanism to anchor proteins to the cell wall or to assemble pili.
EXPERIMENTAL PROCEDURES
Production, Crystallization, and Structure Determination of SrtB-NPQT* Complex
DNA encoding SrtB (residues 31–244) was amplified by PCR from S. aureus genomic DNA, cloned into a pE-SUMO vector (LifeSensors) and transformed into Escherichia coli Rosetta (DE3) pLysS cells (Novagen). Protein expression was induced by addition of 1 mm isopropyl β-d-1-thiogalactopyranoside and allowed to continue for 16 h at 16 °C. Protein was purified by affinity purification using HisPur cobalt resin (Thermo) per the manufacturer's instructions. The His6-SUMO tag was then cleaved by incubating the protein overnight at 4 °C with recombinant ULP1 protease and removed by reapplying the protein mixture to the HisPur cobalt resin. Cbz-NPQT* (where T* is (2R,3S)-3-amino-4-mercapto-2-butanol, and Cbz is a carbobenzyloxy protecting group) was synthesized as in Ref. 34 and added to purified SrtB in modification buffer (10 mm Tris-HCl, pH 7.0, 20 mm NaCl, 1 m l-proline) at a ratio of 10:1 for a final concentration of 1 mm Cbz-NPQT* to 100 μm SrtB. The reaction was first reduced with 1 mm DTT for 4 h, then oxidized by addition of 10 mm CuCl2, and allowed to rock gently at room temperature for 7 days. Production of stable complex was confirmed by MALDI mass spectrometry.
Crystals of the SrtB-NPQT* complex were produced from a stock of 150 μm SrtB-NPQT* in 10 mm Tris-HCl, pH 7.0, 20 mm NaCl. Crystals were grown using the hanging drop, vapor diffusion method in 2.8 m ammonium sulfate, 70 mm sodium citrate, pH 5.0. Data were collected on Beamline 24-ID-C at 100 K at the Advanced Photon Source (λ = 0.964 Å). Three data sets were scaled, integrated, and merged using XDS and XSCALE (35). Using conventional criteria, the resolution boundary for the data set might have been drawn at 2.9 Å given that I/σ in this shell (2.98–2.90 Å) is 2.0, and Rmerge is 72%, with a completeness of 95% and a multiplicity of 5.2. However, recent studies from Karplus and Diederichs (36) have indicated that the CC1/2 statistic has superior properties as an indicator of data precision compared with Rmerge. Moreover, in their study, and in the following work (37), the authors show that high resolution data typically discarded because of high Rmerge values (i.e., over the conventionally acceptable threshold value of ∼60–80%), and low I/σ values (i.e., under the conventionally acceptable threshold value of 2) actually contain information that can improve the quality of the model if used in refinement. Given these results, we thought our model would improve if we used the more generous resolution cutoff (2.5 Å) indicated by the CC1/2 statistic (50.4% in the 2.5 Å shell), rather than a conventionally accepted limit (2.9 Å) indicated by the Rmerge and I/σ statistics. Thus, data extending to 2.5 Å resolution were used for the refinement process, although by conventional standards, the structure should be considered to be resolved at 2.9 Å resolution.
Phases were determined by molecular replacement using the unmodified SrtB structure (28) (Protein Data Bank code 1NG5) as a search model in the program Phaser (38). The NPQT* modifier was modeled into positive density using COOT (39, 40), and the model was prepared through successive iterations of manual adjustment in COOT and refinement in BUSTER (41, 42).
Transpeptidation Assay
Active site mutants were produced using the QuikChange site-directed mutagenesis kit (Stratagene) as per the manufacturer's instructions, confirmed by DNA sequencing, and expressed and purified as described for the wild-type protein. In vitro transpeptidation reactions were performed based on the method developed by Kruger et al. (43). 100 μm SrtB (wild-type or mutant) was incubated with 2 mm GGGGG and 200 μm peptide substrate in 100 μl of assay buffer (300 mm Tris-HCl and 150 mm NaCl) at 37 °C for 24 h. The reactions were quenched by adding 50 μl of 1 m HCl and injected onto a Waters XSelect HSS C18 reversed phase HPLC column. Peptides were eluted by applying a gradient from 3 to 23% acetonitrile (in 0.1% trifluoroacetic acid) over 25 min at a flow rate of 1 ml/min. Elution of the peptides was monitored by absorbance at 215 nm. Peak fractions were collected, and their identities were confirmed by MALDI-TOF mass spectrometry.
Computational Modeling and Molecular Dynamics
Molecular dynamics simulations were performed with NAMD (44), using the AMBER99SB-ILDN force field (45), a 2-fs time step, and the SHAKE algorithm to constrain all hydrogen containing bonds (46). Nonbonded interactions were truncated at 10 Å, with the use of a smoothing function beginning at 9 Å, and long range electrostatics were handled with the particle mesh Ewald method using a maximum grid spacing of 1 Å and a cubic B spline (47). Parameters for the Cys-Thr linkage were generated with GAFF (48, 49), with the charges derived from a RESP fit (48). Constant temperature was maintained through the use of Langevin dynamics with a damping coefficient of 2 ps−1, whereas the barostat was controlled through a Nosè-Hoover method with a target pressure of 1 atm, a piston period of 100 fs, and a damping time of 50 fs (50, 51).
Models of the thioacyl intermediate were originally constructed from the SrtB-NPQT* structure by replacing the disulfide bond with a thioester in PyMOL (52). The models were solvated in a periodic water box with a solvent distance of 10 Å and parameterized in tLeap (53). Models were then energy-minimized and equilibrated in NAMD (44) by slowly removing restraints from the initial atom positions over 1 ns with 2-fs steps. For simulations of SrtA, the NMR structure 2KID was utilized (24).
Potential of mean force calculations were performed using two-dimensional replica-exchange umbrella sampling calculations (54). For the first dimension (the x coordinate in Fig. 6, b–d), a vector was defined based on the difference in positions between residues in the SrtA structure 2KID and the SrtB structure presented here for the heavy atoms in the backbones of residues P1, P2, the catalytic cysteine, and the three residues upstream of it in the sortase molecule, along with the heavy atoms in the backbone of residues P1 and the catalytic cysteine. The second coordinate (the y axis in Fig. 6, b–d) was defined as the radius of gyration for the C atoms in residues 98–100 and 142–144 in SrtA (or residues 171–173 and 235–237 in SrtB) along with the heavy atoms of residues P4 and P3. Restraints for umbrella sampling were evenly spaced every 10 Å from −100 to 100 Å in the first coordinate and every 0.5 Å from 5 to 14 Å in the second coordinate. This created a total of 399 simulation “windows,” each of which were simulated for 10 ns. Positions were exchanged between adjacent windows every 1 ps based upon a Metropolis criteria with a temperature of 300 K. The weighted histogram analysis method was used for computing the potential of mean force based upon the umbrella sampling calculations (55). Analysis of subsamples from these simulations indicate that the overall free energy profiles require on the order of 5 ns to equilibrate; thus the first 5 ns of each window is discarded in the weighted histogram analysis method analysis presented here.
FIGURE 6.

Structures and free energy profiles of the SrtA and SrtB thioacyl complexes. a, selected structures obtained from MD simulations of the SrtA and SrtB thioacyl complexes. The left and middle panels show structures of the SrtA thioacyl complex in which the threonine side chain in the sorting signal either interacts with the active site arginine (left panel, SrtA-LPAT Thr-“In”) or projects away from the active site (middle panel, SrtA-LPAT Thr-“Out”). The right panel shows the structure of the SrtB-NPQT thioacyl complex in which the threonine side chain interacts with the arginine residue (Thr-in). The structures are displayed with the SrtA and SrtB surfaces colored green and blue, respectively. Surface representations of labeled active site residues were calculated independently of the remaining protein surface, which allows for the visualization of SrtB His130 (cyan) behind Leu96 (dark gray) in the right panel, even though this residue would not normally be considered solvent-accessible in this conformation. b–d, free energies were calculated from positions sampled during a Hamiltonian replica exchange simulation for the SrtA-LPAT (b), SrtB-NPQT (c), and SrtB-NPAT (d) thioacyl complexes. The x axis records the position of P1 and P2 residues in the bound sorting signal relative to the active site cysteine residue. The y axis describes the positioning of residues P3 and P4 in the sorting signal relative to the body of the protein.
