

Background: AMPK senses energetic changes and regulates glucose metabolism.
Results: AMPK α2 deficiency aggravated the glucose deprivation and necrosis of the hepatocytes via increased ROS production and decreased mitophagy.
Conclusion: AMPK α2 is essential for attenuation of liver injury during tumor metastasis.
Significance: This is the first time to reveal the mechanism by which glucose/energy competition induced tissue damage in tumor.
Keywords: AMP-activated kinase (AMPK), Glucose, Metastasis, Oxidative Stress, Tumor
Abstract
It is well known that tumors damage affected tissues; however, the specific mechanism underlying such damage remains elusive. AMP-activated protein kinase (AMPK) senses energetic changes and regulates glucose metabolism. In this study, we examined the mechanisms by which AMPK promotes metabolic adaptation in the tumor-bearing liver using a murine model of colon cancer liver metastasis. Knock-out of AMPK α2 significantly enhanced tumor-induced glucose deprivation in the liver and increased the extent of liver injury and hepatocyte death. Mechanistically, we observed that AMPK α2 deficiency resulted in elevated reactive oxygen species, reduced mitophagy, and increased cell death in response to tumors or glucose deprivation in vitro. These results imply that AMPK α2 is essential for attenuation of liver injury during tumor metastasis via hepatic glucose deprivation and mitophagy-mediated inhibition of reactive oxygen species production. Therefore, AMPK α2 might represent an important therapeutic target for colon cancer metastasis-induced liver injury.
Introduction
Tumor-mediated tissue damage, such as cachexia, can result in death. Moreover, reports suggest that tumor-bearing tissues are subjected to energetic stress. The Warburg effect was described decades ago, aerobic glycolysis has recently been accepted as a metabolic hallmark of cancer, and the concomitant increase in glucose uptake has been demonstrated in various tumor types (1, 2). Consequently, increased uptake and metabolization of extra glucose by tumors result in reduced glucose availability for neighboring tissue. The liver is essential for the regulation of glucose metabolism and is the most common site of cancer metastasis. Previous reports have shown an imbalance of glucose and energy metabolism conditions in the livers of tumor-bearing patients and animals (3–5). However, how the redistribution of glucose between tumors and neighboring tissues regulates or affects the function of tumor-bearing tissue remains unclear.
AMPK2 is a heterotrimeric serine-threonine protein kinase that is conserved from yeast to mammals. AMPK is activated by environmental or nutritional stress factors, such as hypoxia, glucose deprivation, and inhibition of mitochondrial oxidative phosphorylation that deplete intracellular ATP levels (6–8). The α subunit of AMPK is the catalytic subunit, which has at least two isoforms (α1 and α2) that can be phosphorylated by several upstream kinases (CaM-dependent protein kinase kinase (9), liver kinase B1 (LKB1) (10), cAMP-dependent kinase (PKA) (11), and Ca2+/CaM-dependent protein kinase II (12). The enrichment of AMPK α1 or α2 is tissue and cell specific, particularly for the AMPK α2 isoform (13). Activated AMPK maintains cellular energy homeostasis by activating energy production and inhibiting energy expenditure in numerous tissues (14). However, the role of AMPK in the regulation of tumor-bearing tissue injury remains unknown.
Similar to AMPK activation, autophagy is also initiated under stress conditions such as nutrient limitation or low energy states. Autophagy acts as a survival mechanism that eliminates damaged organelles and restores the availability of critical metabolic intermediates (15, 16). Autophagy-defective cells accumulate reactive oxygen species (ROS), resulting in cell death (17). Mitochondria produce excessive ROS and release pro-death proteins in response to changes in the intracellular environment, which results in disrupted ATP synthesis and activation of cell death pathways. Mitophagy is mitochondria-specific autophagy that clears damaged mitochondria prior to activation of cell death. Previous studies have shown that AMPK provides a link between energy sensing and mitophagy (18, 19).
In the present study, we investigated the relationship between glucose/energy competition caused by tumors and tumor-bearing liver injury as well as the role of AMPK α2 in this process. We found that liver metastasis was associated with glucose deprivation and necrosis of liver hepatocytes. Knock-out of AMPK α2 significantly aggravated the glucose deprivation and necrosis of hepatocytes via increased ROS production and mitophagy.
MATERIALS AND METHODS
MitoTracker Red CMxRS, chloromethyl-H2DCF-DA, and dihydroethidium (DHE) were purchased from Invitrogen. Antibodies against AMPK α1 and AMPK α2 were from Abcam (Cambridge, MA); antibodies against p-AMPKα (threonine 172), p-mTOR, mTOR, p-4ebp1, 4ebp1, and cytokeratin 18 were from Cell Signaling Biotechnology (Danvers, MA); antibodies against Galectin-3 (Mac-2) and GAPDH were obtained from Santa Cruz Biotechnology Inc. (Santa Cruz, CA). Antibody against LC-3 was from Medical & Biological Laboratories International (Woburn, MA).
Mouse Tumor Models
A liver metastasis model was carried out as described previously (20). SL4, a mouse colon carcinoma cell line with high incidence of experimental hepatic metastasis, was used for this model (20). We transfected the SL4 cell line with a vector expressing luciferase, and G418 was used for screening the stably transfected cell strain. Thus, the intraperitoneal injection of d-luciferin can show the tumor location. Briefly, after the mice were anesthetized by intraperitoneal injection with pentobarbital (100 mg/kg), a longitudinal cut was made below the left flank, and the spleen was exposed; then SL4 cells of the indicated quantity in 100 μl of DMEM/F12 medium were intrasplenically injected into the spleen with a 26-gauge needle. A visible “paling” in the spleen and the lack of bleeding were criteria for successful inoculation. Mice were euthanatized at the indicated day, and the blood, spleen, and liver with tumor were harvested after heart perfusion.
To initiate subcutaneous tumors, the left hind flank region of each mouse was shaved and disinfected. Approximately 107 SL4 cells in 100 μl of DMEM/F12 medium were injected subcutaneously in pentobarbital-anesthetized mice. Mice were euthanatized at the indicated day, the blood and tumor-bearing livers were harvested after heart perfusion.
Glucose Enrichment on Tumor Analysis
Fluorescent probe XenoLight RediJect 2-DeoxyGlucosone 750 (Caliper Life Sciences, Hopkinton, MA) was injected through mouse tail vein. d-Luciferin (15 mg/ml) was intraperitoneal injected at a dose of 10 μl/g of mouse body weight just before analysis. The tumor location and glucose accumulation was analyzed by Xenogen-IVIS® Lumina II device (Caliper Life Sciences) 30 min after injection of 2-DeoxyGlucosone 750.
Liver Glucose Assay
The analysis of liver glucose concentration was performed referred to the report (21) with minor modification. A glucose assay kit was used (Sigma-Aldrich) to measure glucose content in liver remnant. The samples were treated following the kit instructions. Briefly, the remnant liver was weighed and ground with deionized water to aid glucose extraction. Cell lysis fragments were removed by centrifugation at 10,000 × g for 10 min, and the glucose in supernatant was analyzed followed the manual with BioTek SynergyTM 4 multimode microplate reader (BioTek Instruments Inc., Winooski, VT). The data were normalized to the weight of the liver.
