

Abstract
Human recombinant activated factor-VII (rFVIIa) has been used successfully in the treatment of spontaneous intracerebral hemorrhage. In addition, there is increasing interest in its use to treat uncontrolled bleeding of other origins, including trauma. The aim of this study was to evaluate the safety and potential effectiveness of rFVIIa to mitigate bleeding using a clinically relevant model of traumatic brain injury (TBI) in the pig. A double injury model was chosen consisting of (1) an expanding cerebral contusion induced by the application of negative pressure to the exposed cortical surface and (2) a rapid rotational acceleration of the head to induce diffuse axonal injury (DAI). Injuries were performed on 10 anesthetized pigs. Five minutes after injury, 720 μg/kg rFVIIa (n = 5) or vehicle control (n = 5) was administered intravenously. Magnetic resonance imaging (MRI) studies were performed within 30 min and at 3 days post-TBI to determine the temporal expansion of the cerebral contusion. Euthanasia and histopathologic analysis were performed at day 3. This included observations for hippocampal neuronal degeneration, axonal pathology and microclot formation. The expansion of contusion volume over the 3 days post-injury period was reduced significantly in animals treated with rFVIIa compared to vehicle controls. Surprisingly, immunohistochemical analysis demonstrated that the number of dead/dying hippocampal neurons and axonal pathology was reduced substantially by rFVIIa treatment compared to vehicle. In addition, there was no difference in the extent of microthrombi between groups. rFVIIa treatment after TBI in the pig reduced expansion of hemorrhagic cerebral contusion volume without exacerbating the severity of microclot formation. Finally, rFVIIa treatment provided a surprising neuroprotective effect by reducing hippocampal neuron degeneration as well as the extent of DAI.
Keywords: Traumatic brain injury, TBI, rFVIIa, Cerebral contusion, Recombinant Activated Factor VII, Hemostasis, Diffuse axonal injury, Neuroprotection
Introduction
Traumatic brain injury (TBI) is a frequent and potentially disabling event that affects over 1.5 million people in the USA annually (McArthur et al., 2004). It is the leading cause of death in young people and around 2% of the US population live with disabilities as a result of TBI (Ghajar, 2000; Thurman et al., 1999). Traumatic intracranial hemorrhage (tICH), including cerebral contusions, represents a common focal pathology following TBI. Such injuries are characterized by mechanical damage to the parenchyma and its vasculature, resulting in sub-sequent hemorrhage and edema formation. Although clotting may constrain hemorrhage soon after injury, there is considerable evidence to suggest that persistent bleeding can occur in the early hours post-trauma (Servadei et al., 1995; Oertel et al., 2002; Chang et al., 2006; Chieregato et al., 2005; Lobato et al., 1991). Current management of tICH focuses on the identification and surgical evacuation of lesions large enough to generate mass effect and likely result in further neurological deterioration (Bullock et al., 1996). In the case of smaller lesions, the risk versus benefit ratio is such that evacuation may not always be favorable. There are currently no non-invasive therapies licensed for the control of traumatic intraparenchymal hemorrhage.
Recombinant activated factor VII (rFVIIa) is a hemostatic agent currently licensed for the treatment of bleeding in patients with hemophilia with inhibitors to factors VIII or IX. Pharmacological doses of rFVIIa control bleeding by local enhancement of thrombin generation (Monroe et al., 1997), stimulating the formation of a stable hemostatic clot (He et al., 2003). Additionally, rFVIIa has been shown to inhibit fibrinolysis by activation of the thrombin activatable fibrinolysis inhibitor (TAFI) (Lisman et al., 2002). The use of rFVIIa to control bleeding has been reported in various clinical settings, including the treatment of both coagulopathic and hemostatically normal patients peri-surgically (Friederich et al., 2003; Aldouri, 2002), post-trauma (Martinowitz et al., 2001, 2002; Kenet et al., 1999; Kamphuisen et al., 2002; O’Neill et al., 2002; Khan et al., 2005; Filsoufi et al., 2006), in liver failure (Chuansumrit et al., 2000) and in other bleeding conditions refractory to conventional therapy (Mayo et al., 2004). Of particular interest are the encouraging results to date for use of rFVIIa following spontaneous intracerebral hemorrhage (ICH) (Mayer et al., 2005a,b; Steiner et al., 2006) where rFVIIa was reported to decrease hematoma growth, reduce mortality, and improve functional outcome. However, the study of rFVIIa in ICH of traumatic origin is limited to a number of small case series (Zaaroor and Bar-Lavie, 2004; White et al., 2006; Aiyagari et al., 2005; Martinowitz and Michaelson, 2005; Dutton et al., 2004). The successes of rFVIIa in treating spontaneous ICH justify further investigation with respect to its potential use as a treatment for tICH.
In the present study, we evaluated the potential effectiveness and safety of rFVIIa in a combined porcine TBI model of focal cortical contusion and diffuse axonal injury (DAI). Specifically, we examined the effects of rFVIIa on (1) the expansion of cerebral contusion, (2) hippocampal neuronal death, (3) diffuse axonal injury (DAI), and (4) intravascular thrombosis.
Materials and methods
Rationale for animal model
A porcine model was considered appropriate for this study since exceedingly high and potentially toxic doses of rFVIIa are needed to induce hemostasis in other species including rodents (Schreiber et al., 2005). Earlier studies have demonstrated the hemostatic activity of rFVIIa in the pig (Schreiber et al., 2005, 2003, 2002). Since hemorrhagic contusion typically coincides with DAI in humans (Smith and Meaney, 2000), an injury model of DAI and cerebral contusion was selected to evaluate the effects of rFVIIa on both of these pathologies.
Animal preparation
This study was conducted in accordance with the animal welfare guidelines set forth in the Guide for the Care and Use of Laboratory Animals, by the Department of Health and Human Services. All animal procedures were approved by the University of Pennsylvania Institutional Animal Use and Care Committee.
Ten female miniature swine (Hanford strain, Sinclair Research Center, Inc., Columbia, MO) were included in this study. All animals were adult (aged 6–7 months) and weighed between 22–25 kg.
Prior to surgical procedures, animals were fasted for 12 h. Induction of anesthesia was achieved by intramuscular administration of midazolam (400–600 μg/kg) followed by 4% isoflurane gas via snout mask. On reaching a plane of surgical anesthesia, animals were endotracheally intubated and maintained under general anesthesia using spontaneously inhaled isoflurane (1.5–2%). Physiological monitoring included clinical observation, pulse oximetry via the skin of the ear, rectal temperature and intermittent blood pressure measurements using the brachial artery. Monitoring was continuous and documented at 15 min intervals. Previous experience with this model has shown that arterial blood gases, end-tidal CO2 and intracranial pressure (ICP) are well maintained throughout all procedures with only transient changes occurring with the below specified injury parameters.