RESULTS
Crystal Structure of the SrtB-NPQT* Complex and Computational Modeling of the Thioacyl Intermediate
Sortase catalyzed transpeptidation reactions occur via covalent enzyme-substrate acyl and tetrahedral intermediates that are too short-lived to be resolved by NMR or x-ray crystallography (2, 14, 43). To overcome this problem, we synthesized a Cbz-NPQT* sorting signal analog, where Cbz is a carbobenzyloxy protecting group, and T* is a threonine derivative that replaces the carboxyl group with -CH2-SH ((2R,3S)-3-amino-4-mercapto-2-butanol). The peptide contains the sorting signal sequence recognized by the S. aureus SrtB sortase and forms a disulfide bond via its T* moiety to the thiol of Cys223, generating a SrtB-NPQT* complex that structurally mimics the thioacyl catalytic intermediate (Fig. 1a). A soluble version of SrtB lacking its 30-amino acid N-terminal membrane anchor (SrtB, residues 31–244) was disulfide-bonded to the NPQT* substrate analog, and a 2.5 Å crystal structure of the SrtB-NPQT* complex was determined (Table 1). The data were refined to 2.5 Å resolution in accordance compatible with CC1/2 statistics (36, 37), although the data extended only to 2.9 Å using more conventional statistics such as Rmerge and I/σ. In the complex, SrtB adopts an α/β sortase fold containing eight β-strands that are flanked by five α-helices (Fig. 1b). The positioning of the sorting signal substrate is well defined, as evidenced by an Fo − Fc omit map of the complex (Fig. 2a). The solvent-exposed Cbz group at the N terminus of the peptide is only partially modeled in the structure of the complex because its electron density was not strong enough to fully define its position. The signal adopts an L-shaped structure and is connected via the sulfhydryl of the T* moiety to the active site cysteine, Cys223. It is nestled within a narrow groove whose base is formed by residues within strands β4 and β5 and whose walls are formed by residues projecting from loops connecting strands β6 to β7 (the β6/β7 loop), β7 to β8 (the β7/β8 loop), and β2 to β3 (the β2/β3 loop) (Fig. 1, b and c). To facilitate a discussion of the molecular basis of substrate recognition, we henceforth utilize the nomenclature developed by Schechter and Berger (56), where P and P′ refer to amino acids on the N-terminal and C-terminal sides of the scissile peptide bond of the sorting signal, respectively. For the NPQTN sorting signal, the N-terminal N is P4, P is P3, Q is P2, T is P1, and the C-terminal N, occurring after the scissile bond, is P1′. The base of the NPQT binding pocket is defined by residues Asn92, Tyr128, Tyr181, Ile182, and Ser221, with the walls defined by residues Leu96, Thr177, Cys223, Glu224, and Arg233 (Fig. 2, b and c). The side chain of the disulfide linked T* residue is buried inside of a deep groove where its methyl group contacts the side chains of Tyr128 and Ile182. This positions the hydroxyl oxygen on the threonine residue to accept two hydrogen bonds from the side chain of Arg233 in the active site: a 3.0 Å hydrogen bond to the ϵ-nitrogen atom and a 3.3 Å hydrogen bond to the η-nitrogen atom in the guanidino group (Fig. 2, b and c). The side chain of the glutamine residue at position P2 points out of the binding pocket where it is packed against the side chain of Leu96 and donates a hydrogen bond to the backbone carbonyl group of Glu224. Additional enzyme interactions to the backbone of P2 Gln residue stabilize the positioning of the sorting signal. The backbone carbonyl group is held in place by a hydrogen bond from the side chain amide nitrogen of Asn92, and its backbone nitrogen donates a hydrogen bond to a sulfate ion, which in turn is coordinated by the side chain of His93 and the backbone amide of Asn92. The P3 Pro residue rests on top of Ile182 and forms a kink that causes the bound peptide to adopt an L-shaped structure that positions the side chain of the P4 Asn to donate a hydrogen bond to the backbone carbonyl of Thr177 within the β6/β7 loop. Substrate binding induces only small changes in the structure of SrtB as the Cα coordinates of the SrtB-NPQT* complex can be superimposed with the previously determined structure of the unmodified enzyme (28) with a root mean square deviation of 0.44 Å.
FIGURE 1.

Structure of the SrtB-NPQT* complex. a, a comparison of the chemical bonds that join the peptide substrate to the SrtB enzyme in the SrtB-NPQT* complex (left panel) and the SrtB-NPQT thioacyl catalytic intermediate (right panel). Atoms from SrtB are colored red. b, ribbon diagram of the SrtB-NPQT* complex. Green, β2/β3 loop; orange, β6/β7 loop; red, β7/β8 loop. Active site residues and substrate analog are shown as sticks. c, surface representation of SrtB in the complex utilizing the same color scheme as in b. Active site residue Arg233 is highlighted in blue, and Cys223 is in yellow.
TABLE 1.
Crystallographic data collection and refinement statistics
| Parameters | SrtB-NPQTs* |
|---|---|
| Data collection | |
| Space group | P21 |
| Cell dimensions | |
| a, b, c (Å) | 102.6, 59.12, 72.49 |
| Resolution (Å) | 72.49–2.49 (2.58–2.49) |
| Rmerge | 26.6 (212.4) |
| I/σ(I) | 3.8 (0.7) |
| Completeness (%) | 92.2 (94.2) |
| Multiplicity | 5.1 (5.0) |
| CC1/2 | 98.6 (50.4)a |
| CC | 99.6 (81.8)a |
| Refinement | |
| Resolution (Å) | 72.49–2.49 |
| No. Reflections | 28147 |
| Rwork/Rfree | 21.5/27.1 |
| CCwork | 94.4 (76.4)b |
| CCfree | 87.7 (75.5)b |
| No. atoms | |
| Protein | 7248 |
| Ligand/ion | 176 |
| Water | 27 |
| B factors | |
| Mean | 72.5 |
| Wilson | 56.2 |
| Protein | 72.3 |
| Ligand/ion | 87.6 |
| Water | 29.9 |
| Root mean square deviation | |
| Bond lengths (Å) | 0.010 |
| Bond angles (°) | 1.26 |
a The values are reported as percentages (%).
b The values in parentheses are highest resolution shell.
FIGURE 2.