Glucose Mass Spectrometry Imaging
Fresh frozen tumor-bearing liver tissue was cryosectioned at 10-μm thickness. The slices of WT and AMPKα2−/− were transferred onto the same conductive side of indium tin oxide (ITO) slides. Desiccate the slide in vacuum for 30–60 min. After drying, matrix was applied by the Bruker ImagePrep device (Bruker Daltonics, Bremen, Germany). MALDI mass spectra were acquired at negative ion mode using a reflectron geometry MALDI-TOF mass spectrometer (Ultraflextreme; Bruker Daltonics) equipped with a neodymium-doped yttrium aluminum garnet (Nd:YAG)/355-nm laser as the excitation source. Imaging data were analyzed using FlexImaging v3.0 and FlexAnalysis v3.4. The intensity of glucose was shown as false color of image from the mass spectrometry peak at m/z 215.03 (glucose + 35Cl)−. The intensity of the peak at m/z 70.75 (H35Cl + 35Cl)− from matrix was as an internal reference.
Cell Culture and Isolation of Primary Mouse Hepatocyte Cells
SL4 cells were maintained in 1:1 mixture of DMEM/F12 containing 10% heat-inactivated FCS in a humidified atmosphere of 95% air and 5% CO2 at 37 °C. Primary hepatocytes were isolated from 8–10-week-old C57BL/6J mice using the collagenase perfusion technique as described previously (22).
Histological Analysis
Livers from control mice and SL4-injected mice were harvested and then paraffin-sectioned and stained with hematoxylin and eosin for morphological observation. For immunohistochemistry, the livers sections were fixed with 4% paraformaldehyde in PBS, incubated with the primary antibodies, and then incubated with the Dako ChemMateTM EnVision System (Dako, Glostrup, Denmark) for 30 min. Staining was visualized with use of diaminobenzidine and counterstaining with hematoxylin.
For immunofluorescence, cells on slides were fixed in 4% paraformaldehyde for 30 min at room temperature; permeabilized; blocked with 0.1% Triton X-100, 0.2% bovine serum albumin, and 5% normal donkey serum in PBS; incubated with the primary antibodies overnight at 4 °C; and then incubated with FITC- or TRITC-conjugated secondary antibody (Jackson ImmunoResearch Laboratories) at room temperature for 1 h in the dark. After being washed with PBS, the slides were mounted with DAPI-containing medium.
Western Blot Analysis
Samples of liver tissues or cells were homogenized in lysis buffer (Thermo Scientific) with the HALT protease and phosphatase inhibitor cocktails (Thermo Scientific), and then the homogenates were centrifuged at 12,000 × g for 30 min at 4 °C. After protein concentration was determined in supernatants by using the Bio-Rad protein assay based on the method of Bradford, protein samples were loaded on 10% SDS-PAGE and then transferred to a nitrocellulose membrane. Nonspecific proteins were blocked by incubating the membrane with 5% nonfat dried milk in TBS-T for 1 h at room temperature with agitation. The nitrocellulose membrane was incubated with the primary antibodies of anti-AMPK α1 (1:1000 dilution), anti-AMPKα2 (1:1000 dilution), anti-p-AMPK (threonine 172) (1:1000 dilution), anti-p-mTOR (1:1000 dilution), anti-mTOR (1:1000 dilution), anti-p-4ebp1 (1:1000 dilution), anti-4ebp1 (1:1000 dilution), and anti-GAPDH (1:2000 dilution) overnight at 4 °C and then fluorescent secondary antibodies (Alexa Fluor 800; Rockland Immunochemicals, Gilbertsville, PA) for 1 h at room temperature. Protein expression was analyzed with the Odyssey infrared imaging system and Odyssey software and normalized to GAPDH expression.
Alanine Aminotransferase (ALT)/Aspartate Aminotransferase (AST) Activity Measurement
Fasting blood was collected from the heart of mice and then centrifuged at the speed of 1000 × g for 5 min at room temperature. The serum were harvested and stored at −80 °C until analyzed. The activities of AST and ALT were measured by Automatic Analyzer 7020 (Hitachi) with the ALT, AST assay kit (BioSino Biotechnology & Science Inc., Beijing, China).
Measurement of Cell Survival Rate
Hepatocyte death in tumor-metastasized liver was analyzed by using the DeadEndTM fluorometric TUNEL system (Promega, Madison, WI) according to the manufacturer's instructions. The determination of primary hepatocyte survival rates was performed according to the methods used in a previous study (23) by flow cytometry with the parameter: plasma membrane integrity. The plasma membrane integrity was tested by the ability of cells to exclude propidium iodide (PI; Sigma) and Hoechst 33342 (Sigma). Briefly, the primary cultured hepatocytes were trypsinized, washed with PBS, stained in PBS containing 5 μg/ml PI and 10 μg/ml Hoechst 33342 for 10 min, and then analyzed by flow cytometry (MoFlo XDP; Beckman Coulter). The dead cells included the PI-positive cells and the double negative cells of PI and Hoechst 33342 (ghost cells). Manually counting was carried out to confirm the results obtained by flow cytometry.
Electron Microscope
Normal liver and tumor-metastasized liver were harvested and fixed in 2% glutaraldehyde solution buffered with 0.1 mol/liter sodium cacodylate (pH 7.4). All the samples were embedded in Durcupan ACM. Ultrathin sections were viewed under the transmission electron microscope (JEOL-1230; JEOL, Tokyo, Japan) to observe the necrosis and mitophagy of hepatocytes in tumor-bearing liver.
ROS Detection
The oxidative fluorescent dye DHE (Invitrogen) was employed to examine in situ production of superoxide from 7-μm frozen tissue sections (24). Briefly, serial sections of control livers and tumor-metastasized livers were first equilibrated with Krebs-HEPES buffer for 30 min at 37 °C. Fresh buffer containing DHE (2 μmol/liter) was added to each tissue section and incubated for 30 min at 37 °C in a light-protected, humidified chamber. Then the sections were stained with Hoechst 33342 to detect the cell nucleus and viewed by fluorescent microscopy.
The 5-(and-6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate, acetyl ester (CM H2DCFDA) (Invitrogen) was used to detect the ROS production in cultured cells (5 μmol/liter). Its intensity was analyzed by fluorescent microscopy or flow cytometry (FACSCalibur; BD Biosciences).
Adenoviral Infection
The constitutively active AMPK mutant (Ad-AMPK-CA) adenoviral vector was kindly offered by Ming-Hui Zou (25). Briefly, a rat cDNA encoding residues 1–312 of AMPK, which contained an aspartic acid residue substituted for threonine 172 (T172D), was subcloned into a shuttle vector (pShuttle cytomegalovirus; Stratagene, La Jolla, CA). An adenoviral vector expressing green fluorescent protein (Ad-GFP) served as a control. The primary hepatocytes were infected with Ad-GFP or Ad-AMPK-CA overnight in medium supplemented with 2% FCS. The cells were then washed and cultured in medium containing 5% FCS with or without glucose for another 36 h.
Statistical Analysis
The data are expressed as means ± S.D. Statistical analyses involved use of GraphPad Prism v5.00 for Windows (GraphPad Software Inc., San Diego, CA). Comparisons among more than two groups were analyzed by one-way analysis of variance followed by Student-Newman-Keuls test. p < 0.05 was considered statistically significant.