Induction of traumatic brain injury
Traumatic brain injury was induced using a double injury model resulting in both diffuse axonal injury and cerebral contusion. Diffuse axonal injury (DAI) was induced using non-impact rotational acceleration (Smith et al., 1997, 2000). Briefly, the animals’ heads were secured to a padded snout clamp mounted to the linkage assembly of a pneumatic actuator, or HYGE device. This device converts linear motion to angular (rotational) motion and produces pure impulsive head rotation of 110° (20 ms) in the coronal plane, triggered by the release of pressurized nitrogen. The center of rotation is close to the brain’s center of mass. Control of the head rotational acceleration profile was accomplished by adjusting both the hydraulic fluid level within the actuator and the pneumatic pressure delivered to the actuator. The peak angular velocity for coronal plane rotation ranged between 221 and 262 rad/s. Following DAI induction, animals’ heads were immediately released from the clamp. The inertial loading conditions produced by activation of this device have been shown to closely approximate the conditions of inertial brain injury in humans, such as those encountered during automobile crashes (Smith et al., 1997).
Within 30 min of rotational acceleration injury, a cerebral contusion injury was also induced using dynamic cortical deformation (DCD) (Shreiber et al., 1999). An 8 mm burr hole through the skull was placed 1 cm lateral to the sagittal suture, centered between lambda and bregma. An incision was made through the dura and a rigid plastic tube (8 mm external diameter, 6 mm internal diameter) was inserted and sealed with bone wax. The tube was connected to a vacuum pulse generator and 1 atm of negative pressure was applied for 2 seconds to generate a contusion. The craniotomy was left open and the overlying scalp incision sutured.
Blood collection
Blood samples were collected at four time points: pre-injury, 5 min following administration of drug/vehicle, 2 h post-injury and 72 h following injury. All samples were collected through peripheral veins.
Drug administration
Femoral veins were surgically exposed prior to rotational injury. Within 15 min following the cerebral contusion, five animals received an IV bolus via the femoral vein of rFVIIa (720 μg/kg, rFVIIa Novo Nordisk A/S, Bagsvaerd, Denmark, delivered in a solution containing NaCl (83.5 mM), CaCl2 (16.7 mM), glycylglycin (16.7 mM) mannitol (50 mg/ml), 0.017% polysorbate). Five animals received a vehicle solution (NaCl (50 mM), CaCl2 (10 mM), glycylglycin (10 mM) mannitol (30 mg/ml), 0.01% polysorbate 80) of equivalent volume. The rFVIIa dose level was selected based on the experience gained in previously performed porcine models (Schreiber et al., 2003, 2002). Notably, mannitol was included in the delivery solution for both rFVIIa-treated animals and vehicle controls for the purpose of attaining appropriate osmolarity. However, the concentration of mannitol was far below the range used to treat brain swelling(Cloyd et al., 1986). Investigators were blinded to which animals received drug versus the vehicle solution until all analysis was complete.
Magnetic Resonance Imaging (MRI) and analysis of cortical contusion expansion
Acute brain imaging was performed using a 1.5T Siemens MRI system. Scanning was initiated approximately 20 min after the contusion injury. Repeat imaging was performed at 3 days post-injury.
All animals were maintained under general anesthesia during imaging procedures. The swine were placed in the prone position, and a 13-cm surface coil was positioned on the head. Sequences included Magnetization Prepared Rapid Acquisition Gradient Echo (MPRAGE) T1 weighted images (TR/TE1900/5.6 ms, slice thickness 1mm), T2-weighted turbo spin echo (TSE) images (TR/TE 2500/81 ms, slice thickness 3 mm), and T2* weighted gradient echo images (TR/TE 4480/46, slice thickness 5 mm). According to previous studies, T2* weighted gradient echo is the most sensitive sequence to detect hyper-acute hemorrhage in brain and thus was deemed appropriate for measurement of contusion volume immediately post-injury (Gustafsson et al., 1999; Alemany Ripoll et al., 2002, 2004). In contrast, by sacrifice day (day 3), hemorrhagic contusions were best identified by T2 weighted TSE. T2-weighted TSE images are characterized by a hypointense contusion surrounded by hyperintense edema. Measurements of contusion volume were conducted by an experienced neuroradiologist using Leonardo workstation imaging software (Siemens, Germany). Expansion of the cortical contusion was expressed as a ratio of lesion volume on injury day to sacrifice day. Measurements and analysis were conducted blind to the treatment group of the animals.
Coagulation parameters analysis
Plasma samples were assayed for FVIIAg level, prothrombin time (PT), and activated partial thromboplastin time (aPTT) (Organon Teknika, Denmark). A slightly modified PT assay was performed using dilute thromboplastin (Innovin, Dade Behring) at a final reagent dilution of 1/1200 which gives longer clotting times. FVII antigen level was determined using a FVII EIA (Dako A/S, Denmark) according to manufacturer’s specifications.
Tissue preparation
Three days after TBI, all animals were sacrificed. Transcardial perfusion was performed using heparinized saline (4 L) followed by 4% paraformaldehyde (8 L). The brain, lungs and liver were removed and post-fixed in 4% paraformaldehyde for 2 h before being transferred for storage in phosphate buffer solution.
Brains were blocked into 5 mm coronal sections for gross examination and photography. Tissue blocks from some animals were cryoprotected in sucrose. A series of 40 μm frozen sections were cut from the front face of each block and mounted on slides for microscopy. Representative slides from each block were stained with haemotoxylin–eosin (H&E), cresyl violet (CV) and Fluorojade. Blocks from lungs and the liver were processed for paraffin embedding in an automated tissue processor (Shandon Scientific Instruments, Pittsburgh, PA). Serial sections (6 μm) were cut on a Leitz rotary microtome (Leica, Malvern, PA) and mounted on Fisherbrand Superfrost/Plus microscope slides (Fisher Scientific, Pittsburgh).
The volumetric analysis of cerebral contusion by gross pathological examination
To verify the MRI findings, the cerebral contusion volumes were also measured by gross pathological examination. The contusion region in the fixed brain was blocked into 5 mm coronal sections. Each block containing the contusion lesion was photographed and transferred to a personal computer for analysis. A neuropathologist, blinded to treatment group, identified lesion boundaries in the gross specimens, defined as areas of hemorrhage and necrosis using standard imaging software (ImageJ™, NIH). The total volume of cerebral contusion observed was recorded and compared with the findings from the final MRI scan.
Immunohistochemical Methods
To detect axonal pathology, immunohistochemistry was performed on both paraffin embedded and frozen sections using the avidin–biotin immunoperoxidase complex method. Primary mouse monoclonal antibody N52, targeting the 200-kD neurofilament subunits (Sigma, 1:400) was used. The brain sections were incubated with primary antibody overnight at 4 °C and then incubated at room temperature for 1 h with the appropriate secondary antibodies. Antibody complexes were detected using avidin–biotin peroxidase (ABC) histochemistry (Vector Labs). Visualization was achieved using 3,3′ diamino-benzadine (DAB).
