

Background: Although macrophage inflammasome activation has been implicated in metabolic diseases, its regulation is not well understood.
Results: Palmitate and LPS trigger IL-1β release via lysosome- and calcineurin-mediated pathways.
Conclusion: The lysosome is a key regulator of signal 1 and signal 2 in lipotoxic inflammasome activation.
Significance: Molecular pathways involved in lipid-induced inflammasome activation may be useful targets for obesity- and diabetes-related disorders.
Keywords: Calcineurin, Diabetes, Inflammation, Lipids, Lysosomes, Macrophages
Abstract
Macrophage dysfunction and inflammasome activation have been implicated in the pathogenesis of diabetes and its complications. Prolonged inflammation and impaired healing are hallmarks of the diabetic response to tissue injury, and excessive inflammasome activation has been associated in these phenotypes. However, the mechanisms that regulate the inflammasome in response to lipid metabolic and inflammatory stress are incompletely understood. We have shown previously that IL-1β secretion is induced in primary macrophages exposed to the dietary saturated fatty acid palmitate in combination with LPS. In this study, we sought to unravel the mechanisms underlying the activation of this lipotoxic inflammasome. We demonstrate that palmitate-loaded primary macrophages challenged with LPS activate the NLRP3 inflammasome through a mechanism that involves the lysosome. Interestingly, the lysosome was involved in both the regulation of pro-IL-1β levels and its subsequent cleavage/release. The lysosomal protease cathepsin B was required for IL-1β release but not pro-IL-1β production. In contrast, disrupting lysosomal calcium regulation decreased IL-1β release by reducing pro-IL-1β levels. The calcium pathway involved the calcium-activated phosphatase calcineurin, which stabilized IL-1β mRNA. Our findings provide evidence that the lysosome plays a key role in both the priming and assembly phases of the lipostoxic inflammasome. These findings have potential relevance to the hyperinflammatory phenotypes observed in diabetics during tissue damage or infection and identify lysosomes and calcineurin as potential therapeutic targets.
Introduction
The prevalence of obesity and diabetes continues to grow at a staggering rate worldwide. Complications of these metabolic diseases account for significant morbidity, mortality, and health care costs. Evidence is accumulating that macrophage dysfunction in diabetes may contribute to several clinically important sequelae including impaired wound healing, excessive atherosclerosis, adverse cardiac remodeling after myocardial infarction, and increased susceptibility to infection (1–5). However, the mechanisms underlying these inflammatory defects in macrophages are not well understood.
The metabolic perturbations of diabetes likely play a key role in immune cell dysfunction in this disease. In addition to hyperglycemia, diabetes is characterized by substantial lipid metabolic abnormalities including excessive triglyceride and fatty acid levels in circulation and non-adipose tissues. Long chain saturated fatty acids (SFAs)2 such as palmitate are enriched in states of nutrient excess and are thought to be particularly pathogenic in diabetic patients. Consistent with this, the exposure of cells to elevated concentrations of SFAs can produce insulin resistance, ER stress, oxidative stress, and cell death, a phenomenon referred to as lipotoxiciy (6). Consequently, there is significant interest in understanding how lipids modulate macrophage inflammatory and reparative functions in diabetes. Our previous studies have shown that lipid-loaded macrophages exposed to inflammatory stimuli such as LPS undergo a cell death response that occurs via a TRIF-dependent pathway involving lysosome dysfunction (1). In addition, we have demonstrated increased ceramide biosynthesis in lipid-stressed primary macrophages, which augments the release of important pro-inflammatory cytokines such as IL-1β (2).
The processing and release of IL-1β from macrophages is regulated by a multiprotein complex known as the inflammasome (7). Recently, the inflammasome has gained attention as a contributor to several important cardiometabolic diseases including atherosclerosis, post-myocardial infarction remodeling, and diabetes (8–11). Activation of the inflammasome is a multistep process that requires a priming signal to induce the expression of IL-1β (signal 1) and a second “stress” signal to activate cytosolic assembly of the caspase-1-associated proteolytic complex (signal 2) (12). The inflammasome components include the adaptor protein apoptosis-associated speck-like protein containing a caspase activation and recruitment domain, a NOD-like receptor family member (NLRP3, NLRC4, or AIM1), and caspase-1. Regulation of the inflammasome is complex. However, in the case of NLRP3, reactive oxygen species (ROS), calcium signaling, mitochondrial dysfunction, and lysosome damage have all been implicated in the assembly and activation of the complex (13).
Diabetes and hyperlipidemia have been associated with excessive inflammasome activation, raising the possibility that this inflammatory complex may be relevant to diabetic complications (14, 15). It has been proposed that fatty acids mediate inflammasome activation by inhibiting AMP kinase (AMPK)-induced autophagy, leading to the accumulation of dysfunctional and ROS-generating mitochondria (10). However, in our prior studies using elicited primary macrophages, mitochondrial ROS was not increased by LPS in lipid-stressed cells (1). Instead we observed progressive lysosome dysfunction, which ultimately culminated in a cell death response. Therefore, we sought to further investigate the mechanisms of lipotoxic inflammasome activation. Our data reveal selective activation of the NLRP3 inflammasome in LPS-treated lipid stressed macrophages through lysosome- and calcineurin-dependent pathways.
EXPERIMENTAL PROCEDURES
Reagents
Triascin C, CAO74-ME, bafilomycin A, Z-YVAD, Z-VAD, and mito-TEMPO were from Enzo Life Sciences. Dynasore, NED-19, and 2-aminoethoxydiphenyl borate (2-APB) were from Tocris. Ammonium chloride, ATP, actinomycin D, BHA, BHT, Z-FA, apyrase, and α-tubulin antibody were from Sigma. FK506 was from AG Scientific. U18666A, necrostatin, and CN585 were from EMD-Calbiochem. Cyclosporin A and the α-IL-1β antibody were from Cell Signaling. LysoTracker Red, tetramethylrhodamine-dextran (10,000 Mr), tetramethylrhodamine ethyl ester, and calcium-free DMEM were from Invitrogen. Caspase-1 FLICATM was from Immunocytochemistry Technologies. Ultrapure Escherichia coli LPS, PamCSK4, CL075, and silica were from Invivogen. Thioglycollate was from Difco. Fatty acids were from Nu-Chek Prep. Ultrapure bovine serum albumin (BSA) was from Lampire and was tested for TLR ligand contamination prior to use.
Cell Culture
Peritoneal macrophages (pMACs) were isolated from C57BL/6 or the indicated knock-out mice 4 days after an intraperitoneal injection of 3.85% thioglycollate and plated at a density of 0.9–1 × 106 cells/ml in DMEM containing 10% inactivated fetal serum, 50 units/ml penicillin G sodium, and 50 units/ml streptomycin sulfate (pen-strep). Stimulations were performed on the day after harvest. For flow cytometry experiments, peritoneal cells were cultured on low adherence plates (Greiner Bio-One) to facilitate cell harvest. Cells were removed from the plate by washing with PBS followed by 10 min with Cell Stripper (Invitrogen) and then 10 min with EDTA/trypsin (Sigma). Growth medium was supplemented with palmitate, oleate, or stearate complexed to BSA at a 2:1 molar ratio as described previously (16), and BSA-supplemented medium was used as control. For cell stimulations, PBS or LPS (50 ng/ml) were added to BSA- or free fatty acid-containing medium.