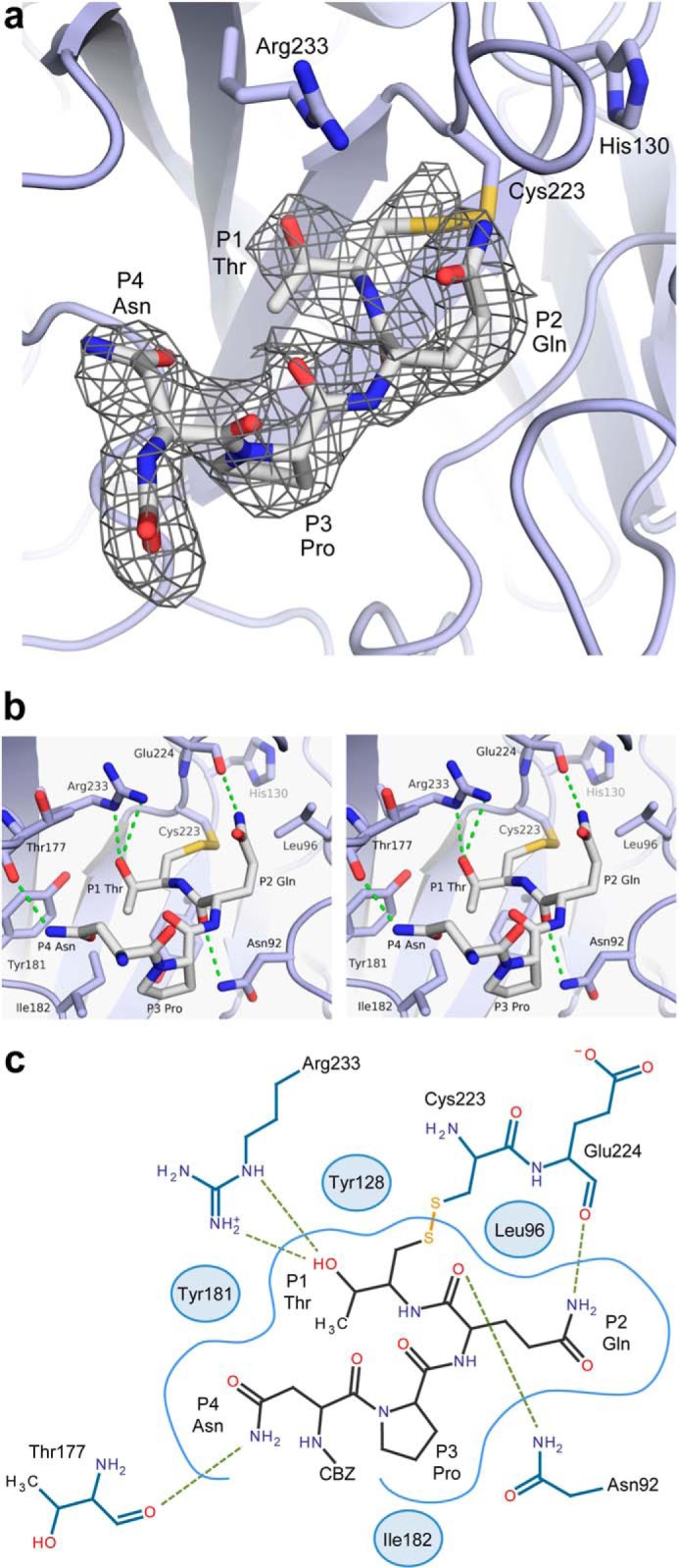
Structure and interactions of the NPQT* modifier in complex with SrtB. a, Fo − Fc map contoured at 3 σ. Electron density (gray mesh) was generated by removing the NPQT* peptide from the final model and repeating refinement. The map shown is an average of the density from all four chains in the asymmetric unit. The peptide extends from the active site cysteine. b, stereo view of the NPQT* peptide (gray sticks) and interacting residues (blue sticks). Hydrogen bonds are indicated by dashed green lines. c, diagram of the interactions between SrtB (blue) and the NPQT* peptide (gray). Hydrogen bonds are indicated by dashed green lines. SrtB residues that make only hydrophobic contacts are depicted as blue circles positioned near their most significant point of contact to the peptide.
To gain insight into how sortase stabilizes reaction intermediates, an energy-minimized model of the thioacyl complex was generated by replacing the disulfide link in the structure of the SrtB-NPQT* complex with a thioacyl bond (Fig. 3). This required only small changes including the removal of the methylene group in between the Cys223 thiol and the P1 Thr residue of the sorting signal that decreased their separation by ∼1 Å. The coordinates of the thioacyl intermediate were then energy-minimized while the restraints on the initial atom positions were gradually removed. To verify that the final model contained the proper orientation of the thioacyl bond, two initial models of the intermediate were energy-minimized, one in which the carbonyl oxygen pointed toward Arg233 and a second model in which the thioacyl bond was rotated by 180° (carbonyl oxygen pointing away from Arg233). After minimization, both starting models converged to nearly identical structures (root mean square deviation = 0.71 Å for all Cα) that closely resembled the SrtB-NPQT* complex. Importantly, in both refined models, the thioacyl linkage adopts a similar conformation in which the P1 threonine residue in the signal is poised to accept hydrogen bonds from the active site Arg233 residue and the backbone amide from Glu224 (Fig. 3). This key sorting signal-enzyme interaction may hold the active site Arg233 side chain in a catalytically competent conformation that also stabilizes higher energy tetrahedral reaction intermediates that form during catalysis (see “Discussion”).
FIGURE 3.
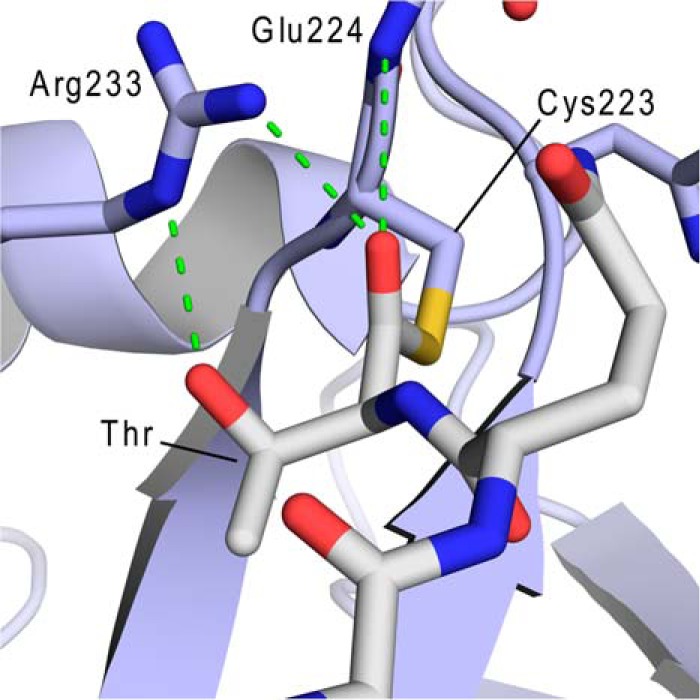
Expanded view of the active site in the energy-minimized model of the SrtB-NPQT thioacyl intermediate. Interactions between SrtB and the threonine residue in the sorting signal are shown. The thioacyl carbonyl oxygen atom is positioned to accept hydrogen bonds from the η-nitrogen atom of Arg233 and the backbone nitrogen atom of Glu224. The side chain of Arg233 is held in position by a hydrogen bond between its ϵ-nitrogen atom and the hydroxyl group on the Thr residue located at the P1 position of the sorting signal.
Identification of Enzyme-Substrate Interactions Required for Catalysis
The catalytic importance of active site and sorting signal amino acids was investigated in vitro using SNKDKVENPQTNAGT (sorting signal in bold) and GGGGG peptides that mimic the sorting signal and secondary lipid II substrates, respectively. HPLC separation of the reaction products indicates that SrtB used for crystallographic studies is fully functional in vitro (kcat and Km values for SrtB of 1.0 × 10−4 s−1 and 1.8 mm, respectively) (Fig. 4a) (57). Based on sequence conservation, residues His130, Cys223, and Arg233 in SrtB have been postulated to form a triad that mediates catalysis. In addition, it has been proposed that the side chain of Asp225, positioned near the active site, may also play a critical role in catalysis by stabilizing and activating His130 (28). To investigate the relative importance of these residues, we purified four single amino acid mutants of SrtB and assayed them for their ability to catalyze transpeptidation. H130A, C223A, and R233A mutant enzymes had no detectable activity after 24 h, whereas D225A exhibited nonspecific proteolytic activity (Fig. 4b). This suggests that unlike the conserved active site residues (His130, Cys223, and Arg233), Asp225 is not required for early steps in transpeptidation that involve the formation of the first thioacyl intermediate. The D225A mutation may disrupt the active site architecture of SrtB, allowing recognition of various sequences as primary substrates, but it does not appear to play a direct role in the catalytic mechanism. Interestingly, similar promiscuous activity has been observed in several SrtA mutants (57, 58), as well as the wild-type SrtB enzyme from Listeria monocytogenes (59).
FIGURE 4.