Histopathological analysis
Quantification of axonal profiles
The length, varying diameter and convoluted nature of damaged axons can pose a significant challenge to the precise quantification of diffuse axonal injury. We therefore used profile counting to determine the extent of pathologic changes. A profile was considered to be a darkly immunoreactive varicose axonal swelling or an axonal bulb. Counting was performed in multiple brain areas which included the basal ganglia, the frontal, parietal and occipital lobes. These areas were selected based on the high density of axonal pathology seen in our previous study (Chen et al., 2004). The specific anatomic location for each area was selected based on a stereotactic atlas of the pig brain (Félix et al., 1999).
For microscopic examination, a 10× objective lens was used to provide a counting frame measuring 1.2 mm2. For each coronal section, a brain area with a total of 80–550 frames (approximately 96–660 mm2) was screened and only profiles with a diameter of greater than 10 μm were counted. Counting was performed manually under light microscopy at ×100 magnification. The total number of profiles per brain area was determined by summing the number of profiles in all of the frames across one coronal section in the specified area. This was performed in four incremental coronal sections (0.5 cm) and the total numbers across these four sections were averaged resulting in a single value for each subject. For the purposes of generating a schematic map of the distribution, the number of profiles per brain region were also ranked in a semiquantitative scale (+= 10 profiles or less,++= greater than 100 profiles and+++= greater than 500 profiles.)
All observations were made blinded to the injury status and drug treatment group of the animals.
Intravascular thrombi counting
The extent of intravascular thrombi was determined by examining sections stained with H&E. Thrombi were identified by their distinct morphology. Counting was performed in 3 random sections each hemisphere in the frontal, parietal and occipital lobes as well as in the basal ganglia. These numbers were summed to give the total number of microclots. Comparisons were made between rFVIIa-treated animals versus vehicle controls in whole brain, whole hemisphere and regionally.
Degenerating neuron counting
For assessment of hippocampal neuronal pathology, five H&E stained brain sections (40 μm apart) were selected bilaterally from the mid-dorsal hippocampus in the coronal plane. The pyramidal cell layer was examined for degenerating neurons (darkly staining shrunken profiles) in three hippocampal subfields (CA1, CA2 and CA3 including the dentate hilus region). In CA1, three frames (1.2 mm2) encompassing an approximate total area of 3.6 mm2 were analyzed. Two frames in each CA2 region and 3 frames in CA3, encompassing an approximate total area of 3.6 mm2 and 7.2 mm2, respectively were analyzed. Light microscopy at ×100 magnification was used for counting. The mean number of degenerating neurons in hippocampal region (CA1, CA2 and CA3) from both hemi-spheres in each group of animals was determined.
Statistical analysis
Statistical analyses were performed using Statistica 4.1 software and Microsoft Excel. Wilcoxon rank–sum tests were used to determine whether there were differences between groups in (1) initial contusion volume at the time of injury, (2) the percent expansion of contusion volume at the time of sacrifice, and (3) the severity and distribution of axonal pathology. Kendall’s tau (λ) was used to determine the association between gross pathology and MRI contusion volumes of samples (Hettmansperger, 1991). Student’s t-test was used to determine differences between groups in degenerating hippocampal neurons and global intravascular thrombi. All values are presented as the mean±SEM. A p-value less than 0.05 was considered significant.
Results
Cerebral contusion expansion analysis
MRI sequences in this study were selected to detect the evolution of hemorrhagic cerebral contusion over three days post-injury. The T2* weighted gradient echo (GE) sequence was used to identify acute blood in the brain. T2* weighted (GE) sequence detected intracerebral hemorrhages in all animals as easily identifiable hypointense areas (Fig. 1) In the acute stage, the lesions were mainly composed of intraparenchymal hemorrhages. Three days after injury, the components of hemorrhagic contusion changed, to include localized tissue necrosis, chromatolysis of subacute blood and surrounding edema. The contusions in all pigs, at 3 days post-injury were markedly visible on T2-weighted TSE imaging, and were characterized by a well-defined hyperintense area surrounded by a high signal region representative of edema (Fig. 1). The initial volume of cerebral contusion shortly after injury was 637 ± 142 mm3 (mean± SD) in the rFVIIa-treated animals and 684± 129 mm3 in the vehicle-treated pigs with no statistically significant differences between treatment groups (p= 1.0) prior to treatment. Contusion volumes measured at 3 days post-injury were 780 ± 354 mm3 in rFVIIa-treated animals versus 1840± 662 mm3 in vehicle controls. This was a significant difference with p =0.013. Thus, the extent of contusion volume expansion (Fig. 2) was 25.54± 6.4% for the rFVIIa-treated group and 256.7± 128.7% for the vehicle-treated group, a statistically significant difference (p<0.01).
Fig. 1.

Representative MRI images at injury and on sacrifice day between the two groups. Cerebral contusions were seen as hypointense signal on T2* weighted gradient echo images acutely post-trauma (black arrows). At 3 days post-trauma contusions were seen as slightly hypointense on T2-weighted TSE images surrounded surrounded by a hyperintense signal (edema).
Fig. 2.
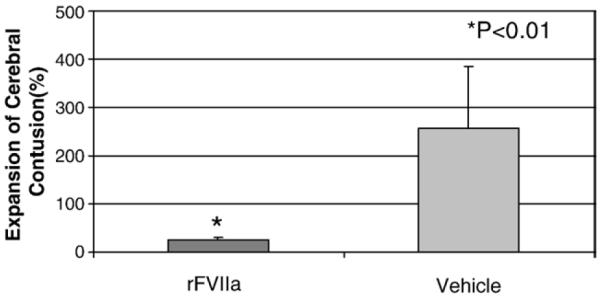
Comparison of contusion expansion (MRI) between rVIIa-treated pigs and vehicle-treated animals.
Gross pathological examination at 3 days post-injury revealed a volume of 760 ± 480 mm3 (mean ± SD) in rFVIIa-treated animals versus 1270 ± 147 mm3 in the control group. Although there was a difference, this did not reach significance with p = 0.15. However, when the volume of cerebral contusion on gross pathology was compared to the MRI results obtained on day 3. A significant correlation between histological findings and MRI images was demonstrated (λ = 0.69, p < 0.005).
Coagulation parameters
In the rFVIIa-treated group, FVII antigen, measured by FVIIAg-EIA, increased significantly (p < 0.05) from baseline levels for the 5 min post-dosing and at 2 h post-injury. This declined to normal levels at three-days post-injury (Fig. 3). The observations are consistent with the half life of rFVIIa (Pusateri et al., 2005).
Fig. 3.
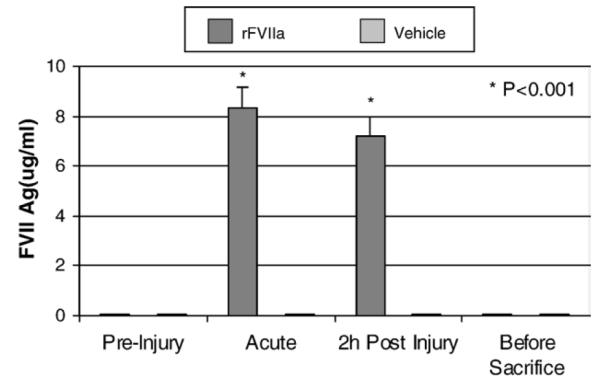
Comparison of FVIIa antigen between the treatment group and the vehicle group at different time-points.