RESULTS
Liver Metastasis of Colon Cancer Resulted in Glucose Deprivation in Liver Remnants
Changes in the glucose/energy storage level between the liver and tumor metastasis was examined in a murine model of liver metastasis (26). Luciferase-expressing mouse colon cancer SL4 cells were injected into the spleens of mice, which were then monitored for tumor development and metastasis. Tumors were detected in liver and spleen sites (Fig. 1, A and B), and they developed in the liver as time progressed (Fig. 1A). Luciferin and 2-DeoxyGlucosone 750 were then injected into the same mice to assess glucose uptake by the tumors. Glucose significantly accumulated in the tumor sites of the liver and spleen of injected versus normal mice, rather than in the brain and heart (Fig. 1, B–D), implying that the Warburg effect was present in our liver metastasis model. Glycogen provides the major energy source in the liver before gluconeogenesis is exploited (27). As shown in Fig. 1E, glycogen storage in tumor-bearing livers was decreased on day 10 after SL4 cell injection and was markedly reduced on day 12. The glucose levels in the liver remnants were then analyzed and were found to decrease over time following tumor metastasis (Fig. 1F). Decreased glucose content in tumor-bearing livers was further confirmed by mass spectrometry imaging on days 8 and 12 after SL4 injection (Fig. 1, G and I). Thus, our results suggested that liver metastasis of colon cancer resulted in glucose/energy deprivation in liver remnants.
FIGURE 1.

Liver metastasis of colon cancer resulted in energy deprivation of liver remnant. A, the spleens of C57 BL/6 WT mice (n = 12) were injected with 5 × 105 SL4 tumor cells that stably expressed luciferase. Tumor-bearing livers and spleens were harvested at 10, 12, 14, and 16 days after injection. B, bioluminescence imaging after d-luciferin injection on 12 day after SL4 injection showed luciferase activity from SL4, indicating the tumor location and size. C, the same mice as B (above) were intravenously injected with a 10-nmol 2-DeoxyGlucosone (DG) 750 probe and imaged at 45 min with IVIS spectrum (excitation, 745 nm; emission, 820 nm) to show the extra accumulation of glucose by the tumor. D, the corresponding tumor-bearing liver and spleen of mice in B and C. E, glycogen staining of liver sections, in feeding condition, 10 and 12 days after SL4 injection and of normal (control) liver. Glycogen storage decreased as the tumor developed in the liver. The dashed line indicates the tumor region. Scale bar, 50 μm. F, glucose levels in liver remnants on days 10 and 14 after SL4 injection and in normal (control) livers. The hepatic glucose level decreased as the tumor developed in the liver (n = 6 each group). G, mass spectrometry imaging of glucose in tumor-bearing liver on days 8 and 12 after SL4 injection. Green is the false color from (glucose + 35Cl)−. It also shows that the hepatic glucose level decreased as the tumor developed in the liver. The blue line indicates the scanned area. The arrow indicates the tumor region. Scale bar, 5 mm. H, the corresponding tumor-bearing liver of G was imaged, whereas the fresh frozen slide was cryosectioned. I, the representative mass spectrometry spectrum of G. * indicates the peak of (glucose + 35Cl)−, m/z 215.03, and # indicates the internal reference peak of (H35Cl + 35Cl)−, m/z 70.75. d, day(s).
Glucose Deprivation in Tumor-bearing Livers Was Associated with Liver Injury and Hepatocyte Necrosis
We then investigated the role of glucose deprivation in liver injury after liver metastasis and found that the activities of AST and ALT were slightly elevated during the early stages and markedly increased by 12 days after SL4 injection (Fig. 2A), indicating the presence of liver damage. However, AST and ALT activity declined rapidly at 16 days (Fig. 2A), corresponding with the limited liver remnant (Fig. 1A). In addition, zonal necrotic cell masses with nucleus loss (thus becoming dead “ghost” cells) were observed in the liver remnants after 12 days (Fig. 2B). These cells were cytokeratin 18-positive, suggesting that they were hepatocytes (Fig. 2C). The ghost-like hepatocytes were also observed among cultured primary hepatocytes under glucose deprivation conditions (see Fig. 4, E and F). Some of these necrotic hepatocytes could still be detected by TUNEL staining (Fig. 2D). Hepatocyte necrosis was further confirmed by electron microscopy (Fig. 2E). The necrotic cells were surrounded by Mac-2-positive macrophages (Fig. 2, F and G). Thus, liver metastasis caused both serious liver glucose deprivation and liver injury.
FIGURE 2.
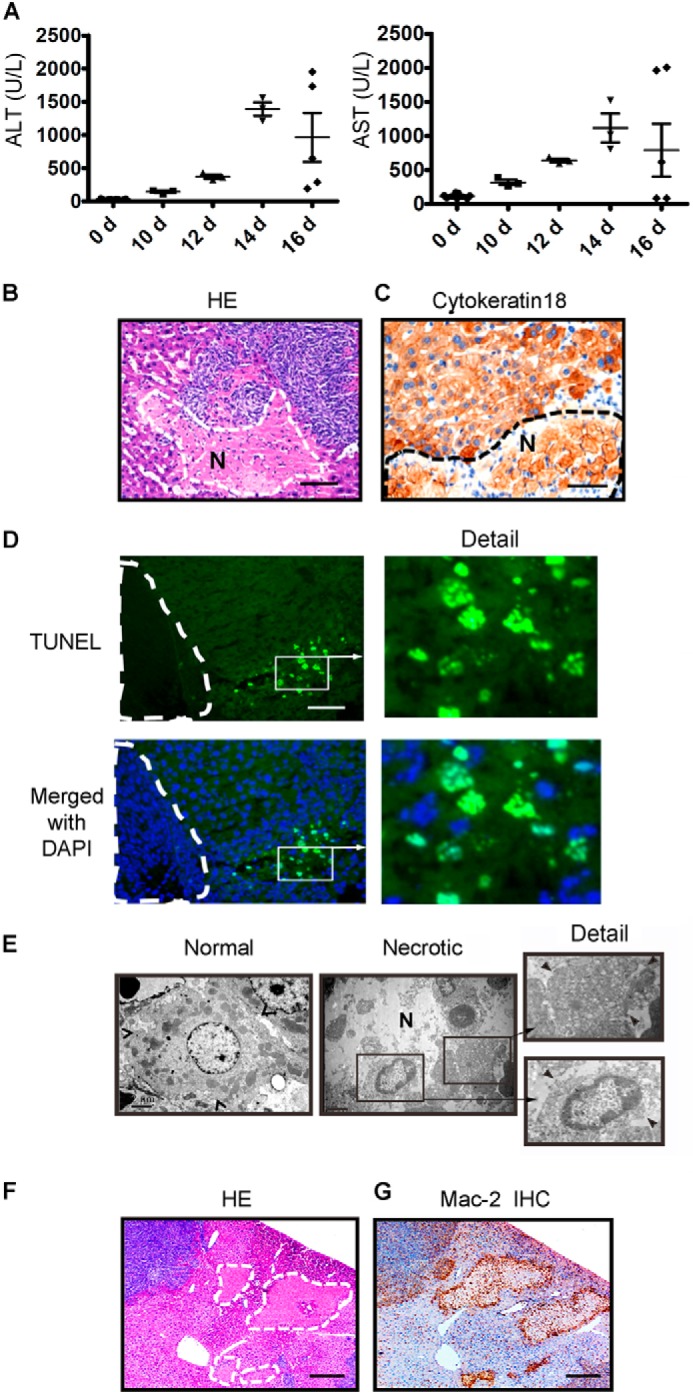
Energy deprivation in the tumor-bearing liver is positively associated with liver injury and hepatocyte necrosis in colon cancer liver metastasis. A, the serum ALT and AST activity mice were determined by immune complex kinase assays at the time indicated in both mice injected with SL4 and mice not injected with SL4. The injury to the liver worsened as the tumor developed in the liver (n = 8). B, the livers were harvested 12 days after SL4 injection, paraffin-sectioned, and subjected to hematoxylin and eosin staining. The dashed line and letter N indicate the necrotic mass with weak staining of cell nuclei. Scale bar, 100 μm. C, the livers were frozen-sectioned and subjected to immunohistochemistry with a specific antibody against cytokeratin 18 that recognized hepatocytes. The dashed line and letter N indicate the necrotic cells of hepatocytes with weak staining of cell nuclei. Scale bar, 100 μm. D, a tumor-bearing liver that was paraffin-sectioned on day 12 was subjected to TUNEL staining to indicate the dead cells. The dashed line indicates the tumor region. Scale bar, 100 μm. E, electron microscopy images show necrotic hepatocytes in the tumor-bearing liver on day 12. The arrowhead indicates the cell border. Scale bar, 100 μm. F and G, hematoxylin and eosin (HE) and IHC staining with Mac-2 antibody show the necrotic masses and the accumulation of macrophages around the masses in the tumor-bearing liver on day 12. The dashed line indicates the necrotic mass. Scale bar, 200 μm. d, day(s).