Mice
Wild type (WT) C57BL/6 mice were obtained from Oriental Bioscience and maintained in our mouse colony. NLRP3 KO mice were purchased from The Jackson Laboratory; ATG5flox × LysM-Cre were a gift from Herbert Virgin (Washington University). All lines were in the C57BL/6 background. Mice were maintained in a pathogen-free facility on a standard chow diet ad libitum (6% fat). All animal experiments were conducted in strict accordance with National Institutes of Health guidelines for humane treatment of animals and were reviewed by the Animal Studies Committee of Washington University School of Medicine.
RNA Isolation and Quantitative RT-PCR
Total cellular RNA was isolated using Qiagen RNeasy columns and reverse-transcribed using a high capacity cDNA reverse transcription kit (Applied Biosystems). Real-time qRT-PCR was performed using SYBR Green reagent (Applied Biosystems) on an ABI 7500 Fast thermocycler. Relative gene expression was determined using the Δ-Δ CT method normalized to 36B4 expression. Mouse primers sequences were as follows (all are 5′-3′): 36B4 (forward, ATC CCT GAC GCA CCG CCG TGA; reverse, TGC ATC TGC TTG GAG CCC ACG TT); TNFα (forward, CAT CTT CTC AAA ATT CGA GTG ACA A; reverse, TGG GAG TAG ACA CAA GGT ACA ACC C); IL-1β (forward, AAG GAG AAC CAA GCA ACG ACA AAA; reverse, TGG GGA ACT CTG CAG ACT CAA ACT); NLRP3 (forward, AAA ATG CCT TGG GAG ACT CA; reverse, AAG TAA GGC CGG AAT TCA CC); NLRC4 (forward, CTG GAA AAG GAT GGG AAT GA; reverse, CCA AGG CAG CAT CAA TGT AG); and CXCL10 (forward, ATC ATC CCT GCG AGC CTA TCC TG; reverse, CGG ATT CAG ACA TCT CTG CTC ATC).
Western Blotting
Total cellular protein was isolated by lysing cells in 150 mm NaCl, 10 mm Tris (pH 8), Triton X-100 1%, and 1× Complete protease inhibitor. Proteins were separated on a TGX gradient gel (4–20%, Bio-Rad) and transferred to a nitrocellulose membrane. Western blotting for pro-IL-1β and tubulin was performed using 40 μg of total cellular protein.
IL-1β and TNFα ELISA
Supernatants were harvested from macrophage cultures after the indicated stimulations. IL-1β and TNFα were quantified using a DuoSet ELISA kit (R&D Systems) according to the manufacturer's instructions.
LDH Release Assay
After stimulation, macrophage supernatants were collected at 20 h, and LDH was quantified using the CytoTox 96 non-radioactive cytotoxicity assay (Promega) per the manufacturer's instructions using a Tecan Infinite M200 plate reader. In prior experiments we showed that total LDH does not vary between treatments, and thus only the supernatant values were determined in this study (1).
Lysosome Imaging
After the indicated stimulations, cells were removed from the plate as described above and then stained with 500 nm LysoTracker Red in tissue culture medium for 15 min at 37 °C. For fluorescent dextran experiments, macrophages were incubated with 500 μg/ml tetramethylrhodamine-dextran for 2 h in regular medium followed by cell stimulations for 16 h. After staining, cells were washed three times with PBS, harvested as described above, and analyzed by flow cytometry.
Caspase-1 FLICA Flow Cytometry
After the indicated stimulations, pMACs were removed from low adherence plates as described above. The caspase-1 FLICA reagent was reconstituted per the manufacturer's instructions and diluted in DMEM plus 10% inactivated fetal serum. Macrophages were incubated in 200 μl of staining solution for 45 min at 37 °C with gentle mixing every 10 min. After the incubation, cells were washed twice in 1 ml of apoptosis wash buffer (provided with FLICA reagent) and analyzed by flow cytometry.
Statistics
Statistical analysis was performed using GraphPad Prism software. All results are expressed as means ± S.E. Groups were compared by paired Student's t test or two-way analysis of variance as appropriate. A value of p ≤ 0.05 was considered significant.
RESULTS
SFA and LPS Activate the Lipotoxic Inflammasome
Elicited pMACs provide a tractable model system to study macrophages that enter and develop within sites of tissue injury. To evaluate the influence of lipid loading on macrophage inflammatory responses, we preincubated pMACs with 250 μm palmitate for 2 h prior to LPS challenge. This stimulation protocol led to a time- and dose-dependent release of IL-1β in a manner that required both stimuli (Fig. 1A and B). IL-1β mRNA and pro-IL-1β protein were strongly induced by LPS in the presence or absence of palmitate (Fig. 1, C and D). An LPS dose response revealed that maximal IL-1β release occurred at 50 ng/ml. The LPS response was significantly more potent than that seen with the TLR2 agonist PamCSK4, even at doses where TNFα production was similar between the two TLR stimuli (Fig. 1, E and F). We also evaluated the ability of other inflammatory ligands, including the TLR7 agonist CL075 and TNFα, to activate the lipotoxic inflammasome (Fig. 1G). LPS was the most potent inflammasome stimulus, and this occurred despite equivalent cellular activation by the TLR2 and TLR7 ligands as indicated by the TNFα mRNA response (Fig. 1H). Although 1L-1β mRNA expression was slightly less in response to PamCSK4 and CL075, this was out of proportion to the marked differences in IL-1β release (Fig. 1, G and H). Of note, TNFα was a weak activator of cytokine production by pMACs (Fig. 1, G and H).
FIGURE 1.

Palmitate and LPS activate the lipotoxic inflammasome in primary macrophages. A, pMACs were preloaded with 250 μm palmitate (palm) or with BSA for 2 h followed by BSA or palmitate ± 50 ng/ml LPS for the indicated times, and IL-1β in the supernatant was quantified by ELISA. B, pMACs were incubated with BSA or the indicated concentrations of palmitate in combination with LPS and IL-1β release at 20 h was determined by ELISA. C, macrophages were stimulated as indicated for 4 h, and IL-1β mRNA was quantified by qRT-PCR. D, pMACs were stimulated as indicated for 16 h, and pro-IL-1β protein was assessed by Western blotting. E and F, pMACs were stimulated with the indicated doses of LPS or PamCSK4 (TLR2 ligand) with BSA or palmitate, and IL-1β (E) or TNFα (F) release was quantified at 20 h. G and H, IL-1β release at 20 h and IL-1β and TNF mRNA levels at 4 h were determined after stimulation with LPS (black bars), PamCSK4 (50 ng/ml, hatched bars), CL075 (TLR7 ligand, 500 ng/ml, gray bars), or TNFα (50 ng/ml, hatched bars) with BSA or palmitate. Bar graphs indicate the means ± S.E. for a minimum of three experiments, each performed in triplicate. *, p < 0.05 for BSA-PBS versus palmitate-LPS or LPS versus other TLR agonists.