Consistent with the increase in rFVII activity, the prothrombin time (PT) decreased significantly (p < 0.05) in the rFVIIa-treatment group for blood sampled both acutely and at two hours post-injury. At 3 days post-injury there were no significant differences in PT between treatment groups (Fig. 4). PT was unchanged in the vehicle-treated group. No changes at any timepoint were found in aPTT levels in any group.
Fig. 4.
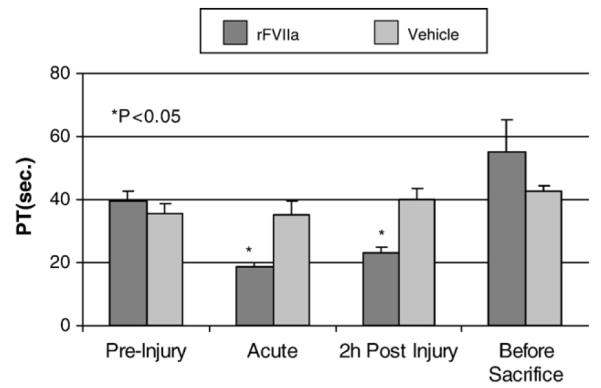
Comparison of PT between the treatment group and the vehicle group at different time-points.
Pathological Findings
At day 3 post-injury, H&E and Fluorojade staining demonstrated degenerating neurons in the CA1, CA2, and CA3 regions, including the dentate hilus of hippocampus bilaterally in the vehicle-treated group. In marked contrast, few degenerating neurons were detected within the three sub-regions in rFVIIa-treated pigs (Fig. 5). Statistically, there was a significant reduction in neuronal degeneration in rFVIIa-treated pigs compared to the vehicle-treated animals in both the ipsilateral and contralateral hippocampus (p < 0.01) (Fig. 6).
Fig. 5.

Representative photomicrographs showing degenerating neurons in the hippocampus at three days post-injury in the pig. H&E and Fluorojade staining demonstrate more degenerating neurons found in the CA1 CA2 and CA3 regions in vehicle-treated animals (A and B, upper) compared with few degenerating neurons in the same regions of hippocampus in rFVIIa-treated animals (A and B, bottom). Bar = 50 μm.
Fig. 6.
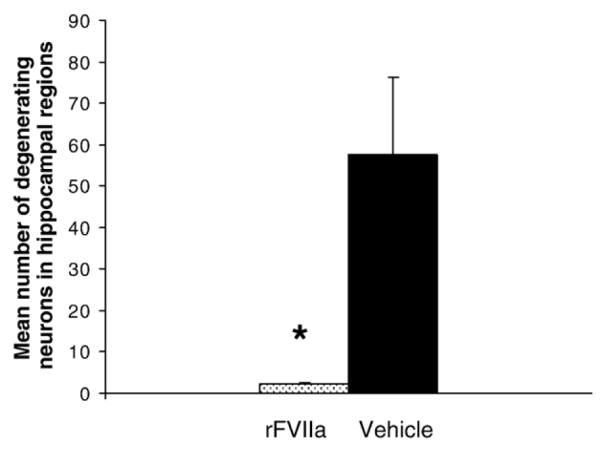
Graph demonstrates the distribution of degenerating neurons in the hippocampus between rFVIIa and vehicle-treated animals following brain trauma. A significant difference between groups was observed with p < 0.01.
Axonal injury identified as axonal bulbs and varicose axonal swellings were found throughout the brain, most notably in the peri-contusional region, the subcortical white matter of the frontal lobe, parietal lobe, occipital lobe and the basal ganglia (Figs. 7, 8 and 9). The most abundant pathology was seen in the parietal lobe (Fig. 9). According to the averaged axonal injury profile counts per 4 brain sections, we found the mean number of axonal profiles in the frontal lobe to be 209.25 ± 107 (mean± SE) in rFVIIa-treated animals versus 562.25 ± 220 in animals receiving vehicle. In the parietal lobe, occipital lobe, and the basal ganglia, respectively, the corresponding values were 552.55 ±202 vs 739.45±294.28, 76.72±28.9 vs 207.11±99.58 and 397.5± 147.34 vs 446.72 ± 185.34. Thus, all animals treated with rFVIIa had a lower mean number of axonal injury profiles compared to the vehicle group throughout the brain. The observed rFVIIa treatment induced reduction in axonal injury reached statistical significance only in the frontal lobe (p < 0.01). However, when the mean numbers were combined for all brain regions, the difference between rFVII treated and vehicle groups was statistically significant (p < 0.05) (Fig. 8).
Fig.7.

Schematic representation of the distribution and severity of axonal pathology in various coronal planes of the brain at 3 days post-injury. In all animals the contusion injury was performed on the left side. (A) septal nuclei and anterior commissure level, (B) rostral-thalamus level, (C) caudal–hippocampal level, (D) occipital cortical brain stem level.
Fig. 8.
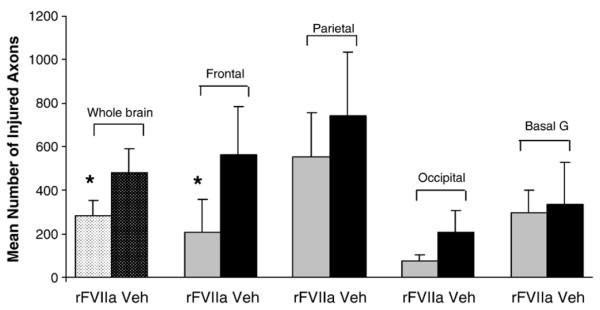
Mean number of axonal profiles representing pathology (axonal bulbs or varicosities) per brain region (bilaterally) in rFVIIa-treated animals versus controls.
Fig. 9.
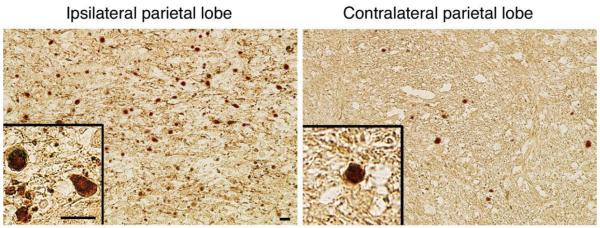
Immunohistochemical staining with antibody to neurofilament, demonstrating axonal pathology in the parietal lobe both ipsilaterally and contralaterally. Multiple positively stained axons are seen with the classic appearance of swollen axon terminals and tortuous varicosities. Note the classic appearance of halos surrounding the swollen axon terminals. This phenomenon occurs secondary to dehydration of the tissue during processing.