FIGURE 4.

AMPK α2 deficiency exacerbated hepatocyte death and elevated ROS. A, fresh liver cryosections were prepared on 12 day after SL4 injection and incubated with 2 μmol/liter DHE for 30 min at 37 °C. Positive staining of the oxidized dye was identified by fluorescence microscopy, indicating the level of ROS in the tumor-bearing liver. Dashed lines show the tumor region. Scale bar, 100 μm. B, the primary hepatocytes were cultured in Williams's medium E with 5% FBS for 24 h and then transferred to 5% FBS DMEM medium with or without glucose and incubated for 36 h. Cells were trypsinized and oxidized to the FITC-CM H2DCFDA for 20 min. The oxidized dye was identified by fluorescence microscopy, indicating the ROS level in cultured hepatocytes after glucose starvation. DCF indicates FITC-CM H2DCFDA; DIC, differential interference contrast. Scale bar, 50 μm. C, the primary hepatocytes were treated as B and then analyzed using a FACSCalibur flow cytometer. AMPK α2 deficiency aggravated the production of ROS after glucose starvation. D, the WT or AMPK α2−/− primary hepatocytes were infected with Ad-GFP or Ad-AMPK-CA overnight. The cells were then glucose-starved for an additional 36 h and then analyzed by flow cytometer. E, the primary hepatocytes were double stained by PI and Hoechst 33342 after glucose starvation for 36 h, indicating hepatocyte death after glucose starvation. Dead cells included ghost hepatocytes and PI-positive cells. The blue arrow indicates a viable cell, the yellow arrow indicates a PI-positive dead cell, and a white arrow indicates the ghost cell. Scale bar, 50 μm. F, the primary hepatocytes were harvested for analysis by electron microscopy after glucose starvation for 36 h with or without the apoptosis inhibitor, z-VAD. Necrotic death of the primary hepatocytes can be observed. G, the primary hepatocytes were trypsinized, washed with PBS, incubated with PI and Hoechst 33342 for 20 min, and analyzed on MoFlo XDP flow cytometer. AMPK α2 deficiency aggravated hepatocyte death after glucose starvation and N-acetyl-l-cysteine (NAC) partially rescued the cells.
AMPKα2 Deficiency Aggravated Both Liver Glucose Deprivation and Liver Injury under Tumor Stress Conditions
It is well known that AMPK can decrease energy consumption and promote energy production to maintain energetic balance under stress conditions (28). AMPK also plays a positive and important role in promoting cell survival under numerous types of cell stress, including glucose deprivation (29). Thus, we examined whether AMPK regulates glucose/energy imbalance and protects against liver injury in response to tumor metastasis. First, it was confirmed that the liver and hepatocytes enrich AMPK α2, but not AMPK α1 (Fig. 3A). Liver AMPK α2 is primarily expressed in hepatocytes, and the level of AMPK α1 was just slightly increased in the liver and primary cultured hepatocytes after AMPK α2 knock-out (Fig. 3A). AMPK phosphorylation in the liver remnant increased as the tumor developed (Fig. 3C). Obvious zonal phosphorylation of AMPK and a necrotic mass were also detected in the paracarcinomatous liver of primary hepatocellular carcinoma (Fig. 3D). The AMPK α2−/− mouse model was then used to investigate the role of host AMPK α2 in liver metastasis. SL4 cells were injected into WT and AMPK α2−/− mice, and then blood and tissues were harvested. To avoid the effect of tumor size on liver injury, we analyzed tumor-bearing livers with similar gross weights (Fig. 3E). The hepatic glucose level was lower in AMPK α2−/− tumor-bearing mice (Fig. 3F), but the serum ALT and AST activities were significantly higher relative to those of WT tumor-bearing mice (Fig. 3G). It has been reported that severe liver damage is associated with more compensatory cell proliferation (24). Our results showed that compensatory cell proliferation was present in the tumor-bearing liver and was enhanced by AMPK α2 deficiency (Fig. 3, H and I). AMPK can exploit numerous pathways to decrease energy consumption, including inhibition of gluconeogenesis and protein synthesis (30). Increased activation of mTOR signaling, which enhanced protein synthesis in the tumor-bearing livers of AMPK α2−/− mice, was also confirmed (see Fig. 5D). Furthermore, the mRNA level of G6Pase, an important gluconeogenic enzyme, was higher in the tumor-bearing livers of AMPK α2−/− mice (Fig. 3J), corresponding to the higher blood glucose levels found in these mice prior to the late stage (Fig. 3K). These results imply that AMPK α2 contributes to glucose regulation during liver tumor metastasis in remnant liver tissue.
FIGURE 3.