Lipotoxic Inflammasome Activation Occurs via a NLRP3 Mechanism
Consistent with inflammasome assembly, active caspase-1 was increased in palmitate-LPS-treated cells when compared with PBS or LPS stimulation (Fig. 2A). Moreover, pMACs incubated with the caspase-1-specific inhibitor Z-YVAD or the pan-caspase inhibitor Z-VAD together with necrostatin 1 (to prevent necroptosis (1)) significantly decreased IL-1β secretion (Fig. 2B). Importantly, caspase-1 inhibition did not reduce LDH release, indicating that IL-1β secretion was not a consequence of passive release from dying cells (Fig. 2C).
FIGURE 2.

Activation of the lipotoxic inflammasome occurs via a NLRP3-dependent mechanism. A, pMACs were stimulated with BSA-PBS, BSA-LPS, or palmitate (palm)-LPS for 16 h, and caspase-1 activation was determined by staining cells with caspase-1 FLICA reagent followed by quantification of FL1 high fluorescence cells by flow cytometry. B and C, macrophages were stimulated as indicated in the presence of Z-YVAD (50 μm, black bars) or Z-VAD/necrostatin 1 (nec1) (25 μm/50 μm, gray bars) for 20 h followed by quantification of IL-1β and LDH release. D, pMACs were pretreated with BSA or palmitate ± LPS for 4 or 16 h, and mRNA expression of NLRP3 and NLRC4 was determined by qRT-PCR. E–G, WT (white bars) or NLRP3 KO (hatched bars) pMACs were stimulated as indicated for 20 h, after which the release of IL-1β (E), TNFα (F), and LDH (G) was quantified. Bar graphs indicate the mean ± S.E. for a minimum of three experiments, each performed in triplicate. *, p < 0.05 for BSA versus palmitate, WT versus KO, or vehicle (veh) versus inhibitor.
Inflammasomes are defined by their cryopeptin component, of which the best characterized family members are NLRP3 and NLRC4 (12). In line with prior observations (17), NLRP3 was rapidly and dramatically up-regulated in pMACs treated with LPS, irrespective of palmitate (Fig. 2D). In contrast, NLRC4 mRNA was only modestly increased at later time points after LPS stimulation, and this induction failed to occur in the presence of palmitate (Fig. 2D). To assess the functional relevance of NLRP3 in the lipotoxic inflammasome response, we stimulated NLRP3-deficient macrophages with palmitate-LPS. As seen in Fig. 2E, IL-1β release was abrogated in NLRP3 KO macrophages. In contrast, TNFα and LDH release were similar between WT and NLRP3 KO pMACs (Fig. 2, F and G). Thus, NLRP3 is required for inflammasome activation in response to lipotoxic stimuli.
Saturated Fatty Acids Are Required for Lipotoxic Inflammasome Activation
To further investigate the upstream requirements for inflammasome assembly, we evaluated other lipid and glycemic stressors. The release of IL-1β occurred in macrophages loaded with the SFAs palmitate and stearate but not the monounsaturated fatty acid oleate (Fig. 3A). IL-1β secretion was also reduced by triascin C, an inhibitor of fatty acid esterification by acyl-CoA synthetase enzymes (Fig. 3B). Thus, IL-1β release is selectively triggered by SFAs through a mechanism that requires fatty acid activation. IL-1β secretion was similar whether macrophages were cultured in high or low glucose medium, indicating that hyperglycemic conditions are not required for the observed phenotype (Fig. 3C).
FIGURE 3.

Lipotoxic inflammasome activation requires saturated fatty acids independently of high glucose or ATP. A–C, IL-1β concentration in the media was determined 20 h after stimulation of pMACs with the indicated FFA (stearate (stear) or oleate (ole) (A)), in the presence or absence of 1 μm triascin C (TC (B)), or in high (4.5 g/liter) or low (1g/liter) glucose medium (C). D, macrophages were treated with palmitate (palm)-LPS for 20 h ± apyrase or primed with LPS for 2 h followed by 5 mm ATP ± apyrase for 60 min, and IL-1β release was quantified by ELISA. E, pMACs were treated with palmitate-LPS simultaneously (S) or pretreated with LPS for the indicated times followed by palmitate incubation for 20 h. As a control, the cells were also treated with LPS for 2 h followed by silica (200 μg/ml) for 6 h. IL-1β concentration in the supernatant was determined by ELISA. Bar graphs indicate the means ± S.E. for a minimum of three experiments, each performed in triplicate. *, p < 0.05 for BSA-PBS versus FFA-LPS, or vehicle (veh) versus inhibitor. ns, not significant.
ATP can be released from dying cells and is one of the best described activators of the NLRP3 inflammasome. To determine whether ATP release was responsible for lipotoxic inflammasome activation, we utilized the compound apyrase, which rapidly degrades ATP in the media. Apyrase ablated IL-1β secretion in response to ATP but had no effect on cells stimulated with palmitate-LPS (Fig. 3D). In most models of the NLRP3 inflammasome, macrophages are pretreated with LPS (signal 1) followed by stimulation with a second “cytoplasmic” stressor (signal 2). Surprisingly, when pMACs were preincubated with LPS for either 2 or 16 h followed by palmitate treatment, IL-1β secretion was markedly reduced compared with cells with simultaneous stimulation (Fig. 3E). In the same experiment, LPS-pretreated macrophages incubated with silica released substantial IL-1β. Thus, the lipotoxic inflammasome is unique among NLRP3 activators in that TLR4 must be activated concomitantly with SFA for a robust response. Together these data demonstrate that activation of the lipotoxic inflammasome by saturated fatty acids occurs via an NLRP3-dependent, ATP-independent mechanism that requires simultaneous TLR4 and lipid stress signals.