Microthrombi
The total number of microthrombi in whole brain in rFVIIa-treated animals was 522.8±168.2 (mean±SE) versus 769.2±214.3 in vehicle controls (p=0.38) (Fig. 10). Upon examining the hemisphere ipsilateral to contusion, there were 258.8±86.4 microthrombi in the rFVIIa-treated group versus 440.4±159.5 in vehicle controls (p=0. 17). Similarly upon examination of the contralateral hemisphere, there were 264.0±85.5 microthrombi in the rFVIIa-treated group versus 328.8±68.5 in vehicle controls (p=0.28). No significant differences were found between the groups in any regional analysis (p>0.05).
Fig. 10.
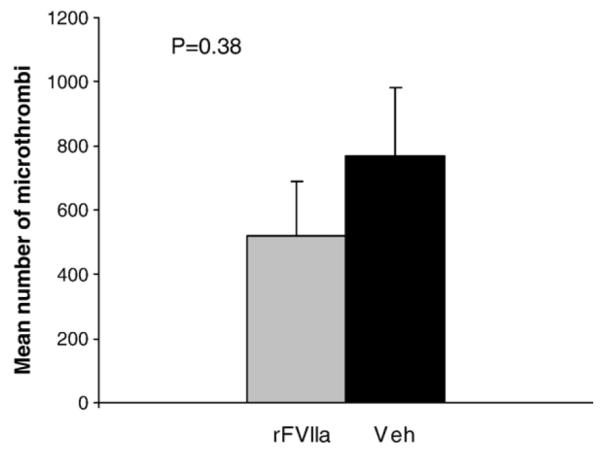
Total number of microclots as determined by H&E staining. Whole brain analysis for rFVIIa-treated animals versus vehicle controls.
Histological examination of lung and liver demonstrated a small number of microthrombi within the tissue of both rFVIIa and the vehicle-treated animals. While the extent of this pathology was not quantified, qualitatively, there was no obvious difference between groups.
Discussion
We provide evidence that early treatment with rFVIIa after TBI significantly reduced the expansion of cerebral contusion as measured 72 h after TBI. Surprisingly, we also discovered that rFVIIa treatment exhibited neuroprotective effects. In particular, profound reductions in axonal pathology were seen within the frontal lobe with an overall significant decrease noted throughout the brain. Hippocampal neuronal death in rFVIIa-treated brain-injured animals was also substantially reduced compared with vehicle-treated animals. The animals displayed no clinical signs suggestive of thromboembolic disease nor was there an increase in microclots detected in brain or other organs (liver and lung).
There are two components to the mechanism by which rFVIIa enhances hemostasis. Recombinant FVIIa binds to exposed tissue factor at the site of tissue and vascular injury and induces the activation of factor X (FX). This in turn activates the extrinsic pathway to promote thrombin synthesis and the generation of fibrin based clots (Hoffman, 2003). In addition, at high concentrations, rFVIIa also binds to activated platelets where it can directly activate FX and factor IX, thus promoting coagulation in the absence of tissue factor (Monroe et al., 1997; Gerotziafas et al., 2004). There is evidence to support the suggestion that such clots formed under the pharmacological influence of rFVIIa are of a more robust architecture than normally formed clots (Banninger et al., 1994; Blomback et al., 1994; Sondeen et al., 2004).
The potential benefit of using a pro-hemostatic drug in TBI patients would be to control intracranial bleeding and reduce the expansion of contusions in the acute post-traumatic phase. Contusion expansion is well documented in both spontaneous and traumatic ICH (Servadei et al., 1995; Oertel et al., 2002; Chang et al., 2006; Chieregato et al., 2005; Lobato et al., 1991; Brott et al., 1997; Fujii et al., 1994; Lobato et al., 1997). A worse clinical outcome following spontaneous ICH is directly associated with the hematoma volume (Mayer et al., 2005a,b; Brott et al., 1997). In TBI, hematoma progression or hemorrhage extension results in greater mortality, slowed recovery, and poorer functional outcome (Stein et al., 1993). Here we demonstrate a reduction in hematoma expansion with the use of rFVIIa in our dynamic contusion model of injury. Gross pathological measurements of contusion volume at day 3 post-TBI correlated well with MRI findings. However, it is noted that volumes determined by MRI were significantly different between drug- and vehicle-treated groups whereas measurement using gross pathological examination, although different, was not significant. This is likely due to the increased sensitivity of MRI in detecting injured and degenerating tissue than would be possible with simple gross examination.
Further studies in TBI patients should include an assessment of the timing of drug delivery on hemorrhage expansion in order to support these preclinical findings and provide model validation of this approach. In addition, the baseline coagulation parameters likely are important in determining the efficacy of the drug. All of our animals had normal coagulation parameters prior to injury and PT retuned to baseline levels in association with the half life of rFVIIa. In addition, there was no evidence of any consumptive coagulopathies that have previously been associated with delayed or progressive bleeding following human TBI (Stein et al., 1992). With appropriate dosing, rFVIIa may confer additional benefit to such coagulopathic patients. In essence, our demonstration of substantial reduction in contusion expansion provides hope that this novel drug could possibly improve outcome in severe tICH. In addition, rFVIIa may provide a non-invasive treatment for smaller bleeds where surgery is not indicated due to the risk–benefit ratio.
The model of injury we chose for this study was dual in nature, representing both cerebral contusion and diffuse axonal injury (DAI). This was deemed appropriate as DAI is a common feature of head trauma, particularly in the severe category (Smith et al., 2003; Adams et al., 1991). Unexpectedly, in this study we found that intravenous rFVIIa resulted in a statistically significant reduction in the degree of axonal pathology when looking at whole brain. In addition, this was found to be profound and with high statistical significance in the frontal lobe. We also demonstrated a profound sparing of hippocampal neurons in bilateral hippocampi in rFVII treated animals versus vehicle controls. The mechanism of how rFVIIa is both neuronally and axonally protective is not clear. In rodents, aggressive cerebral contusion expansion in one hemisphere results in aggravated ipsilateral hippocampal neuronal death (Shreiber et al., 1999). Conversely, arresting contusion growth may decrease hippocampal ipsilateral neuronal death. However, since in the current study we demonstrated bilateral protection, it is possible that rFVIIa exerts direct neuronal and axonal protection. It is known that TBI usually induces the breakdown of the blood–brain barrier (BBB) with accompanying serum protein extravasation into the brain (Tanno et al., 1992a,b,). It is also known that Factor VIIa, when bound to its cell surface receptor (tissue factor), provides cellular protection against apoptosis, primarily through activation of the PI3-kinase/Akt pathway (Sorensen et al., 2003). Thus, it is possible that rFVIIa enters the brain parenchyma as a result of the pathological BBB disruption secondary to trauma and directly exerts its neuroprotective effects via this mechanism. Alternatively, rFVIIa controls bleeding pharmacologically by enhancing thrombin generation on the surface of platelets adherent to the site of vascular injury (Kjalke et al., 2007). Thrombin has been demonstrated to be neurotoxic at high concentrations and neuroprotective at low concentrations (Figueroa et al., 1998; Gorbacheva et al., 2006; Gingrich and Traynelis, 2000). A transient, rFVIIa-mediated, surge in thrombin formation (Kjalke et al., 2007) may limit the extent of neuron exposure to high toxic thrombin concentrations and protract contact with more moderate thrombin concentrations, thus inducing secondary neuroprotection. Further work is required to elucidate the precise mechanisms responsible for the axonal and neuronal protection seen in this study.