AMPK α2 deficiency aggravated the glucose deprivation in livers under tumor stress and was associated with more serious liver injury. A, lysates of mouse primary hepatocytes and mouse bone marrow-derived macrophages were analyzed by Western blot using antibodies against the AMPK α1 and AMPK α2 proteins, and AMPK α2 was found to be enriched in the liver, especially in hepatocytes. The spleens and mouse bone marrow-derived macrophage lysates of WT mice were used as controls for AMPK α1 and AMPK α2 expression. B, the tumor-bearing livers of WT and AMPK α2−/− mice were harvested 12 days after SL4 injection and subjected to IHC staining with AMPK α2 antibodies. The results show that mouse AMPK α2 isoform was express predominantly in hepatocytes but not in SL4 or other liver cells. The dashed line shows the tumor region. Scale bar, 50 μm. C, lysates of tumor-bearing liver were analyzed 10 and 12 days after SL4 injection by Western blot using antibodies against the p-AMPKα threonine 172 and GAPDH. AMPK phosphorylation in the liver remnant was elevated as the tumor developed. D, analysis of a human paraffin-sectioned paracarcinomatous liver with HCC. Panel D1, IHC with AMPK α2 antibody shows the expression of the AMPK α2 isoform in human hepatocytes. Panel D2, IHC with p-AMPKα threonine 172 antibody shows the zonal activation of AMPK in the paracarcinomatous liver with HCC. Panel D3, TUNEL-stained paraffin-sectioned paracarcinomatous liver with HCC. Panel D4, hematoxylin and eosin (HE) staining shows the necrotic mass in the paracarcinomatous liver with HCC. E, a different dose of SL4 was injected into WT (4 × 106 cells/mouse) and AMPK α2−/− (5 × 106 cells/mouse) mice, and the tumor-bearing livers, with similar gross weights, were harvested for subsequent glucose analysis (n = 6). F, the liver glucose assay of WT and AMPK α2−/− mice, with similar gross tumor liver weights on 12 day after SL4 injection, shows that AMPK α2 deficiency aggravated the glucose level in tumor-bearing livers (n = 6). G, analysis of serum ALT and AST in WT and AMPK α2−/− mice, with the similar gross tumor liver weights 12 days after SL4 injection, indicates that the AMPK α2 deficiency aggravated the injury in tumor-bearing livers (n = 6). H and I, proliferating cell nuclear antigen (PCNA) IHC and Western blot staining were used to determine compensatory hepatocyte proliferation in tumor-bearing livers. J, the mRNA of liver remnants were prepared 14 days after injection of SL4 and sequenced by BGI-shenzhen. The mean number of reads/kb/million reads of WT was 1-fold (n = 2). K, mice were fasted for 6 h, and the blood glucose was measured using a blood glucose monitor on the days indicated after SL4 spleen injection (n = 8). The plasma glucose level and the G6Pase mRNA level in the tumor-bearing liver after AMPK α2 deficiency were both higher than in WT mice. d, day(s).
FIGURE 5.

AMPK α2 deficiency enhanced ROS accumulation by decreasing mitophagy, which was related with mTOR signaling pathway. A, liver tissue was harvested 12 days after SL4 injection. Electron microscopy shows the mitophagy in hepatocytes from the tumor-bearing liver. B, the livers were harvested 12 days after SL4 injection, lysed, and subjected to Western blot with antibodies against LC3 and GAPDH. The LC3 II levels decreased in the tumor-bearing livers of AMPK α2−/− mice. C and D, the livers were harvested 10 and 13 days after SL4 injection, lysed, and subjected to Western blot with antibodies against AMPK α2, p-mTOR, mTOR, p-4ebp1, 4ebp1, and GAPDH. The phosphorylation level of mTOR and its substrate, 4ebp1, both increased in AMPK α2-deficient tumor-bearing livers. E, primary hepatocytes underwent 36 h of glucose starvation, with or without autophagy inhibitor (3-MA) treatment, and were harvested for electron microscopic analysis. The mitophagy was reduced by the 3-MA. F, the lysate from primary hepatocytes was analyzed with LC3 antibody by Western blot, after 3 and 8 h of glucose starvation. AMPK α2 deficiency decreased the level of LC3 II. G, primary hepatocytes were cultured with or without glucose for 36 h. Cells were incubated with Mito Tracker for 20 min, and the autophagosome level was then detected by immunofluorescence of the LC3 antibody. Many autophagosomes colocalized with mitochondria, suggesting mitophagy. AMPK α2 deficiency decreased the colocalization. Scale bar, 10 μm. H, localization of ROS and mitochondria were detected by Mito Tracker Red and FITC-CM H2DCFDA. Cells were incubated with Mito Tracker for 20 min, and the fluorescence was recorded 20 min after FITC-CM H2DCFDA was added. A large amount of ROS produced after glucose starvation colocalized with mitochondria. DCF indicates FITC-CM H2DCFDA. Scale bar, 10 μm. I and J, primary hepatocytes were cultured with or without glucose or 3-MA for 36 h. Hepatocytes were then trypsinized and oxidized to the FITC-CM H2DCFDA for 20 min and analyzed using a FACSCalibur flow cytometer. The results indicated that 3-MA enhanced ROS production after glucose starvation. d, day(s).
AMPK α2 Deficiency Exacerbated Hepatocyte Death via ROS Elevation
Increased ROS production is a major contributor to liver damage and/or failure (31). Therefore, the association between the contribution of AMPK α2 to hepatocyte survival and ROS production was examined. An increase in ROS production in tumor-bearing livers was first detected by hydroethidine oxidation (DHE), and cells with a DHE-positive signal showed zonal scattering (Fig. 4A). It was then confirmed that glucose deprivation resulted in ROS accumulation (Fig. 4B) and cell death in cultured primary hepatocytes (Fig. 4E), indicating that ROS is involved in glucose deprivation-induced hepatocyte death. Electron microscopy showed that most of these cells died via necrosis, and treatment with Z-VAD, an apoptosis inhibitor, did not inhibit hepatocyte necrosis (Fig. 4F). Furthermore, AMPK α2 deficiency resulted in increased ROS levels (Fig. 4C) and hepatocyte death (Fig. 4G) under conditions of glucose deprivation. WT or AMPK α2−/− primary hepatocytes were infected with Ad-GFP or Ad-AMPK-CA; then the cells were glucose-starved. Overexpression of Ad-AMPK-CA, but not Ad-GFP, markedly reduced the ROS in primary hepatocytes from AMPK α2−/− mice (Fig. 4D), suggesting that activation of AMPK can effectively suppress ROS production. The ROS inhibitor N-acetyl-l-cysteine partially rescued both WT and AMPK α2-deficient hepatocytes (Fig. 4G). These results suggest that AMPK α2 deficiency enhances glucose deprivation-induced ROS accumulation in hepatocytes, leading to increased cell death.
AMPK α2 Deficiency Enhanced ROS Accumulation via Decreased Mitophagy
Mitochondria are major ROS producers when cells are under energetic stress (32). Mitophagy is initiated as a survival mechanism under starvation conditions, thus eliminating damaged organelles such as mitochondria and reducing ROS production (19, 33). We then examined whether mitophagy is involved in the mechanism of AMPK α2-induced ROS clearance in hepatocytes under tumor stress. Mitophagy was initially detected in tumor-bearing hepatocytes using electron microscopy (Fig. 5A). LC3-II associates with both the outer and inner membranes of the autophagosome (34). As expected, the LC3-II level was decreased in the AMPK α2−/− tumor-bearing liver (Fig. 5B). AMPK activation inhibits mTOR signaling, which inhibits autophagy (35). Increased phosphorylation of mTOR and 4ebp1 were also detected in the AMPK α2−/− tumor-bearing liver (Fig. 5, C and D). Obvious mitophagy was also observed on electron microscopy in glucose-starved primary hepatocytes (Fig. 5E). The autophagy inhibitor 3-methylladenine (3-MA) reduced such mitophagy (Fig. 5E). AMPK α2 deficiency also decreased the LC3-II level in primary cultured hepatocytes under glucose deprivation (Fig. 5F). Furthermore, there was obvious colocalization of LC3-labeled autophagosomes and mitochondria, implying the existence of mitophagy (Fig. 5G). It was also found that the ROS consistently colocalized with MitoTracker-labeled, hepatocytic mitochondria after glucose deprivation (Fig. 5H), and 3-MA treatment boosted the ROS level in response to glucose starvation (Fig. 5, I and J), indicating that mitophagy is involved in ROS depletion in hepatocytes. These results suggest that AMPK α2 deficiency increases ROS production in hepatocytes through inhibition of mitophagy under tumor metastatic stress and glucose deprivation both in vivo and in vitro; this is positively related to activation of the mTOR signaling pathway.
