

Summary
Diet has a substantial impact on cellular metabolism and physiology. Animals must sense different food sources and utilize distinct strategies to adapt to diverse diets. Here we show that C. elegans lifespan is regulated by their adaptive capacity to different diets, which is controlled by alh-6, a conserved proline metabolism gene. alh-6 mutants age prematurely when fed an E. coli OP50 but not HT115 diet. Remarkably, this diet-dependent aging phenotype is determined by exposure to food during development. Mechanistically, alh-6 mutation triggers diet-induced mitochondrial defects and increased generation of ROS, likely due to accumulation of its substrate 1-pyrroline-5-carboxylate. We also identify that neuromedin U receptor signaling is essential for diet-induced mitochondrial changes and premature aging. Moreover, dietary restriction requires alh-6 to induce longevity. Collectively, our data reveal a novel mechanism that animals employ to cope with potential dietary insults and uncover an unprecedented example of lifespan regulation by dietary adaptation.
Introduction
Animals in the wild are exposed to a variety of food resources, which can have a substantial impact on their metabolism and physiology. As such, intricate homeostatic mechanisms have evolved to facilitate adaptation to even subtle changes in diet composition. It is conceivable that adaptation failure could lead to highly deleterious effects on animal physiology. However, the mechanisms facilitating dietary adaptation and an understanding of how disruption of these pathways contributes to complex phenotypes such as aging remain elusive.
Aging is perhaps the most complex physiological phenotype, which is subject to regulation by both intrinsic cues, such as genes (Kenyon, 2010), and environmental cues, such as diet (Fontana et al., 2010). Dietary composition has a great impact on the rate of animal aging (Grandison et al., 2009), however in most cases, animals are capable of living similarly well on different diets (Brooks et al., 2009). There are two possibilities that can explain this: first, these diets simply have no impact on the aging process or second, some of these diets have the potential to be harmful to the organismal rates of aging, but animals employ specific adaptive strategies to cope with those potential insults, thereby ensuring survival. Considering the significant impact of different diets on animal metabolism and physiology (Brooks et al., 2009; Soukas et al., 2009), it is likely that the latter hypothesis is more practical from an evolutionarily perspective.
C. elegans is a well-established organism for studying the mechanisms that govern the rate of aging and dietary effects on animal physiology. In the laboratory, C. elegans can utilize a variety of bacteria strains as their food source (Coolon et al., 2009; Shtonda and Avery, 2006), the most common being the E. coli B strain OP50 and the K-12 strain HT115. Different bacterial diets can influence multiple life history traits of C. elegans, such as development (Coolon et al., 2009; MacNeil et al., 2013; Shtonda and Avery, 2006), reproduction (Coolon et al., 2009), fat metabolism (Brooks et al., 2009; Soukas et al., 2009) and lifespan (Coolon et al., 2009; MacNeil et al., 2013; Maier et al., 2010; Soukas et al., 2009). When fed either of the standard laboratory diets, wild type C. elegans live similarly well, as evidenced by their similar development, reproduction and lifespan (Brooks et al., 2009). However, because these two diets differentially impact metabolism (Brooks et al., 2009; Soukas et al., 2009), we hypothesized that these two different diets may have the potential to differentially affect lifespan, but intrinsic homeostatic mechanisms facilitate dietary adaptation and ensure normal lifespan.
Previous studies on the mechanisms that govern the rate of aging have focused on either the genetic or environmental causes of lifespan determination. In our current study, we report that the rate of aging is regulated by an organisms’ adaptive capacity to diet. We have discovered that a conserved proline metabolic gene alh-6 helps C. elegans adapt to some diets to ensure normal lifespan. We also uncover two fundamental components of this homeostatic response that define this novel adaptive mechanism; first, the preservation of mitochondrial structure and functional homeostasis and second, neuromedin U receptor signaling, which is essential for integrating certain dietary cues. We propose that adaptive capacity to diet may influence multiple aspects of animal physiology, which ultimately converge upon lifespan.