The safety profile of rFVIIa has been followed since its first use in humans in the 1990’s, primarily in patients suffering from hemophilia. The safety profile of rFVIIa in those with initially normal coagulation parameters is less well understood. In the recent phase II spontaneous ICH trial involving 399 patients a non-significant increase in the thromboembolic complication rate from 2% in the placebo group to 7% in the rFVIIa-treated group was reported along with encouraging results with regard to limiting hematoma expansion and improving functional outcomes (Mayer et al., 2005a,b). In this study we found a small number of microclots when tissue from both the liver and lung was examined. Qualitatively, there was no difference in the number of these clots between drug- and vehicle-treated animals. There is some evidence to suggest that patients who experience TBI can develop systemic coagulopathy similar to a mild form of disseminated intravascular coagulation (Stein and Smith, 2004); (Keimowitz and Annis, 1973). Although we found no evidence of the traumatic or consumptive coagulopathy associated with delayed or progressive bleeding in TBI (Stein et al., 1992) it is possible that the clots found in the lungs and liver of both treated and non-treated animals developed secondary to a trauma-induced imbalance in the coagulation system. Notably, however, we found no evidence of an increased tendancy towards extracerebral coagulopathy within the drug treated animals. Further to this, we found no difference between groups with respect to the number of microthrombi in the cortical contusion hemisphere or contralaterally.
Clinically, all animals were well with no signs of thromboembolic disease. The so called ‘cell-based model’ of coagulation suggests systemic hypercoagulability is a dose dependant risk, associated with direct platelet activation secondary to excessively high levels of rFVII and thrombin (Hoffman, 2003). Indeed, our measurement of coagulation parameters, specifically prothrombin time, suggest that there is a transient period of systemic hypercoagulability consistent with the drug half life. Although there was no clinical or pathologically overt evidence of thrombotic events, use of this drug should be coupled with close monitoring for adverse events, particularly as this is a patient subgroup that is already has elevated risk of thromboembolism (Geerts et al., 1994); (Kurtoglu et al., 2004).
In summary we have demonstrated, using a double injury swine model of TBI, that use of rFVIIa not only reduced contusion expansion, but also offers significant protection to axons and hippocampal neurons, in the absence of any significant adverse effects. rFVIIa has the potential to be the first clinically successful medical therapy for TBI.
Acknowledgments
Funding for this study was provided through sponsorship by Novo Nordisk A/S (Bagsvaerd, Denmark) and by NIH grant: NS38104.
References
- Adams JH, et al. Diffuse axonal injury in non-missile head injury. J. Neurol. Neurosurg. Psychiatry. 1991;54(6):481–483. doi: 10.1136/jnnp.54.6.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aiyagari V, Menendez JA, Diringer MN. Treatment of severe coagulopathy after gunshot injury to the head using recombinant activated factor VII. J. Crit. Care. 2005;20(2):176–179. doi: 10.1016/j.jcrc.2005.02.004. [DOI] [PubMed] [Google Scholar]
- Aldouri M. The use of recombinant factor VIIa in controlling surgical bleeding in non-haemophiliac patients. Pathophysiol. Haemost Thromb. 2002;32(Suppl 1):41–46. doi: 10.1159/000057301. [DOI] [PubMed] [Google Scholar]
- Alemany Ripoll M, et al. MR follow-up of small experimental intracranial haemorrhages from hyperacute to subacute phase. Acta Radiol. 2002;43(1):2–9. doi: 10.1080/028418502127347538. [DOI] [PubMed] [Google Scholar]
- Alemany Ripoll M, et al. Detection and appearance of intraparenchymal haematomas of the brain at 1.5 T with spin-echo, FLAIR and GE sequences: poor relationship to the age of the haematoma. Neuroradiology. 2004;46(6):435–443. doi: 10.1007/s00234-004-1191-5. [DOI] [PubMed] [Google Scholar]
- Banninger H, Lammle B, Furlan M. Binding of alpha-thrombin to fibrin depends on the quality of the fibrin network. Biochem J. 1994;298(Pt 1):157–163. doi: 10.1042/bj2980157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blomback B, et al. Fibrin in human plasma: gel architectures governed by rate and nature of fibrinogen activation. Thromb Res. 1994;75(5):521–538. doi: 10.1016/0049-3848(94)90227-5. [DOI] [PubMed] [Google Scholar]
- Brott T, et al. Early hemorrhage growth in patients with intracerebral hemorrhage. Stroke. 1997;28(1):1–5. doi: 10.1161/01.str.28.1.1. [DOI] [PubMed] [Google Scholar]
- Bullock R, et al. Guidelines for the management of severe head injury. Brain Trauma Foundation. Eur. J. Emerg Med. 1996;3(2):109–127. doi: 10.1097/00063110-199606000-00010. [DOI] [PubMed] [Google Scholar]
- Chang EF, Meeker M, Holland MC. Acute traumatic intraparenchymal hemorrhage: risk factors for progression in the early post-injury period. Neurosurgery. 2006;58(4):647–656. doi: 10.1227/01.NEU.0000197101.68538.E6. discussion 647–56. [DOI] [PubMed] [Google Scholar]
- Chen XH, et al. Long-term accumulation of amyloid-beta, beta-secretase, presenilin-1, and caspase-3 in damaged axons following brain trauma. Am. J. Pathol. 2004;165(2):357–371. doi: 10.1016/s0002-9440(10)63303-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chieregato A, et al. Factors associated with neurological outcome and lesion progression in traumatic subarachnoid hemorrhage patients. Neurosurgery. 2005;56(4):671–680. doi: 10.1227/01.neu.0000156200.76331.7a. discussion 671–80. [DOI] [PubMed] [Google Scholar]
- Chuansumrit A, et al. Recombinant activated factor VII in children with acute bleeding resulting from liver failure and disseminated intravascular coagulation. Blood Coagul. Fibrinolysis. 2000;11(Suppl 1):S101–S105. doi: 10.1097/00001721-200004001-00019. [DOI] [PubMed] [Google Scholar]