DISCUSSION
The Warburg effect results in extra energy consumption by tumor cells, which confers a significant growth advantage. However, normal cells also exploit a variety of mechanisms to keep the rates of catabolism and anabolism in balance with energy metabolism. Abnormal glucose/energy metabolism always results in cell death, including apoptosis and necrosis (23, 36). Numerous studies have focused on how tumor cells exploit the Warburg effect to deal with energetic stress, but only a few have analyzed energetic stress and its consequences in tumor-bearing tissues. In the present study, the signaling mechanism of liver injury in response to tumor metastasis was investigated.
First, it was demonstrated that glucose levels are significantly enriched in tumors, including those in the liver, whereas the levels of glycogen and glucose are significantly reduced in remnant liver (Fig. 1). Several reports have shown that glucose deprivation activates a metabolic signaling amplification loop, leading to cell death both in vivo and in vitro (37–39). Our study showed that colon cancer liver metastasis also resulted in glucose deprivation and liver injury during tumor development (Figs. 1F and 2A).
Second, the signaling mechanism that mediates energetic stress and liver cell death was explored by studying the “energy sensor” AMPK, because this protein shuts down energy-consuming processes and facilitates energy-producing processes during various metabolic stresses (28, 29, 40). The AMPK α2 isoform was enriched in hepatocytes (Fig. 3, A and B). We found that AMPK α2 seemed to protect against glucose loss in the tumor-bearing remnant liver, which was associated with reduced liver injury (Fig. 3, F and G). AMPK exploits multiple mechanisms to preserve the glucose balance under energetic stress (6). AMPK inhibits protein synthesis and cell proliferation to decrease mTOR-related energy consumption (5). Results of the current study suggested the presence of increased compensatory cell proliferation (Fig. 3, H and I) and enhanced activation of mTOR signaling in the tumor-bearing liver (Fig. 5C). It has been reported that gluconeogenesis consumes more energy to supply glucose after glycogen is consumed, and hepatic AMPK may reduce this process (27, 41). G6Pase mRNA expression was ∼2.3-fold higher in the tumor-bearing livers of AMPK α2−/− mice than in WT mice (Fig. 3J). AMPK α2 deficiency also resulted in higher blood glucose levels under fasting conditions, which lasted until the late stage of liver metastasis (Fig. 3K), providing further evidence of enhanced gluconeogenesis in the tumor-bearing livers of AMPK α2−/− mice. Thus, the mechanisms exploited by AMPK α2 for reduce glucose deprivation in the tumor-bearing liver have been elucidated in the current study. Indeed, glucose deprivation also caused hepatocyte death, and AMPK α2 promoted hepatocyte survival under glucose stress in vitro (Fig. 4, E–G), consistent with previous studies (29, 42–45). Furthermore, the caspase inhibitor z-VAD did not block cell death, indicating that the hepatocytes underwent necrotic death (Fig. 4E).
Inflammation is not only an important causal factor for both acute and chronic liver injury (46) but also is the result of liver injury (47). It is known that necrotic hepatocytes activate Kupffer cells and trigger inflammation, and activated phagocytes can damage hepatocytes and other liver cells by releasing ROS and cytokines (48). Our results showed that colon cancer liver metastasis resulted in both liver glucose deprivation and liver injury (Figs. 1, F and G, and 2A), whereas AMPK α2 deficiency aggravated these responses (Fig. 3, F and G). We also demonstrated that glucose deprivation triggered hepatocyte death, which was enhanced by AMPK α2 deficiency (Fig. 4, E–G). Thus, liver injury may trigger inflammation, whereas inflammation further promotes liver injury (47).
It has been reported that energetic imbalances cause cell damage because of elevated ROS, and elevated ROS is associated with cell death (23, 24, 49). Increasing evidence suggests that mitochondria are the main target of stress that leads to cell death, and 90% of ROS are generated from mitochondria (17, 19). ROS-induced cell death during inflammation involves the promotion of mitochondrial dysfunction through intracellular oxidative stress in hepatocytes, which results primarily in oncotic necrosis and secondarily in apoptosis (48). In the current study, hepatocyte death occurred via necrosis in the tumor-bearing liver and under glucose deprivation conditions in vitro. Mitochondrial autophagy, or mitophagy, reduces ROS through elimination of injured mitochondria under stress conditions (50, 51). AMPK can promote autophagy/mitophagy by inhibiting mTOR signaling and phosphorylating the core conserved autophagy protein, ULK1, in response to nutrient deprivation in mammals (19). In the present study, we observed that ROS were elevated both in the tumor-bearing liver and in glucose-starved hepatocytes in vitro (Fig. 4), and AMPK α2 deficiency increased the ROS level (Fig. 4). Moreover, a relationship exists between ROS, mitophagy, and AMPK α2 in the tumor-bearing liver and glucose-starved hepatocytes (Fig. 5). Mitophagy was confirmed using electron microscopy in the tumor-bearing liver and glucose-starved hepatocytes. Hepatocytic ROS, under glucose stress, were produced primarily by mitochondria. AMPK α2 deficiency impaired the level of mitophagy in tumor-bearing livers and cultured hepatocytes under glucose deprivation, which elevated ROS levels (Fig. 5, C, F, and G). Recently, it was reported that AMPK regulates NADPH homeostasis to promote cell survival during oxidative stress caused by glucose deprivation (52). Therefore, it is possible that NADPH homeostasis could also be involved in a mechanism in which AMPK protects against liver injury under liver metastatic stress.
In summary, colon cancer liver metastasis resulted in decreased glycogen and glucose storage in the liver remnant, along with liver damage and hepatocyte death. AMPK α2 attenuated glucose deprivation in tumor-bearing livers and antagonized hepatocyte death via promotion of mitophagy and reduction of ROS.
Acknowledgments
We thank Dr. Ming-Hui Zou from University of Oklahoma Health Sciences Center for kindly offering us constitutively active AMPK mutant (Ad-AMPK-CA) adenoviral vector. We thank Dr. Xiaolin Zhang and Dr. Qunsheng Ji from Innovation Centre China, AstraZeneca Innovative Medicine, and Early Development for lots of suggestions and extensive discussion during the progress of this project. We thank Dr. Lun Cai and Dr. Jing Zhang for flow cytometry analysis.
This work was supported by Chinese Ministry of Science and Technology Grants 2009CB522205 and 2012AA02A201 and National Natural Science Foundation of China Grants 81230006, 31090363, and 81000871.