Results
alh-6 mutations accelerate aging without affecting development
SKN-1 is the worm ortholog of the mammalian NRF family of transcription factors that function to defend against exogenous and endogenous cellular stresses including oxidative stress, proteostasis and starvation (An and Blackwell, 2003; Li et al., 2011; Paek et al., 2012; Wang et al., 2010). In C. elegans, SKN-1 activation in two neurons has been shown to promote lifespan extension resulting from caloric restriction (Bishop and Guarente, 2007). In a genetic screen for mutations that activate SKN-1, we identified a complementation group defined by three independent recessive alleles of alh-6 (Figure 1A), which post-developmentally activated SKN-1 in muscle tissue (Figure S1A–S1C). Feeding RNAi targeting alh-6 phenocopied the activation of SKN-1 reporter in muscle (Figure S1D). alh-6 encodes the C. elegans 1-pyrroline-5-carboxylate dehydrogenase (P5CDH), an evolutionarily conserved mitochondrial enzyme catalyzing the second step of proline catabolism, from 1-pyrroline-5-carboxylate (P5C) to glutamate (Figure S1E). Based on ALH-6::GFP expression, ALH-6 is expressed in both pharyngeal and body wall muscle (Figure S1F and S1G). In addition, ALH-6 expression is also observed in some neuronal cells (Figure S1H).
Figure 1. Mutation of alh-6 accelerates C. elegans aging when fed the standard laboratory diet.

(A) Gene structure of alh-6 and location of three isolated mutations. lax102 and lax105 are missense mutations. lax209 changes the splicing between exon 5 and exon 6, and generates a premature stop codon. (B) Lifespan of alh-6 mutant animals fed OP50. (C) Developmental rates. Synchronized L1 worms were added to OP50 plates and cultured at 20°C. The percentages of animals at different stages were counted 48 hours later. (D) Time courses of reproductive output. (E) Total brood sizes through life. (F) lipofuscin accumulation was measured by epifluorescence with DAPI channel (ex350/em445). (G) DIC images of heads of animals at indicative stages. Pharyngeal muscle degeneration was pointed by arrows. (H) Time courses of pumping rate as measured by pharyngeal contraction. (I) Time courses of movement as measured by thrash frequency in liquid droplet. (J) Lifespan of alh-6 transgenic worms fed OP50. Data are presented as mean ± SEM. * P < 0.05, ** P < 0.01, *** P < 0.001 versus wild type controls. See also Figure S1 and Table S1.
Because skn-1 is a well-established longevity gene in C. elegans, which is activated in several longevity mutants (Bishop and Guarente, 2007; Li et al., 2011; Paek et al., 2012; Tullet et al., 2008), we first measured the lifespan of the alh-6 mutants. Unexpectedly, mutations in alh-6, which activate SKN-1, led to a significant ~40% reduction of mean lifespan when animals were raised on the standard laboratory E. coli OP50 diet (Fig. 1B; Table S1). The shortened lifespan phenotype observed is not due to general sickness or developmental defects, because these animals are phenotypically normal until day 2 of adulthood, as measured by developmental rate (Figure 1C) and reproductive output (Figure 1D). Notably, the adult alh-6 mutants also prematurely exhibited other characteristic aging phenotypes. Reproductive decline is a universal hallmark of aging (Hughes et al., 2007) and we found that alh-6 mutants had reduced reproductive output from day 3 of adulthood and accordingly yielded a smaller total brood (Figure 1D and 1E). Lipofuscin pigment accumulation and pharyngeal muscle degeneration also occurred prematurely in alh-6 mutants, appearing by day 4 of adulthood (Figure 1F and 1G). In addition, pharyngeal pumping and movement, two established phenotypes that decline in an age-related manner, decreased prematurely from day 4 of adulthood (Figure 1H and 1I). To further confirm that alh-6 is a bone fide longevity gene, we generated an alh-6 overexpression strain and found that these animals lived longer than their non-transgenic siblings (Figure 1J, Table S1). Thus, we identify alh-6 as a novel regulator of the rate of aging. Upon reaching reproductive maturity, alh-6 mutant animals age rapidly and as such, represent a new model of adult premature aging.
Because activation of SKN-1 canonically leads to longevity, it is unlikely that the observed SKN-1 activation and premature aging phenotypes are causally linked. As expected, skn-1 mutation was able to suppress the muscle SKN-1 activation (Figure S1I) but was unable to significantly reverse the shortened lifespan of alh-6 mutant animals (Figure S2A), thus uncoupling skn-1 from the premature aging phenotype.