Transpeptidation activity of wild-type and mutant SrtB. a, representative HPLC chromatograms showing the reaction products that are produced when SrtB was incubated with SNKDKVENPQTNAGT (sorting signal in bold type) and GGGGG peptides that mimic its sorting signal and secondary lipid II substrates, respectively. Reactions performed in the presence (left panel) and absence (right panel) of SrtB are shown. Only when SrtB is present (left panel) is the appropriate transpeptidation peptide product produced (SNKDKVENPQTGGGGG). b, transpeptidation activity of SrtB mutants. The indicated SrtB mutant was incubated with 200 μm peptide containing an NPQTN sorting signal and pentaglycine and monitored by HPLC as described above. The dark shaded bars indicate the amount of full-length peptide remaining after reaction with the enzyme. The lightly shaded bar indicates the amount of transpeptidation product that was formed. An asterisk indicates that no transpeptidation product could be detected after 24 h. The error bars represent the standard deviation of three reactions. c, transpeptidation activity of sorting signal amino acid mutants. Sorting signal peptides containing select alanine substitutions were assayed for their ability to be utilized by SrtB as a substrate and monitored by HPLC as described above. The amount of transpeptidation product formed for each mutant peptide is expressed as a percentage of the amount formed from reaction of SrtB with the native NPQTN sorting signal.
The importance of enzyme-substrate interactions visualized in the structure of the complex was also tested using the HPLC assay. Sorting signal peptides containing alanine substitutions at sites P1 (NPQAN), P3 (NAQTN), and P4 (APQTN) were unreactive (Fig. 4c), compatible with extensive enzyme contacts to the side chains of these residues in the structure of the complex. In contrast, peptides containing an alanine substitution at either site P2 (NPATN) or P1′ (NPQTA) could be processed by the enzyme to yield transpeptidation products, although they were less reactive than the native signal. The ability of SrtB to process the P2 mutant peptide (NPATN) is compatible with the structure of the complex as the side chain of residue P2 projects out of the binding pocket toward the β7/β8 loop. The SrtA enzyme also processes signals containing a range of amino acids at site P2 in its LPXTG sorting signal (60), suggesting that this promiscuity is evolutionarily conserved.
The structure of the SrtB-NPQT* complex and computational model of the thioacyl intermediate reveal that the threonine side chain within the sorting signal likely plays a key role in catalysis by stabilizing the positioning of Arg233 through hydrogen bonding (Figs. 2b and 3). To determine the importance of this interaction in catalysis, we tested how efficiently NPQSN and NPQVN peptides were used as transpeptidation substrates. These peptides are identical to the native sorting signal peptide but contain threonine to serine and threonine to valine mutations at the P1 position of the signal, respectively. The threonine to serine substitution preserves the hydroxyl group that hydrogen bonds to Arg233 but removes the methyl group of threonine that interacts with the side chains of Tyr128 and Ile182. In contrast, introduction of a valine substitution eliminates the hydroxyl group but does not significantly alter the size or shape of the P1 residue. Both peptides were unreactive, suggesting that each type of substrate-enzyme interaction is important for catalysis (Fig. 4c).
Molecular Dynamics Simulations of SrtA and SrtB Reveal a Conserved Mechanism through Which the Substrate Stabilizes the Positioning of the Active Site Arginine Residue
The atomic structures of only two sortases covalently bound to their substrates have been determined: the NMR structure of SrtA bound to an LPAT* peptide (24) and the structure of the SrtB complex reported here (Fig. 5). A comparison reveals a generally similar mode of binding in which the signals adopt an L-shaped structure enabling extensive enzyme contacts to the side chains of residues located at positions P4 and P3 (Leu-Pro and Asn-Pro in the SrtA and SrtB substrates, respectively). Interestingly, although both bound sorting signals contain a conserved threonine residue at the P1 position, the side chain of this amino acid is oriented differently in the structures of the SrtA-LPAT* and SrtB-NPQT* complexes. As described above, in SrtB the side chain of the P1 Thr residue faces “in” and interacts with the active site arginine, and the side chain of the P2 Gln residue projects “out” toward the β7/β8 loop. In contrast, in the SrtA-LPAT* structure the P1 Thr points out toward the solvent and the P2 Ala residue points in toward the bottom of the binding pocket. This conformational difference occurs because the P1 and P2 residues in each signal have distinct backbone torsional angles; the P2 ϕ and ψ angles are 51 and 98° for SrtA-LPAT*, and −75 and 127° for SrtB-NPQT*, respectively, and the P1 T* pseudo ϕ and ψ angles are −71 and −17° for SrtA-LPAT* and −119 and 25° for SrtB-NPQT*, respectively. This key structural difference is not caused by a lack of coordinate precision in the NMR structure of the SrtA-LPAT* complex because several NOEs define the positioning of the side chains of the P1 and P2 residues (see Fig. 1c in Ref. 24).
FIGURE 5.
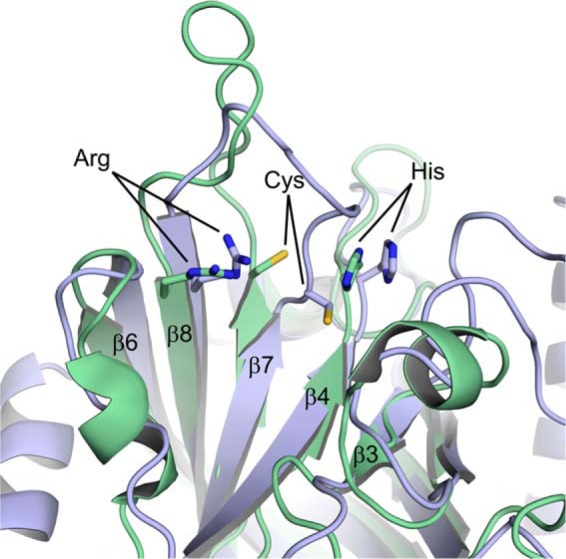
Alignment of SrtB-NPQT* and SrtA-LPAT* from Ref. 24. SrtB is shown as blue ribbons, and SrtA is shown as green ribbons.
It is possible that the sorting signals bound to SrtA and SrtB are flexible and thus capable of undergoing motions in which the P1 Thr side chain moves into, and out of, the active site. To investigate this issue, we performed MD simulations of both complexes using the method of umbrella sampling with Hamiltonian replica exchange (54). The free energy profile for transitions between the Thr-in (SrtB-like) and Thr-out (SrtA-like) states for the bound signals in each complex was then calculated. MD calculations using the coordinates of the SrtA-peptide complex indicate that the peptide can transition from the Thr-out conformation observed in the NMR structure of the SrtA-LPAT* complex (Fig. 6a, middle panel) to a Thr-in orientation observed in the structure of the SrtB-NPQT complex (Fig. 6a, left panel). To investigate the energetics associated with this transition, we calculated the free energies of intermediate structures on this pathway, which are represented by a two-dimensional coordinate system (Fig. 6b). The first coordinate (x axis) reports on the positioning of the P1 and P2 residues, and the second reaction coordinate (y axis) reports on the positioning of the remainder of the sorting signal relative to the enzyme. The former is defined as a collective coordinate that describes the structure of residues P1 and P2 relative to the catalytic cysteine, and the latter is defined as the radius of gyration of residues P3 and P4 with select atoms in the β sheet of the sortase molecule (described further under “Experimental Procedures”). In the free energy profile for the covalent SrtA-LPAT complex, there are three dominant energy wells all with minima within 0.4 kcal/mol of one another. The region we refer to as well 1 corresponds to a sorting signal structure similar to that observed in the SrtB-NPQT* complex in which the sorting signal contacts the arginine (Thr-in) (Fig. 6a, left panel), whereas well 2 conformers resemble the NMR structure of the SrtA-LPAT* complex in which the P1 threonine side chain points away from the active site (Thr-out) (Fig. 6a, middle panel). Interestingly, this analysis reveals that a low energy pathway exists between these two states (Fig. 6b), suggesting that the P1 Thr and P2 Ala residues can alter their conformation within the active site of SrtA. This structural transition is documented in supplemental Video S1, which shows select snapshots from the MD trajectory in which the P2 Thr transitions from the Thr-out to Thr-in state where it engages the active site arginine residue. Interestingly, the Thr-out to Thr-in transition can also occur through a third low energy intermediate (well 3) in which the hydrophobic residues P3 and P4 do not contact SrtA but are instead exposed to solution. This entropically stabilized state is presumably not significantly populated in vivo when the enzyme contacts larger protein substrates that contain a full cell wall sorting signal.