Mitochondrial ROS Primes the Lipotoxic Inflammasome
NLRP3 inflammasome activation is strongly associated with mitochondrial ROS, lysosome destabilization, and calcium signaling. However, the interplay between these elements remains poorly understood (13). We observed previously that mitochondrial ROS decreases by 16 h after palmitate-LPS treatment (1). However, ROS can also influence earlier inflammatory signaling events. To investigate the role of redox signaling, we incubated macrophages with the general antioxidants BHA and BHT, both of which reduced IL-1β with only a modest effect on TNFα release following palmitate-LPS treatment (Fig. 4, A and B). To address mitochondrial ROS specifically, macrophages were incubated with increasing concentrations of the mitochondrial targeted superoxide dismutase mimetic, mito-TEMPO, during palmitate-LPS stimulation. Mito-TEMPO dose-dependently inhibited IL-1β release with only a modest effect on TNFα in the media (Fig. 4, D and E). None of the antioxidants reduced macrophage cell death as determined by LDH release (Fig. 4, C and F). It has been controversial as to whether ROS contributes to signal 1 or signal 2 in NLRP3 activation. In our system mito-TEMPO reduced IL-1β and TNFα mRNA in a manner that paralleled the secretion of these cytokines (Fig. 4G). Moreover, pro-IL-1β protein was significantly reduced by mito-TEMPO, strongly arguing that mitochondrial ROS is a component of signal 1 and thus primes the lipotoxic inflammasome (Fig. 4H).
FIGURE 4.

Mitochondrial ROS primes the lipotoxic inflammasome. A–F, macrophages were stimulated for 20 h with BSA-PBS or palmitate (palm)-LPS ± the general antioxidants BHA (50 μm, black bars) or BHT (50 μm, gray bars) (A–C) or the indicated doses of the mitochondria-specific ROS scavenger mito-TEMPO (MT, hatched bars) (D–F), and the release of IL-1β (A and D), TNFα (B and E), and LDH (C and F) was determined. G, pMACs were treated with palmitate-LPS ± mito-TEMPO for 8 h, and IL-1β or TNFα mRNA was quantified by qRT-PCR. H, macrophages were incubated with palmitate-LPS for 16 h ± mito-TEMPO (500 μm) after which intracellular pro-IL-1β levels were assessed by Western blotting. Tubulin was used as a loading control. Bar graphs indicate the mean ± S.E. for a minimum of three experiments, each performed in triplicate. *, p < 0.05 for vehicle (veh) versus inhibitor.
Activation of the Lipotoxic Inflammasome Occurs via a Lysosome-dependent Mechanism
We have shown previously that lipid-stressed macrophages challenged with LPS develop lysosome dysfunction, lysosomal membrane destabilization, and cathepsin release to the cytosol (1). To investigate the relationship between lysosome damage and caspase-1 activation, we utilized dual color flow cytometry employing tetramethylrhodamine-dextran (lysosome label) and caspase-1 FLICA. Caspase-1 activation selectively occurred in cells where lysosome content was diminished (Fig. 5A). Cathepsin B is a lysosomal protease that has been implicated in inflammasome activation following treatment with lysosome-damaging compounds such as silica and cholesterol crystals (8, 18). The cathepsin B inhibitor CAO74 dose-dependently reduced IL-1β release from palmitate-LPS-treated pMACs, supporting a role for this pathway (Fig. 5B). To further confirm these findings, we used a chemically distinct cathepsin B inhibitor, Z-FA, which produced similar results (Fig. 5C). Despite their profound inhibition of IL-1β, these compounds had only a minimal effect on TNFα release (Fig. 5D). Consistent with a role for cathepsin B in signal 2 of the inflammasome, CAO74 did not reduce pro-IL-1β production (Fig. 5E).
FIGURE 5.

The lysosome is required for lipotoxic inflammasome activation. A, pMACs were loaded with tetramethylrhodamine (TMR)-dextran to label lysosomes followed by stimulation with BSA-PBS or palmitate (palm)-LPS for 16 h. The cells were stained with caspase-1 FLICA and analyzed by flow cytometry. The percentage of cells in each quadrant is indicated. B, the cathepsin B inhibitor CAO74 was added at the indicated doses, and IL-1β release following palmitate-LPS challenge was assessed at 20 h. C and D, macrophages were incubated with CAO74 (black bars) or another cathepsin B inhibitor, Z-FA (50 μm, hatched bars), during stimulation with BSA-PBS or palmitate-LPS for 20 h. IL-1β (C) and TNFα (D) concentrations in the supernatant were determined by ELISA. E, pro-IL-1β levels in palmitate-LPS-treated pMACs ± CAO74 (10 μm) were determined at 16 h by Western blotting. Bar graphs indicate the mean ± S.E. for a minimum of three experiments, each performed in triplicate. *, p < 0.05 for vehicle (veh) versus inhibitor. ns, not significant.
Lysosomal Acidification Inhibitors Suppress Pro-IL-1β Production
To further assess the role of the lysosome, we treated macrophages with the lysosomal acidification inhibitor bafilomycin A (BAF). BAF suppressed IL-1β release from macrophages in a dose-dependent fashion that correlated with its ability to neutralize lysosome pH (Fig. 6, A and B). Similar results were observed when pMACs were treated with ammonium chloride (NH4CL), a compound that neutralizes lysosome pH through an independent mechanism (Fig. 6C). TNFα release was not reduced, and in fact it was slightly increased in response to both BAF and NH4CL (Fig. 6D).
FIGURE 6.

Lysosomal acidification inhibitors block pro-IL-1β synthesis. A and B, pMACs were treated with palmitate (palm)-LPS, and the indicated concentrations of BAF after which IL-1β release (A) or LysoTracker Red (LT-red) staining (B) were assessed by ELISA and flow cytometry, respectively. C and D, the lysosome acidification inhibitors BAF (25 nm, blue bars) or NH4CL (5 mm, gray bars) were added to treated macrophages, and IL-1β (C) or TNFα (D) release was quantified. E, pro-IL-1β protein levels were determined in pMACs treated with palmitate-LPS ± BAF or NH4CL at 16 h by Western blotting. F, pMACs were treated with palmitate-LPS for 8 h in the presence of vehicle (veh, white bars), BAF (blue bars), or NH4CL (gray bars), and mRNA expression of IL-1β, TNFα, NLRP3, and CXCL10 was determined by qRT-PCR. G, palmitate-LPS-treated pMACs were incubated with vehicle, CAO74 (black bars), BAF (blue bars), or Z-YVAD (white bars) either simultaneous with the stimulation (sim) or delayed by 12 h. IL-1β release was quantified by ELISA, and the levels are reported as percent vehicle (where 100% means no inhibition). Bar graphs indicate the mean ± S.E. for a minimum of three experiments, each performed in triplicate. *, p < 0.05 for vehicle versus inhibitor.