alh-6 delays aging in a diet type-dependent manner
While performing RNAi feeding experiments (Figure S1C), we noticed that alh-6 mutant animals appeared much healthier than age matched siblings fed an OP50 E. coli B diet after day 4 of adulthood. Since feeding RNAi utilizes an E. coli K-12 strain HT115, we measured the lifespan of the alh-6 mutants fed HT115. In contrast to the OP50 E. coli B diet, alh-6 mutants exhibited a normal lifespan when fed an HT115 E. coli K-12 diet (Figure 2A; Table S1). These findings suggest that alh-6 may represent a mechanism that confers resistance to certain diet-induced premature aging and importantly, is not simply an essential component of organism survival. Consistent with the diet dependency of alh-6 function, alh-6 overexpression animals lived a similar lifespan as wild type controls when fed HT115 E. coli K-12 diet (Figure 2B, Table S1), in contrast to the longevity phenotype observed on the OP50 E. coli B diet. We also found that SKN-1 activation was observed in alh-6 mutant animals regardless of whether they were fed the OP50 or HT115 diet (Figure S2B), providing additional evidence that the SKN-1 transcriptional activation and the diet-induced shortened lifespan phenotypes are not linked.
Figure 2. alh-6 delays aging in a diet type-dependent manner.

(A) Lifespan of alh-6 mutants fed HT115. (B) Lifespan of alh-6 transgenic worms fed HT115. (C–E) Lifespan of alh-6 mutants fed other E. coli diets, including a K-12 strain HMS174 (C), B strain BL21 (D) and a B x K-12 hybrid stain HB101 (E). See also Figure S2 and Table S1.
OP50 (E. coli B) and HT115 (E. coli K-12) strains have been shown to differentially affect multiple aspects of C. elegans physiology but without an obvious impact on lifespan (Brooks et al., 2009). In light of the significantly different lifespan of alh-6 mutants on these two food sources, we hypothesized that wild type alh-6 confers an adaptive response that promotes animal survival on particular diets. To test this further we examined the lifespan of alh-6 mutants fed other E. coli strains, including BL21 (B strain), HMS174 (K-12 strain), and HB101 (B x K-12 hybrid). Remarkably, alh-6 mutants lived a normal lifespan when fed on HMS174 (K-12 strain) (Figure 2C; Table 1), but exhibited a shortened lifespan when fed BL21 (B strain) and an intermediary lifespan on an HB101 diet (B x K-12 hybrid strain) (Figure 2D and 2E; Table S1). Therefore, alh-6 confers C. elegans the adaptive capacity to survive on certain E. coli diets and disruption of this pathway leads to a shortened lifespan only when exposed to those particular diets. We used OP50 and HT115, the two major laboratory food sources for C. elegans, in the remainder of study to study the underlying mechanism.
Temporal requirements of bacterial diets on lifespan regulation
As demonstrated above, the effect of the OP50 diet on lifespan is uncovered when alh-6 mediated adaptation fails. Genetic factors can regulate lifespan by acting in specific stages of life. Disruption of the mitochondrial electron transport chain (ETC) has been shown to increase C. elegans lifespan only when it is crippled during development, while post-developmental inactivation of some essential genes, including protein synthesis components and the insulin/IGF-I signaling pathway, can lead to longevity (Curran and Ruvkun, 2007; Dillin et al., 2002a, 2002b; Rea et al., 2007). As such, we questioned if there is any timing requirement for diet to regulate lifespan. Specifically, we tested if the OP50 diet accelerated aging in alh-6 mutants continuously through life or at discrete life windows. To do this, alh-6 mutant worms were transferred from HT115 (normal lifespan) to OP50 (short lifespan) diets at different developmental stages (Figure 3E). Populations of alh-6 mutant animals transferred at L2 or L3 stages still exhibited the shortened lifespan phenotype (Figure 3A and 3B; Table S1), whereas mutants transferred at L4 stage lived a normal lifespan as compared to wild type animals (Figure 3C; Table S1). These data suggest that a developmental window between L3 to L4 is particularly important for diet-mediated lifespan regulation. To further test if exposure to OP50 diet during the L3 to L4 stages is sufficient to accelerate aging, alh-6 mutant worms were transferred from the short lifespan inducing OP50 diet to the normal lifespan promoting HT115 diet at the L4 larval stage. We found that these animals exhibited a normal lifespan as wild type worms (Figure 3D; Table S1). Thus, ingestion of E. coli OP50 diet during L3 to L4 stage is essential for initiating the diet-dependent premature aging program, but also requires the continuous exposure to that diet during adulthood.
Figure 3. Timing requirements of the lifespan reduction induced by the OP50 E. coli B diet.

(A–C) Lifespan of alh-6 mutants transferred from HT115 to OP50 diet at L2 (A), L3 (B), or L4 (C) stages. (D) Lifespan of alh-6 mutants transferred from OP50 to HT115 diet at L4 stages. (E) Schematic of lifespan analysis by dietary transfer. See also Table S1.