In contrast to SrtA, MD simulations of the SrtB-NPQT complex reveal only a single, narrow free energy minimum in which the threonine remains projected into the active site (Thr-in) where it contacts Arg233 (Fig. 6, a, right panel, and c). This indicates that the Thr-out conformer observed in SrtA is disfavored in SrtB. The larger size of the P2 residue in the SrtB sorting signal could, in principle, cause this difference. In the SrtA-LPAT complex, the P2 Ala residue adopts a positive ϕ angle, and the side chain projects toward the base of the binding pocket, whereas the larger Gln P2 residue in the SrtB bound peptide adopts a negative ϕ angle and rests on the surface of the enzyme. Because reduced steric stress enables amino acids with smaller side chains to more readily adopt positive ϕ angles, it is possible that the smaller size of the alanine side chain in the LPAT signal facilitates formation of the Thr-out state. To test this hypothesis, a third free energy profile was computed for SrtB bound to NPAT, which changes its P2 residue to alanine (Fig. 6d). This change expanded the range of conformers accessible to the peptide bound to SrtB (Fig. 6, compare c and d), but it did not encourage sampling of the Thr-out state observed in SrtA. This suggests that features of the SrtB-peptide complex other than the identity of its P2 residue are important for dictating how the P1 Thr residue is positioned (described below). In sum, our MD simulations indicate that sorting signals bound to both SrtA and SrtB can adopt conformations in which the P1 Thr side chain is located within the active site for stabilizing interactions with the active site arginine. In SrtB this is the predominant state of the signal, whereas in SrtA, it is one of two possible low energy binding conformations.
DISCUSSION
Members of the sortase superfamily catalyze transpeptidation reactions that covalently attach proteins to the bacterial cell wall or assemble pili (1–3). At present, over a thousand sortase enzymes have been identified that, based on their primary sequences and functions, can be partitioned into at least six distinct families (called class A to F enzymes) (1). The structure of the SrtB-NPQT* complex reveals how class B sortases recognize their substrates. In bacterial pathogens, class B enzymes typically anchor heme-binding proteins to the cell wall that enable the bacterium to utilize host derived heme-iron as a nutrient (1, 10–12). Our results indicate that SrtB recognizes its NPQTN sorting signal via a large groove adjacent to the active site formed by residues in the β6/β7 loop and residues within strands β4 and β7. Structurally, class B enzymes are distinguished from other members of the sortase superfamily by the presence of a large β6/β7 loop that contains a long α-helix. In the complex, this loop plays a major role in signal recognition because it contacts the Asn (P4), Pro (P3), and Thr (P1) residues of the peptide, which are highly conserved in sorting signals recognized by class B enzymes. These substrate-enzyme contacts are important for catalysis because alanine substitutions at these sites in the sorting signal disrupt transpeptidation (Fig. 4c). As in the prototypical SrtA enzyme, site P2 in the SrtB sorting signal is tolerant to alanine substitution, which is consistent with the positioning of the glutamine residue at this site, which rests on the surface of the enzyme. Although not visualized in our structure, the P1′ residue following the scissile peptide bond is also tolerant to alanine mutation (Fig. 4c), distinguishing SrtB from the prototypical class A SrtA enzyme, which only recognizes glycine at the P1′ position (61). Given that the signal sequences and known structures of class B enzymes are highly similar, it is likely that they all recognize their substrates in a fashion similar to what is seen in the SrtB-NPQT* complex. Interestingly, our results indicate that SrtA and SrtB enzymes recognize their cognate sorting signals in a generally similar manner, despite the fact that the enzymes share only 26% sequence identity and that they recognize distinct LPXTG and NPQTN sorting signals, respectively. A comparison of the SrtB-NPQT* and previously reported NMR structure of the SrtA-LPAT* complex reveals that the bound sorting signals both adopt an L-shaped conformation in which the proline residue at position P3 redirects the polypeptide to position the side chain of the P4 residue so that it contacts the β6/β7 loop. This general mode of binding is likely a conserved feature of substrate recognition by sortase enzymes as an inspection of sorting signals predicted to be processed by these enzymes shows that over 90% of them contain a proline at P3 (60).
Computational modeling of the thioacyl enzyme-substrate reaction intermediate suggests that SrtB facilitates catalysis by forming a substrate-stabilized oxyanion hole. In both the experimental and energy-minimized model of this reaction intermediate, the conserved active site arginine (Arg233) is held in place near the scissile bond by donating a hydrogen bond from its ϵ-nitrogen to the hydroxyl group of the threonine located at the P1 position in the sorting signal (Figs. 2b and 3). In the model of the thioacyl complex, this interaction positions the arginine so that its guanidino group and the backbone amide of Glu224 donate hydrogen bonds to the oxygen atom in the thioacyl bond. During catalysis, two oxyanionic transition states form; the first precedes the generation of the thioacyl intermediate emulated by the SrtB-NPQT* structure, and the second occurs after nucleophilic attack of the thioacyl bond by the amino group present in lipid II. It seems likely that Arg233 and Glu224 form an oxyanion hole that stabilizes these high energy reaction intermediates because only small changes in the positioning of the oxygen atom within the thioacyl bond are expected to occur when the carbonyl carbon transitions from its planar sp2 configuration to its tetrahedral sp3 state. This is distinct from the oxyanion hole discovered in penicillin binding proteins that perform a similar transpeptidation reaction but use two backbone amides to stabilize the tetrahedral intermediate (62). The threonine residue in the sorting signal appears to play a significant role in stabilizing this oxyanion hole because even conservative mutations to either serine or valine disrupt transpeptidation (Fig. 4c). Its catalytic role is also substantiated by the high degree of sequence conservation at site P1 in all known SrtB substrates. The putative oxyanion hole reported here is compatible with results obtained by McCafferty and co-workers (22, 23), who demonstrated that the analogous arginine in the SrtA enzyme is intolerant to mutation, except when substituted with citrulline, an arginine isostere that lacks a formal positive charge.
MD simulations indicate that the SrtA enzyme can also form a substrate stabilized oxyanion hole that could facilitate catalysis. In the structures of the SrtB-NPQT* and SrtA-LPAT* complexes, the bound sorting signals adopt similar L-shaped conformations. However, the positioning of the P1 Thr residue in each sorting signal differs substantially; in SrtB-NPQT*, the threonine side chain interacts with the active site arginine residue (Thr-in conformation) (described above), whereas in the SrtA complex, the analogous threonine residue projects away from the active site (Thr-out conformation). Because the threonine in the SrtA complex is not positioned to form stabilizing interactions with the active site arginine residue, we wondered whether the Thr-in conformer observed in the SrtB complex could also be sampled by the bound SrtA sorting signal to stabilize its tetrahedral reaction intermediates. To investigate this issue, MD simulations of the SrtA thioacyl complex were performed, revealing that the P1 threonine residue can transition from the Thr-out to the Thr-in state presumably needed to construct the oxyanion hole (supplemental Video S1 and Fig. 6). Conformations of the SrtA-LPAT complex in which the threonine side chain of the sorting signal forms interactions with the active site arginine were obtained without major rearrangement of the structure of the enzyme. Interestingly, a previous MD study based on the SrtA-LPAT* NMR structure reports that the active site arginine residue functions only to position the sorting signal substrate by hydrogen bonding to its backbone carbonyl atoms at positions P2 and P4 (25). Our work is compatible with this conclusion but suggests that this stabilizing interaction will only occur when the P1 Thr samples the out position that is presumably not catalytically active. Thus, based on primary sequence conservation and the demonstrated importance of the P1 Thr in catalysis in both SrtA and SrtB, we conclude that the Thr-in conformation observed in the SrtB structure, and accessible to SrtA, represents a catalytically competent form of the peptide that is essential for stabilizing the tetrahedral transition state.