- AMPK
- AMP-activated protein kinase
- ROS
- reactive oxygen species
- DHE
- dihydroethidium
- TRITC
- tetramethylrhodamine isothiocyanate
- ALT
- alanine aminotransferase
- AST
- aspartate aminotransferase
- TUNEL
- terminal deoxynucleotidyltransferase-mediated dUTP nick end labeling
- PI
- propidium iodide
- 3-MA
- 3-methylladenine
- z
- benzyloxycarbonyl
- IHC
- immunohistochemistry
- CM H2DCFDA
- 5-(and-6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate, acetyl ester.
REFERENCES
- 1. Hsu P. P., Sabatini D. M. (2008) Cancer cell metabolism: Warburg and beyond. Cell 134, 703–707 [DOI] [PubMed] [Google Scholar]
- 2. Koppenol W. H., Bounds P. L., Dang C. V. (2011) Otto Warburg's contributions to current concepts of cancer metabolism. Nat. Rev. Cancer 11, 325–337 [DOI] [PubMed] [Google Scholar]
- 3. Schneeberger A. L., Thompson R. T., Driedger A. A., Finley R. J., Inculet R. I. (1989) Effect of cancer on the in vivo energy state of rat liver and skeletal muscle. Cancer Res. 49, 1160–1164 [PubMed] [Google Scholar]
- 4. Leij-Halfwerk S., Dagneli P. C., Kappert P., Oudkerk M., Sijens P. E. (2000) Decreased energy and phosphorylation status in the liver of lung cancer patients with weight loss. J. Hepatol. 32, 887–892 [DOI] [PubMed] [Google Scholar]
- 5. Tsuburaya A., Blumberg D., Burt M., Brennan M. F. (1995) Energy depletion in the liver and in isolated hepatocytes of tumor-bearing animals. J. Surg. Res. 59, 421–427 [DOI] [PubMed] [Google Scholar]
- 6. Hardie D. G. (2007) AMP-activated/SNF1 protein kinases: conserved guardians of cellular energy. Nat. Rev. Mol. Cell Biol. 8, 774–785 [DOI] [PubMed] [Google Scholar]
- 7. Oakhill J. S., Steel R., Chen Z. P., Scott J. W., Ling N., Tam S., Kemp B. E. (2011) AMPK is a direct adenylate charge-regulated protein kinase. Science 332, 1433–1435 [DOI] [PubMed] [Google Scholar]
- 8. Xiao B., Sanders M. J., Underwood E., Heath R., Mayer F. V., Carmena D., Jing C., Walker P. A., Eccleston J. F., Haire L. F., Saiu P., Howell S. A., Aasland R., Martin S. R., Carling D., Gamblin S. J. (2011) Structure of mammalian AMPK and its regulation by ADP. Nature 472, 230–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jensen T. E., Rose A. J., JØrgensen S. B., Brandt N., Schjerling P., Wojtaszewski J. F., Richter E. A. (2007) Possible CaMKK-dependent regulation of AMPK phosphorylation and glucose uptake at the onset of mild tetanic skeletal muscle contraction. Am. J. Physiol. Endocrinol. Metab. 292, E1308–E1317 [DOI] [PubMed] [Google Scholar]
- 10. Lizcano J. M., Göransson O., Toth R., Deak M., Morrice N. A., Boudeau J., Hawley S. A., Udd L., Mäkelä T. P., Hardie D. G., Alessi D. R. (2004) LKB1 is a master kinase that activates 13 kinases of the AMPK subfamily, including MARK/PAR-1. EMBO J. 23, 833–843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Siwiak M., Edelman A., Zielenkiewicz P. (2012) Structural models of CFTR-AMPK and CFTR-PKA interactions: R-domain flexibility is a key factor in CFTR regulation. J. Mol. Model. 18, 83–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Raney M. A., Turcotte L. P. (2008) Evidence for the involvement of CaMKII and AMPK in Ca2+-dependent signaling pathways regulating FA uptake and oxidation in contracting rodent muscle. J. Appl. Physiol. 104, 1366–1373 [DOI] [PubMed] [Google Scholar]
- 13. Yang Z., Kahn B. B., Shi H., Xue B. Z. (2010) Macrophage α1 AMP-activated protein kinase (α1AMPK) antagonizes fatty acid-induced inflammation through SIRT1. J. Biol. Chem. 285, 19051–19059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mihaylova M. M., Shaw R. J. (2011) The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat. Cell Biol. 13, 1016–1023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Levine B., Mizushima N., Virgin H. W. (2011) Autophagy in immunity and inflammation. Nature 469, 323–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. White E., Karp C., Strohecker A. M., Guo Y., Mathew R. (2010) Role of autophagy in suppression of inflammation and cancer. Curr. Opin. Cell Biol. 22, 212–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Suzuki S. W., Onodera J., Ohsumi Y. (2011) Starvation induced cell death in autophagy-defective yeast mutants is caused by mitochondria dysfunction. PLoS One 6, e17412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Neufeld T. P. (2010) TOR-dependent control of autophagy: biting the hand that feeds. Curr. Opin. Cell Biol. 22, 157–168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Egan D. F., Shackelford D. B., Mihaylova M. M., Gelino S., Kohnz R. A., Mair W., Vasquez D. S., Joshi A., Gwinn D. M., Taylor R., Asara J. M., Fitzpatrick J., Dillin A., Viollet B., Kundu M., Hansen M., Shaw R. J. (2011) Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 331, 456–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zheng J., Yang M., Shao J., Miao Y., Han J., Du J. (2013) Chemokine receptor CX3CR1 contributes to macrophage survival in tumor metastasis. Mol. Cancer 12, 141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Niewoehner C. B., Nuttall F. Q. (1988) Relationship of hepatic glucose uptake to intrahepatic glucose concentration in fasted rats after glucose load. Diabetes 37, 1559–1566 [DOI] [PubMed] [Google Scholar]
- 22. Matsutani T., Kang S. C., Miyashita M., Sasajima K., Choudhry M. A., Bland K. I., Chaudry I. H. (2007) Liver cytokine production and ICAM-1 expression following bone fracture, tissue trauma, and hemorrhage in middle-aged mice. Am. J. Physiol. Gastrointest. Liver Physiol. 292, G268–G274 [DOI] [PubMed] [Google Scholar]
- 23. Zhang D. W., Shao J., Lin J., Zhang N., Lu B. J., Lin S. C., Dong M. Q., Han J. (2009) RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science 325, 332–336 [DOI] [PubMed] [Google Scholar]
- 24. Sakurai T., He G., Matsuzawa A., Yu G. Y., Maeda S., Hardiman G., Karin M. (2008) Hepatocyte necrosis induced by oxidative stress and IL-1α release mediate carcinogen-induced compensatory proliferation and liver tumorigenesis. Cancer Cell 14, 156–165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Dong Y., Zhang M., Liang B., Xie Z., Zhao Z., Asfa S., Choi H. C., Zou M.-H. (2010) Reduction of AMP-activated protein kinase α2 increases endoplasmic reticulum stress and atherosclerosis in vivo. Circulation 121, 792–803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Morimoto-Tomita M., Ohashi Y., Matsubara A., Tsuiji M., Irimura T. (2005) Mouse colon carcinoma cells established for high incidence of experimental hepatic metastasis exhibit accelerated and anchorage-independent growth. Clin. Exp. Metastasis 22, 513–521 [DOI] [PubMed] [Google Scholar]