- Cloyd JC, et al. Mannitol pharmacokinetics and serum osmolality in dogs and humans. J. Pharmacol Exp. Ther. 1986;236(2):301–306. [PubMed] [Google Scholar]
- Dutton RP, et al. Factor VIIa for correction of traumatic coagulopathy. J. Trauma. 2004;57(4):709–718. doi: 10.1097/01.ta.0000140646.66852.ab. discussion 718–9. [DOI] [PubMed] [Google Scholar]
- Félix B, Léger M-E, Albe-Fessard D. Stereotaxic Atlas of the Pig Brain. Elsevier Science B.V; Amsterdam: 1999. [DOI] [PubMed] [Google Scholar]
- Figueroa BE, et al. Plasminogen activators potentiate thrombin-induced brain injury. Stroke. 1998;29(6):1202–1207. doi: 10.1161/01.str.29.6.1202. discussion 1208. [DOI] [PubMed] [Google Scholar]
- Filsoufi F, et al. Effective management of refractory postcardiotomy bleeding with the use of recombinant activated factor VII. Ann. Thorac Surg. 2006;82(5):1779–1783. doi: 10.1016/j.athoracsur.2006.05.076. [DOI] [PubMed] [Google Scholar]
- Friederich PW, et al. Effect of recombinant activated factor VII on perioperative blood loss in patients undergoing retropubic prostatectomy: a double-blind placebo-controlled randomised trial. Lancet. 2003;361(9353):201–205. doi: 10.1016/S0140-6736(03)12268-4. [DOI] [PubMed] [Google Scholar]
- Fujii Y, et al. Hematoma enlargement in spontaneous intracerebral hemorrhage. J. Neurosurg. 1994;80(1):51–57. doi: 10.3171/jns.1994.80.1.0051. [DOI] [PubMed] [Google Scholar]
- Geerts WH, et al. A prospective study of venous thromboembolism after major trauma. N. Engl. J. Med. 1994;331(24):1601–1606. doi: 10.1056/NEJM199412153312401. [DOI] [PubMed] [Google Scholar]
- Gerotziafas GT, et al. The role of platelets and recombinant factor VIIa on thrombin generation, platelet activation and clot formation. Thromb Haemost. 2004;91(5):977–985. doi: 10.1160/TH03-10-0638. [DOI] [PubMed] [Google Scholar]
- Ghajar J. Traumatic brain injury. Lancet. 2000;356(9233):923–929. doi: 10.1016/S0140-6736(00)02689-1. [DOI] [PubMed] [Google Scholar]
- Gingrich MB, Traynelis SF. Serine proteases and brain damage— is there a link? Trends Neurosci. 2000;23(9):399–407. doi: 10.1016/s0166-2236(00)01617-9. [DOI] [PubMed] [Google Scholar]
- Gorbacheva LR, et al. Modulation of hippocampal neuron survival by thrombin and factor Xa. Biochemistry (Mosc) 2006;71(10):1082–1089. doi: 10.1134/s000629790610004x. [DOI] [PubMed] [Google Scholar]
- Gustafsson O, et al. MR imaging of experimentally induced intra-cranial hemorrhage in rabbits during the first 6 hours. Acta Radiol. 1999;40(4):360–368. doi: 10.3109/02841859909177748. [DOI] [PubMed] [Google Scholar]
- He S, et al. The role of recombinant factor VIIa (FVIIa) in fibrin structure in the absence of FVIII/FIX. J. Thromb Haemost. 2003;1(6):1215–1219. doi: 10.1046/j.1538-7836.2003.00242.x. [DOI] [PubMed] [Google Scholar]
- Hettmansperger TP. Statistical Inference Based on Ranks. Krieger Publishing Company; Malabar, Florida: 1991. [Google Scholar]
- Hoffman M. A cell-based model of coagulation and the role of factor VIIa. Blood Rev. 2003;17(Suppl 1):S1–S5. doi: 10.1016/s0268-960x(03)90000-2. [DOI] [PubMed] [Google Scholar]
- Kamphuisen PW, et al. Control of life-threatening pulmonary bleeding with activated recombinant factor VII. Am. J. Med. 2002;112(4):332–333. doi: 10.1016/s0002-9343(01)01116-0. [DOI] [PubMed] [Google Scholar]
- Keimowitz RM, Annis BL. Disseminated intravascular coagulation associated with massive brain injury. J. Neurosurg. 1973;39(2):178–180. doi: 10.3171/jns.1973.39.2.0178. [DOI] [PubMed] [Google Scholar]
- Kenet G, et al. Treatment of traumatic bleeding with recombinant factor VIIa. Lancet. 1999;354(9193):1879. doi: 10.1016/S0140-6736(99)05155-7. [DOI] [PubMed] [Google Scholar]
- Khan AZ, et al. Recombinant factor VIIa for the treatment of severe postoperative and traumatic hemorrhage. Am. J. Surg. 2005;189(3):331–334. doi: 10.1016/j.amjsurg.2004.11.021. [DOI] [PubMed] [Google Scholar]
- Kjalke M, Kjellev S, Rojkjaer R. Preferential localization of recombinant factor VIIa to platelets activated with a combination of thrombin and a glycoprotein VI receptor agonist. J. Thromb Haemost. 2007;5(4):774–780. doi: 10.1111/j.1538-7836.2007.02389.x. [DOI] [PubMed] [Google Scholar]
- Kurtoglu M, et al. Venous thromboembolism prophylaxis after head and spinal trauma: intermittent pneumatic compression devices versus low molecular weight heparin. World J. Surg. 2004;28(8):807–811. doi: 10.1007/s00268-004-7295-6. [DOI] [PubMed] [Google Scholar]
- Lisman T, et al. Inhibition of fibrinolysis by recombinant factor VIIa in plasma from patients with severe hemophilia A. Blood. 2002;99(1):175–179. doi: 10.1182/blood.v99.1.175. [DOI] [PubMed] [Google Scholar]
- Lobato RD, et al. Head-injured patients who talk and deteriorate into coma. Analysis of 211 cases studied with computerized tomography. J. Neurosurg. 1991;75(2):256–261. doi: 10.3171/jns.1991.75.2.0256. [DOI] [PubMed] [Google Scholar]
- Lobato RD, et al. Sequential computerized tomography changes and related final outcome in severe head injury patients. Acta Neurochir. (Wien) 1997;139(5):385–391. doi: 10.1007/BF01808871. [DOI] [PubMed] [Google Scholar]
- Martinowitz U, Michaelson M. Guidelines for the use of recombinant activated factor VII (rFVIIa) in uncontrolled bleeding: a report by the Israeli Multidisciplinary rFVIIa Task Force. J. Thromb Haemost. 2005;3(4):640–648. doi: 10.1111/j.1538-7836.2005.01203.x. [DOI] [PubMed] [Google Scholar]
- Martinowitz U, et al. Recombinant activated factor VII for adjunctive hemorrhage control in trauma. J. Trauma. 2001;51(3):431–438. doi: 10.1097/00005373-200109000-00002. discussion 438–9. [DOI] [PubMed] [Google Scholar]
- Martinowitz U, et al. Possible role of recombinant activated factor VII (rFVIIa) in the control of hemorrhage associated with massive trauma. Can J. Anaesth. 2002;49(10):S15–S20. [PubMed] [Google Scholar]
- Mayer SA, et al. Recombinant activated factor VII for acute intracerebral hemorrhage. N. Engl. J. Med. 2005a;352(8):777–785. doi: 10.1056/NEJMoa042991. [DOI] [PubMed] [Google Scholar]