The active site of SrtB appears to be more conformationally restrictive than SrtA because MD simulations of the SrtB-NPQT thioacyl complex reveal that only the Thr-in conformer is accessible to the bound peptide. This is compatible with structural and NMR relaxation data, which have shown that the active site of SrtA contains a flexible β6/β7 loop that becomes ordered upon substrate binding (24, 63), whereas SrtB contains a preformed, rigid binding pocket for its sorting signal substrate. Interestingly, inspection of the MD data suggests that SrtA can form unique contacts to the sorting signal that may stabilize the Thr-out conformer. In the NMR structure of the SrtA-LPAT* structure, the conserved active site His101 residue is positioned to form a 3.1 Å hydrogen bond from its ϵ-nitrogen to the P1 Thr hydroxyl. In contrast, although the SrtB active site histidine is about the same distance from the active site cysteine as the analogous residues in SrtA (∼5.2 Å from His δ-N to Cys S), this potential stabilizing interaction for the Thr-out conformer is obstructed by the side chain of Leu96, which is inserted between His130 and Cys223 in SrtB. Thus, the more restrictive active site of SrtB and the lack of stabilizing interactions may prevent the signal bound to SrtB from adopting the catalytically nonproductive Thr-out conformer. It is unlikely that the ability of the SrtA peptide to sample the less reactive Thr-out state impacts the kinetics of transpeptidation because the half-life of the long-lived thioacyl intermediate presumably far exceeds the time needed for the threonine residue to transition between its Thr-in and Thr-out conformers.
It seems likely that nearly all members of the sortase superfamily will employ a substrate stabilized oxyanion hole to anchor proteins to the cell wall or to assemble pili. Based on structural and mutagenesis data, the transpeptidation reaction will be initiated when the sorting signal of the partially secreted protein substrate binds to the groove on sortase formed by residues in the β6/β7 loop and residues within strands β4 and β7. Most sorting signals can be expected to adopt an L-shaped structure when bound to the enzyme because they contain a proline residue at position P3 (∼90% of predicted sorting signals) (60). For catalysis to proceed, the sortase must contain a properly charged active site in which the cysteine and histidine residues are in their thiolate and imidazolium ionization states, respectively. In isolation, only a small fraction of the enzyme may be properly ionized based on pKa measurements of the SrtA enzymes from S. aureus and Bacillus anthracis (14, 29, 64). If properly ionized, the cysteine thiolate attacks the carbonyl carbon of the P1 residue, forming the first tetrahedral intermediate. Our results suggest that the oxyanion in this intermediate is stabilized by hydrogen bonding from the active site arginine residue and a backbone amide group located in the β7/β8 loop (in SrtB Arg233 and the backbone amide of Glu224) (Fig. 7). The threonine residue within the sorting signal, at position P1, plays a key role in constructing the oxyanion hole by stabilizing the positioning of the arginine side chain via hydrogen bonding. This oxyanion hole is presumably used by most sortase enzymes to facilitate catalysis because they all contain conserved active site arginine residues, and ∼95% of their predicted sorting signal substrates contain a threonine at the P1 position (60). Breakage of the scissile bond is then facilitated by protonation of the amide group by His130, resulting in a semistable thioacyl intermediate. It remains unclear where the amino nucleophile on lipid II enters the active site in SrtB. A crystal structure of SrtB noncovalently bound to a triglycine peptide, meant to mimic the lipid II substrate, localized the binding site for the peptide to the β7/β8 loop (33). However, the specificity of this interaction is suspect because the peptide is expected to bind weakly (Km for GGGGG binding is 140 μm (43)) and because the complex was not co-crystallized (the peptide was soaked into the crystal). It is also important to note that in this and all crystal structures of SrtB solved to date, side chains from residues Asp225–Tyr227 in the β7/β8 loop are involved in crystal lattice contacts to symmetry related molecules in the crystal. Although it is possible that these contacts simply reinforce the existing, predominant conformation of this loop, it is also possible that they have captured one of many possible conformations that could be involved in mediating substrate access to the active site. Alternatively, as originally proposed by Joachimiak and co-workers (28), the pentaglycine peptide in lipid II may enter the active site via a groove located between the β7/β8 and β2/β3 loops. This is compatible with NMR chemical shift perturbation studies of SrtA (24), high resolution crystal structures of other sortase enzymes, which also contain a similarly positioned groove (30, 32), and the presence of the highly conserved histidine residue in this groove that has been proposed to function as a general base that deprotonates lipid II. Our model of the thioacyl intermediate does not rule out either of these entry points. However, it is most compatible with lipid II entering via the groove between the β7/β8 and β2/β3 loops because from this direction, attack of the carbonyl carbon by the amino nucleophile will generate a tetrahedral intermediate whose negative charge is positioned to be stabilized by the oxyanion hole formed by Arg233. The transpeptidation reaction would then be completed by the collapse of the second tetrahedral intermediate into the final, covalently linked, protein-lipid II product. Additional hybrid quantum mechanics/molecular mechanics simulations are currently underway to quantitatively investigate the role of the oxyanion hole in catalysis. Beyond providing fundamental insight into the process of protein display and pilin assembly in bacteria, the new mechanistic insights reported in this paper could guide the rational design of therapeutically useful transition state analog inhibitors of sortases and facilitate protein engineering efforts to expand the utility of sortases as biochemical reagents.
FIGURE 7.

The SrtB transpeptidation mechanism showing how the sorting signal may stabilize the oxyanion hole. a, the SrtB active site with His130 in its imidazolium form and Cys223 in its thiolate form. b, the NPQTN substrate binds with its P1 Thr residue in the in position within the active site. The Cys223 thiolate performs a nucleophilic attack on the P1 Thr carbonyl carbon. c, the first tetrahedral intermediate is formed. The P1 Thr residue interacts with Arg233 to construct a substrate-stabilized oxyanion hole in which the side chain of Arg233 and the backbone amide of Glu224 hydrogen bond to the oxyanion. The His130 imidazolium group donates a proton to the leaving group to complete breakage of scissile bond. d, the P1 Thr residue maintains hydrogen bonds with Arg233 to stabilize its interaction with the thioacyl intermediate. e, the incoming GGGGG peptide from lipid II acts as a nucleophile that attacks the carbonyl carbon of the thioacyl bond. f, the second tetrahedral intermediate is formed and is again stabilized by the substrate-stabilized oxyanion hole. This intermediate then collapses, releasing the NPQTGGGGG transpeptidation product and returning the enzyme to its active form (a).
Supplementary Material
This work was supported, in whole or in part, by National Institutes of Health Grants AI52217 (to R. T. C. and M. E. J.), GM31749 (to J. A. M.), and T32 GM007185 (to A. W. J. and G. L. H.). This work was also supported by the National Science Foundation, the Howard Hughes Medical Institute, the Center for Theoretical Biological Physics, the National Biomedical Computation Resource, and the National Science Foundation Supercomputer Centers.
The atomic coordinates and structure factors (code 4LFD) have been deposited in the Protein Data Bank (http://wwpdb.org/).

This article contains supplemental Video S1.
- SrtA
- S. aureus sortase A
- SrtB
- S. aureus sortase B
- MD
- molecular dynamics.