Given the results of prior studies (8, 18, 19) and our cathepsin B data, it was expected that the lysosome acidification inhibitors were acting at the level of inflammasome complex assembly (signal 2). Therefore, we were surprised to discover that BAF and NH4CL significantly reduced pro-IL-1β protein levels in palmitate-LPS-treated macrophages (Fig. 6E). IL-1β mRNA levels were also markedly suppressed by both BAF and NH4CL, whereas TNFα and NLRP3 mRNA levels were largely unaffected (Fig. 6F). The TRIF target gene, CXCL10, was also unaffected by BAF and NH4CL, arguing against disruption of TLR4-TRIF signaling, which occurs from the endolysosomal compartment (20), as a cause for this phenotype (Fig. 6F). To provide further evidence that lysosome acidification inhibitors and cathepsin inhibitors act at different stages of inflammasome activation, we compared the ability of CAO74 and BAF to suppress IL-1β release when the compounds were given simultaneously with LPS or delayed for 12 h. As a control, cells were treated with the caspase-1 inhibitor Z-YVAD (which should be equally inhibitory whether added early or delayed). Consistent with our predictions, the inhibitory effect of BAF was lost when it was added 12 h after stimulation, confirming its role in blocking early signaling events (Fig. 6G). In contrast, CAO74 and Z-YVAD retained their inhibitory ability at both time points (Fig. 6G). Therefore, disrupting lysosome function affects both early signaling relevant to the generation of pro-IL-1β and later events related to inflammasome complex assembly.
Impaired lysosome function also blocks autophagy, which has been implicated previously in inflammasome activation (10, 21). Therefore, we asked whether LPS stimulation of autophagy-deficient cells would be sufficient to reproduce the palmitate-LPS phenotype. To address this question, macrophages were isolated from myeloid-specific ATG5 KO mice. ATG5 KO cells treated with LPS released more IL-1β compared with WT cells; however, the magnitude of this response was small in comparison with palmitate-LPS stimulation (48 versus 2000 pg/ml, respectively) (Fig. 7A). Therefore, impaired autophagic flux does not fully explain the lipotoxic inflammasome phenotype. In fact, ATG5 KO macrophages stimulated with the combination of palmitate and LPS consistently released less IL-1β than WT littermate cells, despite similar TNFα and LDH release (Fig. 7, B and C).
FIGURE 7.
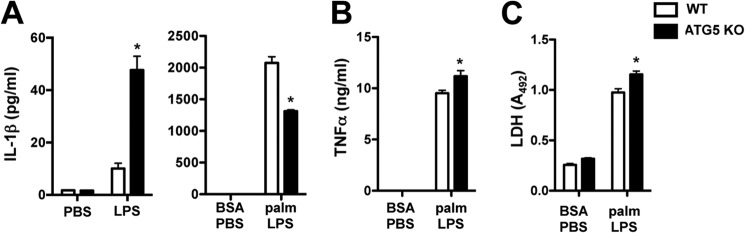
Loss of the autophagy protein ATG5 reduces IL-1β release in response to lipotoxic stress. A, IL-1β release was determined in pMACs from myeloid-specific ATG5-deficient animals (ATG5 KO, black bars) or WT littermate controls (white bars) that were treated with PBS versus LPS (left panel) or BSA-PBS versus palmitate (palm)-LPS (right panel) for 20 h. B and C, the release of TNFα and LDH was quantified in WT versus ATG5 KO macrophages. Bar graphs indicate the mean ± S.E. for a minimum of three experiments, each performed in triplicate. *, p < 0.05 for WT versus KO.
Intracellular Calcium Stores Are Required for IL-1β Release in Response to Palmitate and LPS
Our data with BAF and NH4CL suggested that increasing lysosomal pH could disrupt signaling events that regulate IL-1β expression. Moreover, it is well known that lysosomal acidification inhibitors deplete acidic calcium stores (22). To explore a role for intracellular calcium in lipotoxic inflammasome activation, we initially used 2-APB, which blocks the release of calcium from intracellular stores (ER and lysosomes). 2-APB significantly decreased palmitate-LPS-induced IL-1β secretion, with only a modest effect on TNFα (Fig. 8, A and B). In contrast, pMACs stimulated with palmitate-LPS in calcium-free media released equivalent amounts of IL-1β, indicating that extracellular calcium is dispensable for this phenomenon (Fig. 8C).
FIGURE 8.

Intracellular calcium stores are required for full activation of the lipotoxic inflammasome. A and B, pMACs were stimulated as indicated in the presence of vehicle (veh, white bars) or 2-APB (100 μm, black bars) for 20 h after which IL-1β (A) and TNFα (B) release was quantified. C, cells were treated with the indicated stimuli in standard medium (std, white bars) or calcium-free DMEM (Ca free, hatched bars), and IL-1β release was assessed at 20 h by ELISA. D–F, stimulated macrophages were incubated with vehicle (white bars) versus U18666A (1 μg/ml, black bars), and IL-1β secretion (D), 8-h IL-1β mRNA (E), and TNFα release (F) were quantified. G–I, pMACs were stimulated as indicated in the presence of vehicle (white bars) or NED-19 (100 μm, black bars), and IL-1β secretion (G), 8-h IL-1β mRNA (H), and TNFα release (I) were determined. Bar graphs indicate the mean ± S.E. for a minimum of three experiments, each performed in triplicate. *, p < 0.05 for vehicle versus inhibitor. palm, palmitate. ns, not significant.
The bulk of intracellular calcium resides in the ER and lysosomes, both of which have channels that are inhibited by 2-APB. To more specifically address the contribution of acidic calcium stores, we utilized the compound U18666A, which rapidly depletes lysosomal calcium (23). As shown in Fig. 8, cells treated with U18666A produced significantly less IL-1β than controls (D), and this occurred in concert with reduced IL-1β mRNA (E). TNFα release was slightly augmented in cells treated with this compound (Fig. 8F). To further investigate the role of lysosomal calcium in this response, we took advantage of the compound NED-19, which selectively inhibits the NAADP-activated calcium channel that resides on the lysosome (24). NED-19-treated pMACs also secreted less IL-1β and produced less IL-1β mRNA compared with vehicle-treated cells, providing further evidence that lysosomal calcium contributes to lipotoxic inflammasome activation (Fig. 8, G–I). Together these data argue that acidic calcium stores contribute to signal 1 of inflammasome activation via effects on the production and/or stabilization of the IL-1β transcript.
Calcineurin Inhibitors Suppress Activation of the Lipotoxic Inflammasome
The evidence that calcium signaling influences IL-1β mRNA levels led us to investigate calcineurin, a calcium-activated phosphatase that can modulate cytokine expression. Interestingly, prior studies have demonstrated that inhibition of calcineurin by cyclosporine (CSA) reduces ATP-induced inflammasome activation. However, this was thought to occur via a mechanism that involved CSA-mediated inhibition of the mitochondrial permeability transition pore (MPTP) (25, 26). To investigate the role of calcineurin pathways in the lipotoxic inflammasome, we first assessed the effect of CSA on palmitate-LPS-induced IL-1β release. CSA dose-dependently reduced IL-1β release without significantly impacting TNFα or LDH release (Fig. 9, A–C). To address whether CSA was acting via calcineurin, we treated macrophages with FK506, a calcineurin inhibitor that has no effect on the MPTP. FK506 behaved similarly to CSA in its ability to reduce IL-1β, despite having no effect on baseline MMP as assessed by tetramethylrhodamine ethyl ester staining (TMRE, Fig. 9, D–F). To further confirm these observations, we utilized the potent calcineurin inhibitor CN585, which interacts directly with calcineurin (27). Similar to CSA, treatment with CN585 dose-dependently reduced IL-1β secretion with a minimal effect on TNFα or LDH release (Fig. 9, G–I). These data strongly argue that calcineurin is required for full activation of the lipotoxic inflammasome.