- 27. Hundal R. S., Krssak M., Dufour S., Laurent D., Lebon V., Chandramouli V., Inzucchi S. E., Schumann W. C., Petersen K. F., Landau B. R., Shulman G. I. (2000) Mechanism by which metformin reduces glucose production in type 2 diabetes. Diabetes 49, 2063–2069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Steinberg G. R., Kemp B. E. (2009) AMPK in health and disease. Physiol. Rev. 89, 1025–1078 [DOI] [PubMed] [Google Scholar]
- 29. Bungard D., Fuerth B. J., Zeng P. Y., Faubert B., Maas N. L., Viollet B., Carling D., Thompson C. B., Jones R. G., Berger S. L. (2010) Signaling kinase AMPK activates stress-promoted transcription via histone H2B phosphorylation. Science 329, 1201–1205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hardie D. G., Ross F. A., Hawley S. A. (2012) AMP-activated protein kinase: a target for drugs both ancient and modern. Chem. Biol. 19, 1222–1236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kamata H., Honda S., Maeda S., Chang L., Hirata H., Karin M. (2005) Reactive oxygen species promote TNFα-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell 120, 649–661 [DOI] [PubMed] [Google Scholar]
- 32. Sena L. A., Chandel N. S. (2012) Physiological roles of mitochondrial reactive oxygen species. Mol. cell 48, 158–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kubli D. A., Gustafsson Å. B. (2012) Mitochondria and mitophagy: the yin and yang of cell death control. Circ. Res. 111, 1208–1221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mizushima N., Yoshimori T., Levine B. (2010) Methods in mammalian autophagy research. Cell 140, 313–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ravikumar B., Vacher C., Berger Z., Davies J. E., Luo S., Oroz L. G., Scaravilli F., Easton D. F., Duden R., O'Kane C. J., Rubinsztein D. C. (2004) Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat. Genet. 36, 585–595 [DOI] [PubMed] [Google Scholar]
- 36. Yaglom J. A., Ekhterae D., Gabai V. L., Sherman M. Y. (2003) Regulation of necrosis of H9c2 myogenic cells upon transient energy deprivation. Rapid deenergization of mitochondria precedes necrosis and is controlled by reactive oxygen species, stress kinase JNK, HSP72 and ARC. J. Biol. Chem. 278, 50483–50496 [DOI] [PubMed] [Google Scholar]
- 37. Palorini R., Cammarata F. P., Balestrieri C., Monestiroli A., Vasso M., Gelfi C., Alberghina L., Chiaradonna F. (2013) Glucose starvation induces cell death in K-ras-transformed cells by interfering with the hexosamine biosynthesis pathway and activating the unfolded protein response. Cell Death Dis. 4, e732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Graham N. A., Tahmasian M., Kohli B., Komisopoulou E., Zhu M., Vivanco I., Teitell M. A., Wu H., Ribas A., Lo R. S., Mellinghoff I. K., Mischel P. S., Graeber T. G. (2012) Glucose deprivation activates a metabolic and signaling amplification loop leading to cell death. Mol. Syst. Biol. 8, 589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Caro-Maldonado A., Tait S. W., Ramírez-Peinado S., Ricci J. E., Fabregat I., Green D. R., Muñoz-Pinedo C. (2010) Glucose deprivation induces an atypical form of apoptosis mediated by caspase-8 in Bax-, Bak-deficient cells. Cell Death Differ. 17, 1335–1344 [DOI] [PubMed] [Google Scholar]
- 40. Shang L., Chen S., Du F., Li S., Zhao L., Wang X. (2011) Nutrient starvation elicits an acute autophagic response mediated by Ulk1 dephosphorylation and its subsequent dissociation from AMPK. Proc. Natl. Acad. Sci. U.S.A. 108, 4788–4793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Andreelli F., Foretz M., Knauf C., Cani P. D., Perrin C., Iglesias M. A., Pillot B., Bado A., Tronche F., Mithieux G., Vaulont S., Burcelin R., Viollet B. (2006) Liver adenosine monophosphate-activated kinase-α2 catalytic subunit is a key target for the control of hepatic glucose production by adiponectin and leptin but not insulin. Endocrinology 147, 2432–2441 [DOI] [PubMed] [Google Scholar]
- 42. Varela-Rey M., Beraza N., Lu S. C., Mato J. M., Martinez-Chantar M. L. (2011) Role of AMP-activated protein kinase in the control of hepatocyte priming and proliferation during liver regeneration. Exp. Biol. Med. (Maywood) 236, 402–408 [DOI] [PubMed] [Google Scholar]
- 43. Peng W., Zhang Y., Zhu W., Cao C. M., Xiao R. P. (2009) AMPK and TNF-α at the crossroad of cell survival and death in ischaemic heart. Cardiovasc. Res. 84, 1–3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kewalramani G., Puthanveetil P., Wang F., Kim M. S., Deppe S., Abrahani A., Luciani D. S., Johnson J. D., Rodrigues B. (2009) AMP-activated protein kinase confers protection against TNF-α-induced cardiac cell death. Cardiovasc. Res. 84, 42–53 [DOI] [PubMed] [Google Scholar]
- 45. Hashimoto K., Kato K., Imamura K., Kishimoto A., Yoshikawa H., Taketani Y., Esumi H. (2002) 5-Amino-4-imidazolecarboxamide riboside confers strong tolerance to glucose starvation in a 5′-AMP-activated protein kinase-dependent fashion. Biochem. Biophys. Res. Commun. 290, 263–267 [DOI] [PubMed] [Google Scholar]
- 46. Malhi H., Gores G. J. (2008) Cellular and molecular mechanisms of liver injury. Gastroenterology 134, 1641–1654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lohse A. W., Weiler-Normann C., Tiegs G. (2010) Immune-mediated liver injury. J. Hepatol. 52, 136–144 [DOI] [PubMed] [Google Scholar]
- 48. Jaeschke H. (2011) Reactive oxygen and mechanisms of inflammatory liver injury: present concepts. J. Gastroenterol. Hepatol. 26, 173–179 [DOI] [PubMed] [Google Scholar]
- 49. Monticone M., Bisio A., Daga A., Giannoni P., Giaretti W., Maffei M., Pfeffer U., Romeo F., Quarto R., Romussi G., Corte G., Castagnola P. (2010) Demethyl fruticulin A (SCO-1) causes apoptosis by inducing reactive oxygen species in mitochondria. J. Cell. Biochem. 111, 1149–1159 [DOI] [PubMed] [Google Scholar]
- 50. Roy A., Ganguly A., BoseDasgupta S., Das B. B., Pal C., Jaisankar P., Majumder H. K. (2008) Mitochondria-dependent reactive oxygen species-mediated programmed cell death induced by 3,3′-diindolylmethane through inhibition of F0F1-ATP synthase in unicellular protozoan parasite Leishmania donovani. Mol. Pharmacol. 74, 1292–1307 [DOI] [PubMed] [Google Scholar]
- 51. Ali S. S., Marcondes M. C., Bajova H., Dugan L. L., Conti B. (2010) Metabolic depression and increased reactive oxygen species production by isolated mitochondria at moderately lower temperatures. J. Biol. Chem. 285, 32522–32528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Jeon S. M., Chandel N. S., Hay N. (2012) AMPK regulates NADPH homeostasis to promote tumour cell survival during energy stress. Nature 485, 661–665 [DOI] [PMC free article] [PubMed] [Google Scholar]