REFERENCES
- 1. Spirig T., Weiner E. M., Clubb R. T. (2011) Sortase enzymes in Gram-positive bacteria. Mol. Microbiol. 82, 1044–1059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Clancy K. W., Melvin J. A., McCafferty D. G. (2010) Sortase transpeptidases: insights into mechanism, substrate specificity, and inhibition. Biopolymers 94, 385–396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Navarre W. W., Schneewind O. (1999) Surface proteins of Gram-positive bacteria and mechanisms of their targeting to the cell wall envelope. Microbiol. Mol. Biol. Rev. 63, 174–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Proft T. (2010) Sortase-mediated protein ligation: an emerging biotechnology tool for protein modification and immobilisation. Biotechnol. Lett. 32, 1–10 [DOI] [PubMed] [Google Scholar]
- 5. Popp M. W., Ploegh H. L. (2011) Making and breaking peptide bonds: protein engineering using sortase. Angew. Chem. Int. Ed. Engl. 50, 5024–5032 [DOI] [PubMed] [Google Scholar]
- 6. Tsukiji S., Nagamune T. (2009) Sortase-mediated ligation: a gift from Gram-positive bacteria to protein engineering. ChemBioChem 10, 787–798 [DOI] [PubMed] [Google Scholar]
- 7. Mazmanian S. K., Liu G., Jensen E. R., Lenoy E., Schneewind O. (2000) Staphylococcus aureus sortase mutants defective in the display of surface proteins and in the pathogenesis of animal infections. Proc. Natl. Acad. Sci. U.S.A. 97, 5510–5515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mazmanian S. K., Ton-That H., Schneewind O. (2001) Sortase-catalysed anchoring of surface proteins to the cell wall of Staphylococcus aureus. Mol. Microbiol. 40, 1049–1057 [DOI] [PubMed] [Google Scholar]
- 9. Weiss W. J., Lenoy E., Murphy T., Tardio L., Burgio P., Projan S. J., Schneewind O., Alksne L. (2004) Effect of srtA and srtB gene expression on the virulence of Staphylococcus aureus in animal models of infection. J. Antimicrob. Chemother. 53, 480–486 [DOI] [PubMed] [Google Scholar]
- 10. Maresso A. W., Schneewind O. (2006) Iron acquisition and transport in Staphylococcus aureus. Biometals 19, 193–203 [DOI] [PubMed] [Google Scholar]
- 11. Maresso A. W., Chapa T. J., Schneewind O. (2006) Surface protein IsdC and sortase B are required for heme-iron scavenging of Bacillus anthracis. J. Bacteriol. 188, 8145–8152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mazmanian S. K., Ton-That H., Su K., Schneewind O. (2002) An iron-regulated sortase anchors a class of surface protein during Staphylococcus aureus pathogenesis. Proc. Natl. Acad. Sci. U.S.A. 99, 2293–2298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Schneewind O., Model P., Fischetti V. A. (1992) Sorting of protein A to the staphylococcal cell wall. Cell 70, 267–281 [DOI] [PubMed] [Google Scholar]
- 14. Frankel B. A., Kruger R. G., Robinson D. E., Kelleher N. L., McCafferty D. G. (2005) Staphylococcus aureus sortase transpeptidase SrtA: insight into the kinetic mechanism and evidence for a reverse protonation catalytic mechanism. Biochemistry 44, 11188–11200 [DOI] [PubMed] [Google Scholar]
- 15. Huang X., Aulabaugh A., Ding W., Kapoor B., Alksne L., Tabei K., Ellestad G. (2003) Kinetic mechanism of Staphylococcus aureus sortase SrtA. Biochemistry 42, 11307–11315 [DOI] [PubMed] [Google Scholar]
- 16. Perry A. M., Ton-That H., Mazmanian S. K., Schneewind O. (2002) Anchoring of surface proteins to the cell wall of Staphylococcus aureus. III. Lipid II is an in vivo peptidoglycan substrate for sortase-catalyzed surface protein anchoring. J. Biol. Chem. 277, 16241–16248 [DOI] [PubMed] [Google Scholar]
- 17. Ruzin A., Severin A., Ritacco F., Tabei K., Singh G., Bradford P. A., Siegel M. M., Projan S. J., Shlaes D. M. (2002) Further evidence that a cell wall precursor [C(55)-MurNAc-(peptide)-GlcNAc] serves as an acceptor in a sorting reaction. J. Bacteriol. 184, 2141–2147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Schneewind O., Fowler A., Faull K. F. (1995) Structure of the cell wall anchor of surface proteins in Staphylococcus aureus. Science 268, 103–106 [DOI] [PubMed] [Google Scholar]
- 19. Marraffini L. A., Schneewind O. (2005) Anchor structure of staphylococcal surface proteins: V. anchor structure of the sortase b substrate IsdC. J. Biol. Chem. 280, 16263–16271 [DOI] [PubMed] [Google Scholar]
- 20. Mazmanian S. K., Skaar E. P., Gaspar A. H., Humayun M., Gornicki P., Jelenska J., Joachmiak A., Missiakas D. M., Schneewind O. (2003) Passage of heme-iron across the envelope of Staphylococcus aureus. Science 299, 906–909 [DOI] [PubMed] [Google Scholar]
- 21. Ton-That H., Mazmanian S. K., Alksne L., Schneewind O. (2002) Anchoring of surface proteins to the cell wall of Staphylococcus aureus. Cysteine 184 and histidine 120 of sortase form a thiolate-imidazolium ion pair for catalysis. J. Biol. Chem. 277, 7447–7452 [DOI] [PubMed] [Google Scholar]
- 22. Frankel B. A., Tong Y., Bentley M. L., Fitzgerald M. C., McCafferty D. G. (2007) Mutational analysis of active site residues in the Staphylococcus aureus transpeptidase SrtA. Biochemistry 46, 7269–7278 [DOI] [PubMed] [Google Scholar]
- 23. Bentley M. L., Lamb E. C., McCafferty D. G. (2008) Mutagenesis studies of substrate recognition and catalysis in the sortase A transpeptidase from Staphylococcus aureus. J. Biol. Chem. 283, 14762–14771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Suree N., Liew C. K., Villareal V. A., Thieu W., Fadeev E. A., Clemens J. J., Jung M. E., Clubb R. T. (2009) The structure of the Staphylococcus aureus sortase-substrate complex reveals how the universally conserved LPXTG sorting signal is recognized. J. Biol. Chem. 284, 24465–24477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tian B.-X., Eriksson L. A. (2011) Catalytic mechanism and roles of Arg197 and Thr183 in the Staphylococcus aureus sortase A enzyme. J. Phys. Chem. B 115, 13003–13011 [DOI] [PubMed] [Google Scholar]
- 26. Liew C. K., Smith B. T., Pilpa R., Suree N., Ilangovan U., Connolly K. M., Jung M. E., Clubb R. T. (2004) Localization and mutagenesis of the sorting signal binding site on sortase A from Staphylococcus aureus. FEBS Lett. 571, 221–226 [DOI] [PubMed] [Google Scholar]
- 27. Marraffini L. A., Ton-That H., Zong Y., Narayana S. V., Schneewind O. (2004) Anchoring of surface proteins to the cell wall of Staphylococcus aureus. A conserved arginine residue is required for efficient catalysis of sortase A. J. Biol. Chem. 279, 37763–37770 [DOI] [PubMed] [Google Scholar]
- 28. Zhang R., Wu R., Joachimiak G., Mazmanian S. K., Missiakas D. M., Gornicki P., Schneewind O., Joachimiak A. (2004) Structures of sortase B from Staphylococcus aureus and Bacillus anthracis reveal catalytic amino acid triad in the active site. Structure 12, 1147–1156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Weiner E. M., Robson S., Marohn M., Clubb R. T. (2010) The sortase A enzyme that attaches proteins to the cell wall of Bacillus anthracis contains an unusual active site architecture. J. Biol. Chem. 285, 23433–23443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Race P. R., Bentley M. L., Melvin J. A., Crow A., Hughes R. K., Smith W. D., Sessions R. B., Kehoe M. A., McCafferty D. G., Banfield M. J. (2009) Crystal structure of Streptococcus pyogenes sortase A. J. Biol. Chem. 284, 6924–6933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ilangovan U., Ton-That H., Iwahara J., Schneewind O., Clubb R. T. (2001) Structure of sortase, the transpeptidase that anchors proteins to the cell wall of Staphylococcus aureus. Proc. Natl. Acad. Sci. 98, 6056–6061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Khare B., Krishnan V., Rajashankar K. R., I-Hsiu H., Xin M., Ton-That H., Narayana S. V. (2011) Structural differences between the Streptococcus agalactiae housekeeping and pilus-specific sortases: SrtA and SrtC1. PLoS One 6, e22995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zong Y., Mazmanian S. K., Schneewind O., Narayana S. V. (2004) The structure of sortase B, a cysteine transpeptidase that tethers surface protein to the Staphylococcus aureus cell wall. Structure 12, 105–112 [DOI] [PubMed] [Google Scholar]