FIGURE 9.

Calcineurin inhibitors suppress IL-1β release during activation of the lipotoxic inflammasome. A–C, stimulated macrophages were incubated with the indicated concentrations of CSA, and IL-1β (A), TNFα (B), and LDH release (C) were quantified 20 h later. D and E, pMACs were incubated with vehicle (veh, red bars), CSA (black bars), or FK506 (FK, gray bars) during 20 h of stimulation with BSA-PBS or palmitate (palm)-LPS after which IL-1β (D) and TNFα (E) concentrations in the supernatant were determined by ELISA. F, mitochondrial membrane potential was assessed by tetramethylrhodamine ethyl ester (TMRE) staining followed by flow cytometry in pMACs incubated with vehicle (red line), CSA (black line), or FK506 (gray line) for 20 h. G–I, macrophages were stimulated in the presence of the indicated concentrations of CN585, and IL-1β (G), TNFα (H), and LDH release (I) were quantified 20 h later. Bar graphs indicate the mean ± S.E. for a minimum of three experiments, each performed in triplicate. *, p < 0.05 for vehicle versus inhibitor. ns, not significant.
Calcineurin Inhibitors Reduce IL-1β mRNA Stability
In light of our observations that intracellular calcium pathways regulate pro-IL-1β production, we evaluated the effect of CSA on IL-1β mRNA levels at 4, 8, and 16 h after stimulation with palmitate-LPS. Compared with vehicle, CSA had minimal effect on the early induction of IL-1β mRNA by palmitate-LPS. However, by 8 h IL-1β mRNA levels were reduced by ∼50%, and this inhibitory effect persisted out to 16 h (Fig. 10A). Similar to our data obtained from the other calcium pathway inhibitors, TNFα and NLRP3 mRNA levels were not reduced at any time point (Fig. 10, B and C). Pro-IL-1β protein levels in palmitate-LPS-treated pMACs were also significantly reduced at 16 h in CSA-treated cells compared with controls (Fig. 10, D and E).
FIGURE 10.

Inhibition of calcineurin reduces IL-1β mRNA stability and protein levels in response to lipotoxic stimulation. A–C, pMACs were stimulated with palmitate-LPS ± 5 μm CSA, and mRNA expression of IL-1β (A), TNFα (B), and NLRP3 (C) was assessed at the indicated time points by qRT-PCR. D, the expression of pro-IL-1β protein was determined by Western blotting from macrophages stimulated with palmitate (palm)-LPS for 16 h in the presence of vehicle (veh) or CSA. E, quantification of pro-IL-1β protein expression normalized to tubulin. F, pMACs were activated with palmitate-LPS, and vehicle, CSA, or Z-YVAD (50 μm) was added simultaneously with the stimulation (sim) or delayed by 12 h. IL-1β release was quantified by ELISA, and the levels are reported as % vehicle (where 100% means no inhibition). G, macrophages were treated with palmitate-LPS ± CSA or pretreated with LPS (for 2 h) followed by 5 mm ATP ± CSA for 60 min. IL-1β release was quantified by ELISA. H, pMACs were treated with palmitate-LPS for 4 h after which actinomycin D (ActD, 5 μm) was added to the cells in the presence (black bars) or absence (white bars) of CSA (5 μm). IL-1β and TNFα mRNA levels were quantified at the indicated time points by qRT-PCR. Bar graphs indicate the mean ± S.E. for a minimum of three experiments, each performed in triplicate. *, p < 0.05 for vehicle versus inhibitor or early versus delayed stimulations.
To confirm functionally that the inhibitory effects of CSA were related to early transcriptional events rather than later mitochondrial effects, we again performed a delayed inhibitor experiment in which CSA was added to cells at time 0 or delayed for 12 h, and IL-1β release was assessed. Similar to our experiments with lysosome inhibitors, the caspase-1 inhibitor Z-YVAD was used as a control. In line with our transcriptional data, macrophages treated with CSA at the time of palmitate-LPS stimulation produced dramatically less IL-1β than cells receiving CSA at 12 h (20 versus 67% of control levels, respectively) (Fig. 10F). In contrast, Z-YVAD had a similar efficacy in reducing IL-1β release, ∼25% of control, whether added early or delayed (Fig. 10F). To test this concept another way, we also evaluated the effect of CSA on IL-1β release from LPS-primed macrophages treated with ATP. The rapid kinetics of NLRP3 activation by ATP (30–60 min) precludes CSA from significant impacting IL-1β transcript and/or protein levels. Consistent with its role in IL-1β mRNA regulation, CSA did not inhibit the ATP inflammasome (Fig. 10G). In fact, IL-1β release was increased in CSA-treated cells. Collectively, these findings suggest that calcineurin modulates IL-1β release via its effects on pro-IL-1β mRNA and protein.
Because the early induction of IL-1β mRNA was not affected by CSA, we hypothesized that CSA may act through reducing mRNA stability. To test this possibility, we treated pMACs with palmitate-LPS for 4 h to induce IL-1 β mRNA, after which the transcriptional inhibitor actinomycin D was added in the presence or absence of CSA. IL-1β mRNA levels were subsequently assessed at 1 and 6 h. By 6 h after the addition of actinomycin D, cells treated with CSA had a 31% reduction in IL-1 β mRNA compared with vehicle controls (Fig. 10H). TNFα mRNA stability was unaffected by CSA. Thus, calcineurin inhibitors prevent IL-1β release at the level of signal 1, and this occurs, at least in part, by reducing the stability of IL-1β mRNA.
DISCUSSION
Metabolic stress has been linked to inflammasome activation and the development of insulin resistance in the setting of obesity (10, 11). Given the central role of macrophage dysfunction in complications of metabolic disease and our previous data demonstrating markedly increased IL-1β release from lipid-stressed macrophages (2), we sought to elucidate the regulatory pathways controlling lipotoxic inflammasome activation. To more closely simulate the biology of lipid-overloaded macrophages encountering an inflammatory stimulus, elicited peritoneal macrophages were loaded with lipids prior to the addition of LPS. Using this system we observed robust IL-1β release in palmitate-enriched macrophages treated with LPS; and this was not seen with LPS or palmitate alone. IL-1β release required NLRP3 and caspase-1 activation via a mechanism that was dependent on the lysosome. Interestingly, we provide evidence to support a novel role for lysosomal pathways in both signal 1 and signal 2 of inflammasome activation. Specifically, acidic calcium stores and calcineurin signals modulate the production of pro-IL-1β, whereas lysosomal proteases contribute to its subsequent release.