Activation of proline catabolism causes premature aging in alh-6 mutants fed OP50
How do mutations of alh-6 cause premature aging in a diet-dependent manner? Because alh-6 is a conserved proline catabolic gene, we hypothesized that proline catabolism may be differentially affected by OP50 and HT115 diets, which would differentially regulate lifespan in the context of the alh-6 mutation. Besides ALH-6/P5CDH, the only other enzyme in the proline catabolism pathway is proline dehydrogenase (PRODH/B0513.5) (Figure S1J) that catalyzes the formation of P5C from proline. We found that the expression of prodh/B0513.5 was differentially regulated upon feeding the OP50 or HT115 diet. Worms fed OP50 expressed higher levels of prodh than those fed a HT115 diet (Figure 4A), indicating the activation of proline catabolism by OP50 diet. The expression of alh-6 however was comparable between worms fed either diet (Figure 4A).
Figure 4. Activation of proline catabolism is responsible for the premature aging phenotype of alh-6 mutants.
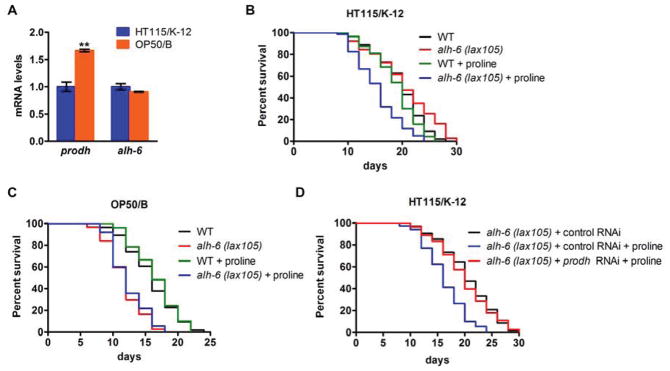
(A) Expression of prodh and alh-6 in wild type animals fed an OP50 or HT115 diet. (B–C) Lifespan of alh-6 mutants fed an HT115 (B) or OP50 (C) diet with proline supplement. (D) Lifespan of alh-6 mutants fed a control or prodh RNAi HT115 diet with proline supplement. See also Table S1.
We next asked if activation of proline catabolism is the reason for premature aging in alh-6 mutant animals. As such, we added proline to the diet of wild type and alh-6 mutant worms fed OP50 and HT115 dietary regimes, and examined the effects of proline supplementation on lifespan. Remarkably, proline supplementation significantly reduced the lifespan of alh-6 mutant worms fed HT115 diet, but not wild type controls fed the same diet (Figure 4B, Table S1), indicating that activation of proline catabolism is sufficient to shorten lifespan in the context of alh-6 mutation. In addition, when fed the OP50 diet, alh-6 mutant animals lived a similarly short lifespan, regardless of proline supplementation or not (Figure 4C, Table S1). These data indicate that the OP50 diet and proline supplementation may act in the same pathway to reduce lifespan. These findings also support the model that activation of proline catabolism due to OP50 diet intake leads to premature aging in C. elegans mutants unable to complete the terminal step of proline catabolism via ALH-6.
Activation of proline catabolism would lead to P5C accumulation in the context of alh-6 mutation, which we hypothesized is responsible for observed aging phenotype. If it is the case, knockdown of prodh should block the generation of P5C and therefore reverse the shortened lifespan of alh-6 animals fed a HT115 diet supplemented with exogenous proline. We found that, without prodh to initiate the production of P5C from proline, alh-6 mutant animals are no longer short-lived on the HT115 + proline diet (Figure 4D, Table S1). Thus, the accumulation of the alh-6 substrate P5C, a toxic intermediate of proline catabolism, is the likely reason to initiate the premature aging phenotype.
alh-6 preserves mitochondrial homeostasis in response to OP50 diet
What is the cellular basis of diet-dependent premature aging of alh-6 mutants? As demonstrated above, accumulation of alh-6 substrate P5C leads to the premature aging phenotype. Intriguingly, P5C has been reported to affect mitochondrial homeostasis across phylogeny (Graaff et al., 2004; Miller et al., 2009; Nomura and Takagi, 2004; Yoon et al., 2004). As such, we hypothesized that alh-6 might regulate mitochondrial homeostasis in a diet-dependent manner. An examination of mitochondrial morphology in developing alh-6 mutant larval animals revealed normal filamentous mitochondria when fed either an OP50 or HT115 diet (Figure S3A). Remarkably, adult alh-6 mutant worms fed an OP50 but not a HT115 diet exhibited a more spherical shaped mitochondrial morphology (Figure 5A), a phenotype usually associated with mitochondrial stress (Ahmad et al., 2013). Consistently, mitochondrial function, as measured by ATP production, was only impaired in adult alh-6 mutants fed an OP50 diet when compared to wild-type controls (Figure 5B and Figure S3B). These data suggest that alh-6 promotes mitochondrial preservation during adulthood in response to the OP50 diet, which links diet to mitochondrial homeostasis.