- 34. Jung M. E., Clemens J. J., Suree N., Liew C. K., Pilpa R., Campbell D. O., Clubb R. T. (2005) Synthesis of (2R,3S)-3-amino-4-mercapto-2-butanol, a threonine analogue for covalent inhibition of sortases. Bioorg. Med. Chem. Lett. 15, 5076–5079 [DOI] [PubMed] [Google Scholar]
- 35. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Karplus P. A., Diederichs K. (2012) Linking crystallographic model and data quality. Science 336, 1030–1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Diederichs K., Karplus P. A. (2013) Better models by discarding data? Acta Crystallogr. D Biol. Crystallogr. 69, 1215–1222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Emsley P., Lohkamp B., Scott W. G., Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Emsley P., Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 41. Smart O. S., Womack T. O., Flensburg C., Keller P., Paciorek W., Sharff A., Vonrhein C., Bricogne G. (2012) Exploiting structure similarity in refinement: automated NCS and target-structure restraints in BUSTER. Acta Crystallogr. D Biol. Crystallogr. 68, 368–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bricogne G. (1993) Direct phase determination by entropy maximization and likelihood ranking: status report and perspectives. Acta Crystallogr. D Biol. Crystallogr. 49, 37–60 [DOI] [PubMed] [Google Scholar]
- 43. Kruger R. G., Dostal P., McCafferty D. G. (2004) Development of a high-performance liquid chromatography assay and revision of kinetic parameters for the Staphylococcus aureus sortase transpeptidase SrtA. Anal. Biochem. 326, 42–48 [DOI] [PubMed] [Google Scholar]
- 44. Phillips J. C., Braun R., Wang W., Gumbart J., Tajkhorshid E., Villa E., Chipot C., Skeel R. D., Kalé L., Schulten K. (2005) Scalable molecular dynamics with NAMD. J. Comput. Chem. 26, 1781–1802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lindorff-Larsen K., Piana S., Palmo K., Maragakis P., Klepeis J. L., Dror R. O., Shaw D. E. (2010) Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins 78, 1950–1958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kräutler V., van Gunsteren W. F., Hünenberger P. H. (2001) A fast SHAKE algorithm to solve distance constraint equations for small molecules in molecular dynamics simulations. J. Comput. Chem. 22, 501–508 [Google Scholar]
- 47. Darden T., York D., Pedersen L. (1993) Particle mesh Ewald: an N log(N) method for Ewald sums in large systems. J. Chem. Phys. 98, 10089–10092 [Google Scholar]
- 48. Wang J., Wang W., Kollman P. A., Case D. A. (2006) Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model. 25, 247–260 [DOI] [PubMed] [Google Scholar]
- 49. Wang J., Wolf R. M., Caldwell J. W., Kollman P. A., Case D. A. (2004) Development and testing of a general amber force field. J. Comput. Chem. 25, 1157–1174 [DOI] [PubMed] [Google Scholar]
- 50. Feller S. E., Zhang Y., Pastor R. W., Brooks B. R. (1995) Constant pressure molecular dynamics simulation: the Langevin piston method. J. Chem. Phys. 103, 4613–4621 [Google Scholar]
- 51. Martyna G. J., Tobias D. J., Klein M. L. (1994) Constant pressure molecular dynamics algorithms. J. Chem. Phys. 101, 4177–4189 [Google Scholar]
- 52. Delano W. L. (2010) The PyMOL Molecular Graphics System, Version 1.3r1, Schrödinger, LLC [Google Scholar]
- 53. Case D. A., Darden T. A., Cheatham T. E., III, Simmerling C. L., Wang J., Duke R. E., Luo R., Walker R. C., Zhang W., Merz K. M., Roberts B., Hayik S., Roitberg A., Seabra G., Swails J., Goetz A. W., Kolossváry I., Wong K. F., Paesani F., Vanicek J., Wolf R. M., Liu J., Wu X., Brozell S. R., Steinbrecher T., Gohlke H., Cai Q., Ye X., Wang J., Hsieh M.-J., Cui G., Roe D. R., Mathews D. H., Seetin M. G., Salomon-Ferrer R., Sagui C., Babin V., Luchko T., Gusarov S., Kovalenko A., Kollman P. A. (2012) AMBER 12, University of California, San Francisco [Google Scholar]
- 54. Jiang W., Luo Y., Maragliano L., Roux B. (2012) Calculation of free energy landscape in multi-dimensions with Hamiltonian-exchange umbrella sampling on petascale supercomputer. J. Chem. Theory Comput. 8, 4672–4680 [DOI] [PubMed] [Google Scholar]
- 55. Souaille M., Roux B. (2001) Extension to the weighted histogram analysis method: combining umbrella sampling with free energy calculations. Comput. Phys. Commun. 135, 40–57 [Google Scholar]
- 56. Schechter I., Berger A. (1967) On the size of the active site in proteases: I. papain. Biochem. Biophys. Res. Commun. 27, 157–162 [DOI] [PubMed] [Google Scholar]
- 57. Bentley M. L., Gaweska H., Kielec J. M., McCafferty D. G. (2007) Engineering the substrate specificity of Staphylococcus aureus sortase A: the β6/β7 loop from SrtB confers NPQTN recognition to SrtA. J. Biol. Chem. 282, 6571–6581 [DOI] [PubMed] [Google Scholar]
- 58. Piotukh K., Geltinger B., Heinrich N., Gerth F., Beyermann M., Freund C., Schwarzer D. (2011) Directed evolution of sortase A mutants with altered substrate selectivity profiles. J. Am. Chem. Soc. 133, 17536–17539 [DOI] [PubMed] [Google Scholar]
- 59. Mariscotti J. F., García-del Portillo F., Pucciarelli M. G. (2009) The Listeria monocytogenes sortase-B recognizes varied amino acids at position 2 of the sorting motif. J. Biol. Chem. 284, 6140–6146 [DOI] [PubMed] [Google Scholar]
- 60. Comfort D., Clubb R. T. (2004) A comparative genome analysis identifies distinct sorting pathways in Gram-positive bacteria. Infect. Immun. 72, 2710–2722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kruger R. G., Otvos B., Frankel B. A., Bentley M., Dostal P., McCafferty D. G. (2004) Analysis of the substrate specificity of the Staphylococcus aureus sortase transpeptidase SrtA. Biochemistry 43, 1541–1551 [DOI] [PubMed] [Google Scholar]
- 62. Nicola G., Peddi S., Stefanova M., Nicholas R. A., Gutheil W. G., Davies C. (2005) Crystal structure of Escherichia coli penicillin-binding protein 5 bound to a tripeptide boronic acid inhibitor: a role for Ser-110 in deacylation. Biochemistry 44, 8207–8217 [DOI] [PubMed] [Google Scholar]
- 63. Naik M. T., Suree N., Ilangovan U., Liew C. K., Thieu W., Campbell D. O., Clemens J. J., Jung M. E., Clubb R. T. (2006) Staphylococcus aureus sortase A transpeptidase: calcium promotes sorting signal binding by altering the mobility and structure of an active site loop. J. Biol. Chem. 281, 1817–1826 [DOI] [PubMed] [Google Scholar]
- 64. Connolly K. M., Smith B. T., Pilpa R., Ilangovan U., Jung M. E., Clubb R. T. (2003) Sortase from Staphylococcus aureus does not contain a thiolate-imidazolium ion pair in its active site. J. Biol. Chem. 278, 34061–34065 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.