NLRP3 inflammasome activation in response to particulate stimuli such as silica, aluminum, uric acid crystals, and cholesterol crystals is thought to occur via a lysosomal damage-mediated mechanism (8, 18, 19). We had observed previously that macrophages treated with palmitate and LPS develop progressive lysosomal destabilization (1), leading us to investigate the role of this pathway in the inflammasome response. A number of our findings support a role for the lysosome in lipotoxic inflammasome activation: 1) the kinetics of IL-1β release correlated with the progression of lysosome dysfunction (1); 2) caspase-1 activation was observed specifically within cells with lysosome impairment; 3) two chemically distinct cathepsin B inhibitors and two different lysosomal acidification inhibitors all led to profound suppression of IL-1β release without affecting TNFα; and 4) TLR agonists that activate MyD88 only (PamCSK4, TLR2; CL075, TLR7) are weak inducers of lysosome dysfunction (1) and IL-1β release.
Although autophagy is also impaired by lysosome dysfunction, this alone did not account for the observed phenotype. In fact, ATG5 KO macrophages treated with palmitate-LPS released less IL-1β compared with WT littermate controls. This surprising finding may be attributable to the role of ATG5 in the unconventional secretion pathway for IL-1β release (28). Of interest, our lysosome, autophagy, and ROS data are not consistent with the findings of a prior study, in which it was suggested that palmitate mediates inflammasome activation via a lysosome-independent mechanism that involves autophagy inhibition and mitochondrial ROS accumulation (10). These differences may relate to variances in the stimulation protocol (i.e. the order of LPS and palmitate addition) or intrinsic differences between bone marrow-derived and peritoneal macrophages. Further investigation will be necessary to explore these possibilities.
Lysosome damage would be predicted to deliver signal 2 of the inflammasome pathway (13), so we were surprised to discover that unlike the cathepsin inhibitors, lysosomal acidification inhibitors reduced IL-1β release by selectively decreasing IL-1β mRNA and protein levels. Germane to this observation, lysosomal acidification inhibitors have also been shown to deplete acidic calcium stores, raising the possibility that calcium signaling may influence IL-1β mRNA levels (22, 29). Consistent with this notion, IL-1β release was reduced significantly by 2-APB, U18666A, and NED-19, all of which disrupt the release or handling of intracellular calcium stores. U18666A and NED-19 specifically modulate acidic calcium stores (23, 24). Similar to the lysosomal acidification inhibitors, these calcium-modulating compounds reduced the level of IL-1β mRNA. Together these data implicate lysosomal calcium signaling pathways in the regulation of pro-IL-1β mRNA levels; however, they do not exclude a contribution from ER calcium stores.
Calcineurin is a calcium-regulated phosphatase with known effects on inflammatory cytokine regulation. Prior studies have shown that the calcineurin inhibitor CSA reduces IL-1β release from activated macrophages, but this has been attributed to MPTP inhibition (25, 26). In this study we found that CSA reduced IL-1β release from macrophages in response to palmitate-LPS, but this action was related to calcineurin inhibition and not blockade of the MPTP. Consistent with the other calcium-signaling inhibitors, CSA also reduced the level of IL-1β mRNA and protein (signal 1). Kinetic mRNA quantification revealed that CSA did not impact the induction of IL-1β transcription but rather reduced transcript levels over time. This action of CSA was selective for IL-1β, as this compound had no effect on TNFα or NLRP3 mRNA levels at any time point. In line with our data from the kinetic mRNA analysis, we demonstrated that calcineurin inhibition decreased IL-1β mRNA stability, accounting for at least part of the observed phenotype. Thus, calcineurin appears to be one of the important calcium-regulated proteins that modulate the expression of pro-IL-1β. Further evidence supporting the relevance of this pathway comes from a recent report demonstrating that calcineurin overexpression in cardiac myocytes also leads to enhanced IL-1β release with activation of the NLRP3 inflammasome (30).
In summary, the current study identifies the lysosome as a central player in lipotoxic inflammasome activation in primary macrophages. Early signaling events that emanate from intracellular and lysosomal calcium stores regulate the production of pro-IL-1β via a mechanism involving calcineurin and stabilization of IL-1β mRNA (signal 1). At later time points, lysosome destabilization contributes to cathepsin B-mediated activation of the NLRP3-caspase-1 complex (signal 2). Macrophage dysfunction is highly relevant to the complications of obesity and diabetes, which are frequently associated with prolonged inflammation and impaired resolution/repair. The inflammasome has been linked to this disease phenotypes (31). Our data add to the mechanistic understanding of how lipids modulate macrophage biology to affect inflammation. Future work exploring lysosome and calcineurin pathway modulators in the context of in vivo models of diabetic inflammation is of significant translational interest for the treatment of obesity/diabetic complications.
This work was supported, in whole or in part, by National Institutes of Health Grant K08HL098373 (to J. D. S.).
- SFA
- saturated fatty acid
- ER
- endoplasmic reticulum
- ROS
- reactive oxygen species
- BHA
- butylated hydroxyanisole
- BHT
- butylated hydroxytolulene
- pMAC
- peritoneal macrophage
- qRT-PCR
- quantitative RT-PCR
- LDH
- lactate dehydrogenase
- Z-
- benzyloxycarbonyl-
- BAF
- bafilomycin A
- 2-APB
- 2-aminoethoxydiphenyl borate
- CSA
- cyclosporine
- MPTP
- mitochondrial permeability transition pore
- TRIF
- TIR-domain-containing adapter-inducing interferon-β
- TLR
- Toll-like receptor.