Figure 5. alh-6 preserves mitochondrial and ROS homeostasis in response to the OP50 E. coli B diet.
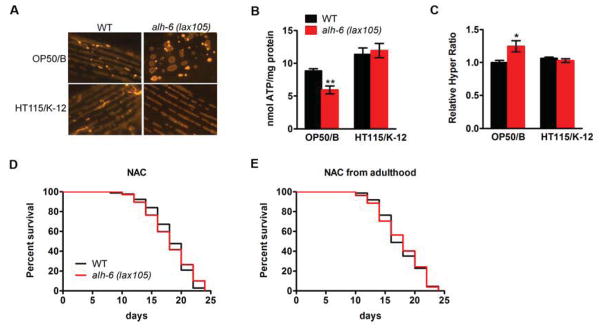
(A) Mitochondrial morphology as revealed by muscle mitochondrial targeted RFP in day 5 adult worms. (B) ATP production from day 5 adult animals fed an OP50 or HT115 diet. (C) The ratio of oxidized/reduced HyPer in day 5 adult animals fed an OP50 or HT115 diet. (D–E) lifespan of OP50 feeding alh-6 mutants treated with NAC from hatching (D) or adulthood (E). Data are presented as mean ± SEM. *P < 0.05, **P < 0.01 versus respective controls. See also Figure S3 and Table S1.
Reactive oxygen species contribute to diet-induced premature aging
Impaired mitochondrial function, such as defects in the mitochondrial ETC, are associated with increased reactive oxygen species (ROS) production in C. elegans (Lee et al., 2010; Yang and Hekimi, 2010). We predicted that the dysfunctional mitochondria of adult alh-6 mutants fed an OP50 diet would generate more ROS. We measured, in vivo, the ratio of oxidized/reduced HyPer, an established indicator of hydrogen peroxide levels (Back et al., 2012; Ocampo et al., 2012), and found that alh-6 mutants fed an OP50 diet exhibited higher ROS levels only during adulthood (Figure 5C and Figure S3C), which correlates with the mitochondrial morphology and ATP production changes.
ROS plays a complicated and controversial role in aging. Low and transient production of ROS benefits longevity (Lee et al., 2010; Yang and Hekimi, 2010; Zarse et al., 2012), whereas high levels of ROS can be deleterious (Melov et al., 2000; Van Raamsdonk and Hekimi, 2012). If the induction of ROS in alh-6 mutants fed an OP50 diet was essential for the observed lifespan reduction, then removal of these reactive species should result in a normal lifespan. To test this, we quenched ROS by feeding the antioxidants N-acetylcysteine (NAC) and Vitamin C to the alh-6 mutants on the OP50 diet. In support of the ROS-dependent aging model, both NAC and Vitamin C treatment completely reversed the shortened lifespan of alh-6 mutants fed an OP50 diet (Figure 5D and Figure S3D; Table S1). Since increased ROS was only observed during adulthood, we also treated worms with NAC post-developmentally. Strikingly, adult-specific treatment with antioxidants was capable of fully reversing the shortened lifespan of alh-6 mutants (Figure 5E; Table S1), suggesting that the adult-onset of ROS production is responsible for the accelerated aging phenotype. In conjunction with the conserved role for P5C in mitochondrial homeostasis in eukaryotes (Graaff et al., 2004; Miller et al., 2009; Nomura and Takagi, 2004; Yoon et al., 2004), our data suggest that alh-6 maintains mitochondrial and ROS homeostasis during adulthood, likely through removal of P5C, thus delaying aging in response to the E. coli OP50 diet. Interestingly, we also found that antioxidant NAC treatment had no effect on muscle SKN-1 activation (Figure S2C), again uncoupling SKN-1 activation from the observed aging phenotypes.