- Mayer SA, et al. Safety and feasibility of recombinant factor VIIa for acute intracerebral hemorrhage. Stroke. 2005b;36(1):74–79. doi: 10.1161/01.STR.0000149628.80251.b8. [DOI] [PubMed] [Google Scholar]
- Mayo A, et al. Recombinant activated factor VII (NovoSeven): addition to replacement therapy in acute, uncontrolled and life-threatening bleeding. Vox Sang. 2004;87(1):34–40. doi: 10.1111/j.1423-0410.2004.00533.x. [DOI] [PubMed] [Google Scholar]
- McArthur DL, Chute DJ, Villablanca JP. Moderate and severe traumatic brain injury: epidemiologic, imaging and neuropathologic perspectives. Brain Pathol. 2004;14(2):185–194. doi: 10.1111/j.1750-3639.2004.tb00052.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monroe DM, et al. Platelet activity of high-dose factor VIIa is independent of tissue factor. Br. J. Haematol. 1997;99(3):542–547. doi: 10.1046/j.1365-2141.1997.4463256.x. [DOI] [PubMed] [Google Scholar]
- Oertel M, et al. Progressive hemorrhage after head trauma: predictors and consequences of the evolving injury. J. Neurosurg. 2002;96(1):109–116. doi: 10.3171/jns.2002.96.1.0109. [DOI] [PubMed] [Google Scholar]
- O’Neill PA, et al. Successful use of recombinant activated factor VII for trauma-associated hemorrhage in a patient without preexisting coagulopathy. J. Trauma. 2002;52(2):400–405. doi: 10.1097/00005373-200202000-00034. [DOI] [PubMed] [Google Scholar]
- Pusateri AE, et al. Effects of increasing doses of activated recombinant factor VII on haemostatic parameters in swine. Thromb Haemost. 2005;93(2):275–283. doi: 10.1160/TH04-03-0200. [DOI] [PubMed] [Google Scholar]
- Schreiber MA, et al. The effect of recombinant factor VIIa on coagulopathic pigs with grade V liver injuries. J. Trauma. 2002;53(2):252–257. doi: 10.1097/00005373-200208000-00011. discussion 257–9. [DOI] [PubMed] [Google Scholar]
- Schreiber MA, et al. The effect of recombinant factor VIIa on non-coagulopathic pigs with grade V liver injuries. J. Am. Coll Surg. 2003;196(5):691–697. doi: 10.1016/S1072-7515(02)01835-5. [DOI] [PubMed] [Google Scholar]
- Schreiber MA, Holcomb JB, Rojkjaer R. Preclinical trauma studies of recombinant factor VIIa. Crit Care. 2005;9(Suppl 5):S25–S28. doi: 10.1186/cc3782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Servadei F, et al. Evolving brain lesions in the first 12 hours after head injury: analysis of 37 comatose patients. Neurosurgery. 1995;37(5):899–906. doi: 10.1227/00006123-199511000-00008. discussion 906–7. [DOI] [PubMed] [Google Scholar]
- Shreiber DI, et al. Experimental investigation of cerebral contusion: histopathological and immunohistochemical evaluation of dynamic cortical deformation. J. Neuropathol. Exp. Neurol. 1999;58(2):153–164. doi: 10.1097/00005072-199902000-00005. [DOI] [PubMed] [Google Scholar]
- Smith DH, et al. Characterization of diffuse axonal pathology and selective hippocampal damage following inertial brain trauma in the pig. J. Neuropathol. Exp. Neurol. 1997;56(7):822–834. [PubMed] [Google Scholar]
- Smith DH, et al. Immediate coma following inertial brain injury dependent on axonal damage in the brainstem. J. Neurosurg. 2000;93(2):315–322. doi: 10.3171/jns.2000.93.2.0315. [DOI] [PubMed] [Google Scholar]
- Smith DH, Meaney DF, Shull WH. Diffuse axonal injury in head trauma. J. Head Trauma Rehabil. 2003;18(4):307–316. doi: 10.1097/00001199-200307000-00003. [DOI] [PubMed] [Google Scholar]
- Smith D, Meaney DF. Axonal damage in traumatic brain injury. Neuroscientist. 2000;6(6):483–495. [Google Scholar]
- Sondeen JL, et al. Recombinant factor VIIa increases the pressure at which rebleeding occurs in porcine uncontrolled aortic hemorrhage model. Shock. 2004;22(2):163–168. doi: 10.1097/01.shk.0000129202.76706.bd. [DOI] [PubMed] [Google Scholar]
- Sorensen BB, et al. Antiapoptotic effect of coagulation factor VIIa. Blood. 2003;102(5):1708–1715. doi: 10.1182/blood-2003-01-0157. [DOI] [PubMed] [Google Scholar]
- Steiner T, et al. Dynamics of intraventricular hemorrhage in patients with spontaneous intracerebral hemorrhage: risk factors, clinical impact, and effect of hemostatic therapy with recombinant activated factor VII. Neurosurgery. 2006;59(4):767–773. doi: 10.1227/01.NEU.0000232837.34992.32. discussion 773–4. [DOI] [PubMed] [Google Scholar]
- Stein SC, Smith DH. Coagulopathy in traumatic brain injury. Neurocrit. Care. 2004;1(4):479–488. doi: 10.1385/NCC:1:4:479. [DOI] [PubMed] [Google Scholar]
- Stein SC, et al. Delayed brain injury after head trauma: significance of coagulopathy. Neurosurgery. 1992;30(2):160–165. doi: 10.1227/00006123-199202000-00002. [DOI] [PubMed] [Google Scholar]
- Stein SC, et al. Delayed and progressive brain injury in closed-head trauma: radiological demonstration. Neurosurgery. 1993;32(1):25–30. doi: 10.1227/00006123-199301000-00004. discussion 30–1. [DOI] [PubMed] [Google Scholar]
- Tanno H, et al. Breakdown of the blood–brain barrier after fluid percussive brain injury in the rat. Part 1: distribution and time course of protein extravasation. J. Neurotrauma. 1992a;9(1):21–32. doi: 10.1089/neu.1992.9.21. [DOI] [PubMed] [Google Scholar]
- Tanno H, et al. Breakdown of the blood–brain barrier after fluid percussion brain injury in the rat: Part 2: effect of hypoxia on permeability to plasma proteins. J. Neurotrauma. 1992b;9(4):335–347. doi: 10.1089/neu.1992.9.335. [DOI] [PubMed] [Google Scholar]
- Thurman DJ, et al. Traumatic brain injury in the United States: a public health perspective. J. Head Trauma Rehabil. 1999;14(6):602–615. doi: 10.1097/00001199-199912000-00009. [DOI] [PubMed] [Google Scholar]
- White CE, et al. Effects of recombinant activated factor VII in traumatic nonsurgical intracranial hemorrhage. Curr. Surg. 2006;63(5):310–317. doi: 10.1016/j.cursur.2006.04.013. [DOI] [PubMed] [Google Scholar]
- Zaaroor M, Bar-Lavie Y. The use of recombinant factor VIIa in head injury: a report of five cases. Semin Hematol. 2004;41(Supplement 1):175–176. [Google Scholar]