REFERENCES
- 1. Schilling J. D., Machkovech H. M., He L., Diwan A., Schaffer J. E. (2013) TLR4 activation under lipotoxic conditions leads to synergistic macrophage cell death through a TRIF-dependent pathway. J. Immunol. 190, 1285–1296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Schilling J. D., Machkovech H. M., He L., Sidhu R., Fujiwara H., Weber K., Ory D. S., Schaffer J. E. (2013) Palmitate and lipopolysaccharide trigger synergistic ceramide production in primary macrophages. J. Biol. Chem. 288, 2923–2932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Khanna S., Biswas S., Shang Y., Collard E., Azad A., Kauh C., Bhasker V., Gordillo G. M., Sen C. K., Roy S. (2010) Macrophage dysfunction impairs resolution of inflammation in the wounds of diabetic mice. PLoS One 5, e9539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mirza R., Koh T. J. (2011) Dysregulation of monocyte/macrophage phenotype in wounds of diabetic mice. Cytokine 56, 256–264 [DOI] [PubMed] [Google Scholar]
- 5. Kanter J. E., Kramer F., Barnhart S., Averill M. M., Vivekanandan-Giri A., Vickery T., Li L. O., Becker L., Yuan W., Chait A., Braun K. R., Potter-Perigo S., Sanda S., Wight T. N., Pennathur S., Serhan C. N., Heinecke J. W., Coleman R. A., Bornfeldt K. E. (2012) Diabetes promotes an inflammatory macrophage phenotype and atherosclerosis through acyl-CoA synthetase 1. Proc. Natl. Acad. Sci. U.S.A. 109, E715–E724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Brookheart R. T., Michel C. I., Schaffer J. E. (2009) As a matter of fat. Cell Metab. 10, 9–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Henao-Mejia J., Elinav E., Strowig T., Flavell R. A. (2012) Inflammasomes: far beyond inflammation. Nat. Immunol. 13, 321–324 [DOI] [PubMed] [Google Scholar]
- 8. Duewell P., Kono H., Rayner K. J., Sirois C. M., Vladimer G., Bauernfeind F. G., Abela G. S., Franchi L., Nuñez G., Schnurr M., Espevik T., Lien E., Fitzgerald K. A., Rock K. L., Moore K. J., Wright S. D., Hornung V., Latz E. (2010) NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 464, 1357–1361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mezzaroma E., Toldo S., Farkas D., Seropian I. M., Van Tassell B. W., Salloum F. N., Kannan H. R., Menna A. C., Voelkel N. F., Abbate A. (2011) The inflammasome promotes adverse cardiac remodeling following acute myocardial infarction in the mouse. Proc. Natl. Acad. Sci. U.S.A. 108, 19725–19730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wen H., Gris D., Lei Y., Jha S., Zhang L., Huang M. T., Brickey W. J., Ting J. P. (2011) Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat. Immunol. 12, 408–415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Vandanmagsar B., Youm Y. H., Ravussin A., Galgani J. E., Stadler K., Mynatt R. L., Ravussin E., Stephens J. M., Dixit V. D. (2011) The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat. Med. 17, 179–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Strowig T., Henao-Mejia J., Elinav E., Flavell R. (2012) Inflammasomes in health and disease. Nature 481, 278–286 [DOI] [PubMed] [Google Scholar]
- 13. Latz E., Xiao T. S., Stutz A. (2013) Activation and regulation of the inflammasomes. Nat. Rev. Immunol. 13, 397–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. De Nardo D., Latz E. (2011) NLRP3 inflammasomes link inflammation and metabolic disease. Trends Immunol. 32, 373–379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lee H. M., Kim J. J., Kim H. J., Shong M., Ku B. J., Jo E. K. (2013) Upregulated NLRP3 inflammasome activation in patients with type 2 diabetes. Diabetes 62, 194–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Listenberger L. L., Ory D. S., Schaffer J. E. (2001) Palmitate-induced apoptosis can occur through a ceramide-independent pathway. J. Biol. Chem. 276, 14890–14895 [DOI] [PubMed] [Google Scholar]
- 17. Bauernfeind F. G., Horvath G., Stutz A., Alnemri E. S., MacDonald K., Speert D., Fernandes-Alnemri T., Wu J., Monks B. G., Fitzgerald K. A., Hornung V., Latz E. (2009) Cutting edge: NF-κB-activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J. Immunol. 183, 787–791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hornung V., Bauernfeind F., Halle A., Samstad E. O., Kono H., Rock K. L., Fitzgerald K. A., Latz E. (2008) Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat. Immunol. 9, 847–856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Martinon F., Pétrilli V., Mayor A., Tardivel A., Tschopp J. (2006) Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 440, 237–241 [DOI] [PubMed] [Google Scholar]
- 20. Kagan J. C., Su T., Horng T., Chow A., Akira S., Medzhitov R. (2008) TRAM couples endocytosis of Toll-like receptor 4 to the induction of interferon-beta. Nat. Immunol. 9, 361–368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Saitoh T., Fujita N., Jang M. H., Uematsu S., Yang B. G., Satoh T., Omori H., Noda T., Yamamoto N., Komatsu M., Tanaka K., Kawai T., Tsujimura T., Takeuchi O., Yoshimori T., Akira S. (2008) Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1β production. Nature 456, 264–268 [DOI] [PubMed] [Google Scholar]
- 22. Christensen K. A., Myers J. T., Swanson J. A. (2002) pH-dependent regulation of lysosomal calcium in macrophages. J. Cell Sci. 115, 599–607 [DOI] [PubMed] [Google Scholar]
- 23. Lloyd-Evans E., Morgan A. J., He X., Smith D. A., Elliot-Smith E., Sillence D. J., Churchill G. C., Schuchman E. H., Galione A., Platt F. M. (2008) Niemann-Pick disease type C1 is a sphingosine storage disease that causes deregulation of lysosomal calcium. Nat. Med. 14, 1247–1255 [DOI] [PubMed] [Google Scholar]
- 24. Pitt S. J., Funnell T. M., Sitsapesan M., Venturi E., Rietdorf K., Ruas M., Ganesan A., Gosain R., Churchill G. C., Zhu M. X., Parrington J., Galione A., Sitsapesan R. (2010) TPC2 is a novel NAADP-sensitive Ca2+ release channel, operating as a dual sensor of luminal pH and Ca2+. J. Biol. Chem. 285, 35039–35046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Shimada K., Crother T. R., Karlin J., Dagvadorj J., Chiba N., Chen S., Ramanujan V. K., Wolf A. J., Vergnes L., Ojcius D. M., Rentsendorj A., Vargas M., Guerrero C., Wang Y., Fitzgerald K. A., Underhill D. M., Town T., Arditi M. (2012) Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 36, 401–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nakahira K., Haspel J. A., Rathinam V. A., Lee S. J., Dolinay T., Lam H. C., Englert J. A., Rabinovitch M., Cernadas M., Kim H. P., Fitzgerald K. A., Ryter S. W., Choi A. M. (2011) Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 12, 222–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Erdmann F., Weiwad M., Kilka S., Karanik M., Pätzel M., Baumgrass R., Liebscher J., Fischer G. (2010) The novel calcineurin inhibitor CN585 has potent immunosuppressive properties in stimulated human T cells. J. Biol. Chem. 285, 1888–1898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dupont N., Jiang S., Pilli M., Ornatowski W., Bhattacharya D., Deretic V. (2011) Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1β. EMBO J. 30, 4701–4711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Patel S., Docampo R. (2010) Acidic calcium stores open for business: expanding the potential for intracellular Ca2+ signaling. Trends Cell Biol. 20, 277–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bracey N. A., Beck P. L., Muruve D. A., Hirota S. A., Guo J., Jabagi H., Wright J. R., Jr., Macdonald J. A., Lees-Miller J. P., Roach D., Semeniuk L. M., Duff H. J. (2013) The Nlrp3 inflammasome promotes myocardial dysfunction in structural cardiomyopathy through interleukin-1β. Exp. Physiol. 98, 462–472 [DOI] [PubMed] [Google Scholar]
- 31. Mirza R. E., Fang M. M., Ennis W. J., Koh T. J. (2013) Blocking interleukin-1β induces a healing-associated wound macrophage phenotype and improves healing in type 2 diabetes. Diabetes 62, 2579–2587 [DOI] [PMC free article] [PubMed] [Google Scholar]