NMUR-1 signaling is required for the diet-induced mitochondrial and aging phenotypes
Because the observed mitochondrial and aging phenotypes are dependent on the OP50 diet, it is conceivable that C. elegans employ specific mechanisms to integrate and process information derived from OP50 diet and initiates the downstream mitochondrial and aging responses. C. elegans sense changes to their environment through a simple yet elegant neuroendocrine system. We asked if neuronal signaling participates in the decoding and processing of different dietary cues, and is required for the mitochondrial and aging phenotypes of alh-6 mutants on the OP50 diet. The neuromedin U receptor (NMUR) family functions to regulate many food-associated activities of invertebrates (Melcher and Pankratz, 2005) and mammals (Hanada et al., 2004; Howard et al., 2000). NMUR-1 is the worm homolog of mammalian NMUR, which is expressed in neurons and processes dietary cues to influence lifespan (Maier et al., 2010). We thus tested if NMUR-1 signaling was required for the mitochondrial response in alh-6 mutants. If a neuroendocrine circuit were required, then nmur-1 deficiency would block the cellular and organismal response from OP50 diet and thus abrogate the mitochondrial and aging phenotypes. In support of this hypothesis, without functional NMUR-1, adult alh-6 mutants fed an OP50 diet no longer exhibited mitochondrial morphology defects (Figure 6A) or functional decline, as measured by ATP production (Figure 6B). Consistently, in the absence of nmur-1, ROS levels in adults were comparable between wild type and alh-6 mutant animals fed OP50 diet (Figure 6C). Most importantly, we found that nmur-1; alh-6 double mutants lived a normal lifespan as nmur-1 mutants (Figure 6D; Table S1). Together, we conclude that NMUR-1 signaling is essential for processing OP50 dietary information and initiating the mitochondrial response, which in turn influences lifespan.
Figure 6. nmur-1 mediates dietary effects on mitochondrial function and lifespan in alh-6 mutants.

(A–C) Effect of nmur-1 loss on mitochondrial morphology (A), ATP production (B) and HyPer ratio (C) of adult alh-6 mutants fed an OP50 diet. (D) Mutation of nmur-1 reverses shortened lifespan of alh-6 mutants fed an OP50 diet. Data are presented as mean ± SEM. *P < 0.05 versus nmur-1 control. See also Table S1.
alh-6 is required for longevity induced by dietary restriction
Finally, we evaluated the genetic interactions between alh-6 and several well-known longevity models, including insulin/IGF-1 daf-2 pathways (Kenyon et al., 1993; Kimura et al., 1997), germline loss model in glp-1 mutants (Arantes-Oliveira et al., 2002) and a genetic model of dietary restriction (DR) with eat-2 mutant animals (Lakowski and Hekimi, 1998). We crossed alh-6 mutants to these longevity model animals, and tested their resulting lifespan. We found that daf-2 and glp-1 mutations were still able to induce longevity in the alh-6 mutant background (Figure 7A and 7B, Table S1), indicating the dispensability of alh-6 on those longevity pathways. By contrast, the eat-2 mutation failed to increase lifespan of alh-6 mutant animals (Figure 7C, Table S1), suggesting that alh-6 is specifically required for lifespan extension mediated by DR, which is consistent with alh-6 playing a central role in organismal response to changes in diet.
Figure 7. alh-6 is specifically required for lifespan extension mediated by DR.

(A–C) Lifespan of daf-2 (A), glp-1 (B) and eat-2 (C) longevity mutants in the background of alh-6 mutation. (D) Model for aging regulation by dietary adaptation: Information from an OP50 diet is processed by NMUR-1 signaling. In wild type C. elegans, proline catabolism is activated and ALH-6 facilitates the efficient catabolism of P5C, thereby preserving mitochondrial function and promoting survival. Without functional ALH-6, harmful P5C accumulates that affects mitochondrial homeostasis and ROS production, eventually accelerating organismal aging. See also Table S1.
Discussion
Our data reveal a novel mechanism utilized by an organism to maintain physiological homeostasis when challenged by exposure to different diets. This response signals mitochondrial adaptation through the coordination of neuronal and metabolic tissues. Food is essential for life as an energy source, but its composition may have potential negative effects on animal physiology (Gracida and Eckmann, 2013; Grandison et al., 2009). Here we show that diet can impose highly adverse effects on the rate of animal aging when adaptation is compromised, highlighting that an optimal balance between these conflicting cues is critical for animal survival. Wild-type C. elegans can develop and live similar lifespan on the OP50 or HT115 diet, indicating an efficient adaptive response to these bacterial food sources. We propose that, in response to the OP50 diet, proline catabolism is activated and ALH-6 facilitates the efficient metabolism of P5C, thereby preserving mitochondrial homeostasis and ensuring a normal rate of aging. Without functional ALH-6, the difference(s) between OP50 and HT115 strains is unmasked, P5C is accumulated, mitochondrial homeostasis is disrupted, and worms age rapidly (Figure 7D).
Aging is a complex phenotype that is modulated by both environmental and intrinsic cues, such as diets and genes, respectively. During past decades, the genetic study of aging has greatly advanced our knowledge on the biological mechanisms that influence the rate of animal aging. Several conserved pathways have been identified and characterized, such as insulin/IGF-1 signaling, mitochondrial respiration and germline signaling pathways (Kenyon, 2010). All these aging pathways represent intrinsic mechanisms, which can affect the rate of aging regardless of environmental input. However, animals in the wild face a much more challenging environment and must adapt to changes in the abundance and composition of diverse diets to ensure survival. Animals that can successfully utilize multiple food sources are at an evolutionary advantage as they can adapt to more confined environments when their optimal food source is depleted. Our findings uncover a novel type of aging modulator, which impacts the rate of aging in a diet type-dependent manner and mechanistically defines a new gene-environment interaction pair. The function of genes, such as alh-6, can only be uncovered under particular dietary conditions. As such, some current methods for aging research may be less valuable for identifying them. For example, genomic RNAi screen in C. elegans would be incapable of pulling out these genes, because the E. coli diet used for RNAi is a K-12 strain HT115. Generating alternative feeding RNAi diet like E. coli B strain will be valuable for future aging studies.
Metabolism is a central node for the aging process. Both glucose and lipid metabolism have been implicated in modulation of aging (Lee et al., 2009; Schulz et al., 2007; Wang et al., 2008). Amino acids represent another essential pool of nutrients that can be utilized for cellular energy production. As such, it is not surprising that amino acid metabolism, similar to carbohydrate and lipid metabolism, plays an equally important role in aging. For example, methionine has been proved to be a critical nutrient in lifespan regulation by CR (Cabreiro et al., 2013; Grandison et al., 2009). More interestingly, enzymes involved in amino acid metabolism have been reported to affect the dietary response in C. elegans (Watson et al., 2013). Here, we propose that proline metabolism significantly impacts the aging process in a diet-type dependent manner. We find that the proline catabolism enzyme ALH-6 modulates diet-dependent mitochondrial homeostasis and the rate of aging, likely through removal of its substrate P5C. Recently, the expression of prodh, another proline catabolism enzyme generating P5C, is found to be increased in insulin/IGF-1 mutant C. elegans and lead to transient ROS upregulation and longevity (Zarse et al., 2012). Our study supports the importance of proline catabolism in lifespan regulation and points to P5C as a possible critical regulator of mitochondrial ROS homeostasis and aging. Therefore, proline is a unique amino acid, the catabolism of which is extremely important for mitochondrial homeostasis and the rate of aging, which must be under tight regulation.
Mitochondrial quality and function are under strict genetic regulation that is essential for the rate of aging (Curran and Ruvkun, 2007; Dillin et al., 2002b; Lee et al., 2003). Our finding that alh-6 links diet to mitochondrial homeostasis highlights that mitochondrial quality control during aging is also subject to regulation by environmental cues such as diet type. ROS generated from mitochondria is an important player of the aging process. The free radical theory of aging states that animal aging results from ROS accumulation over time (Harman, 1956). This theory however has recently been challenged as a series of studies point to a beneficial role for ROS in longevity (Lee et al., 2010; Yang and Hekimi, 2010; Zarse et al., 2012). A possible explanation is that ROS has dual roles on aging: low and transient production of ROS benefits longevity, whereas uncontrolled high levels of ROS can cause accelerated aging (Hekimi et al., 2011; Van Raamsdonk and Hekimi, 2012). Our finding that ROS induction contributes to observed premature aging adds new weight to this model and highlights that ROS levels must be under tight control during aging.
Another interesting finding of our study is that exposure to the OP50 diet only at specific stages of development in combination with continued adult exposure to that diet is critical for impacting the rate of adult aging. This is divergent from the current caloric restriction (CR) dogma observed in some model organisms such as D. melanogaster, where CR can increase lifespan at any stage of life (Mair et al., 2003). In C. elegans, initiation of DR at either L4/Young adult (Bishop and Guarente, 2007; Panowski et al., 2007) or later after reproduction (Greer et al., 2007) is able to induce a longevity phenotype. Notably, mitochondrial ETC regulation during the exact same developmental stages is essential for determining the rate of aging in later life (Dillin et al., 2002b; Rea et al., 2007). Thus, it is possible that a shared mechanism between the two, exists and represents an event that is established during developmental stages and impacts mitochondrial homeostasis and aging in later life. However, our observed diet-induced aging phenotype requires constant exposure to the pro-aging diet even after the developmental window is closed, while inactivation of ETC can extend lifespan even when knock-down of ETC component is restored in adulthood (Dillin et al., 2002b; Rea et al., 2007), indicating an important distinction between these mechanisms on regulating lifespan.
Dietary differences can be decoded by animals through either simple discrimination of nutrient compositions by metabolic tissues, or neuronal sensing of specific dietary cues. Neuronal systems integrate environmental information and communicate to peripheral tissues. The essential role for nmur-1 in observed mitochondrial and aging phenotypes establishes a neuronal circuit required for communicating dietary information and promoting the downstream mitochondrial response. Further work studying the mechanisms linking NMUR-1 to this mitochondrial response will provide valuable insight into the regulation of mitochondrial homeostasis in response to diet changes.
In summary, we have uncovered an unprecedented example of lifespan regulation by a novel gene and environment interaction: gene-mediated adaptive capacity to specific diet regulates lifespan. This is a novel diet-induced adult-premature aging model. Because NMUR-1 and proline catabolism pathways are highly conserved throughout evolution, we propose that metazoan lifespan and dietary adaptation could be governed by shared mechanisms.
Experimental Procedures
C. elegans and E. coli strains utilized in this study
C. elegans were cultured using standard techniques (Brenner, 1974). The following strains were used: wild type N2 Bristol, CB4856 (HW), SPC320[alh-6 (lax102)], SPC321[alh-6 (lax105)], SPC322[alh-6 (lax209)], RB1288[nmur-1 (ok1387)], CL2166[gst4-p::gfp], the HyPer expression strain jrIs1[Prpl-17::HyPer] (Back et al., 2012), Muscle mitochondrial targeted red fluorescent protein (RFP) strain [myo-3p::TOM20::mRFP], DR1572[daf-2 (e1368)], CF1903[glp-1 (e2141)], DA1113[eat-2 (ad1113)], VC1772[skn-1 (ok2315) IV/nTi[qIs51] (IV; V)]. SPC326[alh-6p::alh-6::gfp] transgenic animals were constructed by standard cloning and microinjection techniques. Double and triple mutants were generated by standard genetic techniques.
E. coli strains used: OP50 (Brenner, 1974), HT115(DE3) [F- mcrA mcrB IN(rrnD-rrnE)1 lambda- rnc14::Tn10 λ(DE3)] (Timmons et al., 2001), HMS174(DE3) [F- recA1 hsdR(rK12- mK12+) λ(DE3)], BL21(DE3)[F– ompT gal dcm lon hsdSB(rB− mB−) λ(DE3)], HB101[F− mcrB mrr hsdS20(rB− mB−) recA13 leuB6 ara-14 proA2 lacY1 galK2 xyl-5 mtl-1 rpsL20(SmR) glnV44 λ−].
Mapping of alh-6 mutations
A complementation group containing three recessive alleles (lax102, lax105 and lax209) that activated SKN-1transcriptional activity reporter gst-4p::gfp in muscle were isolated in a Ethyl methanesulfonate (EMS) screen (Paek et al., 2012). The mutations were mapped to the left arm of chromosome II by standard SNP mapping. Deep sequencing of genomes revealed that only one genetic locus in that region contained mutations changing amino acid sequences in all three mutants. The genetic locus corresponds to gene alh-6. RNAi targeting alh-6 confirmed that loss of function of alh-6 activated muscle SKN-1.
Lifespan analysis
Lifespan assays were performed as previously described (Paek et al., 2012). Briefly, synchronized eggs were added to NGM plates seeded with different E. coli strains. The worms were kept at 20°C and transferred everyday during reproductive period. Worms that died of vulva burst, bagging, or crawling off the plates were censored. For NAC and Vitamin C treatment, concentrated NAC or Vitamin C (Sigma) were added to NGM plates at a final concentration of 5mM NAC or 10mM Vitamin C. For proline supplementation, proline was added to plates at a final concentration of 40mM.
ROS (Hydrogen peroxide) measurement
Worms of indicated genotypes and developmental stages were mounted on slides. The oxidized and reduced HyPer of individual worm was excited with green fluorescent protein (GFP) and cyan fluorescent protein (CFP) channel, respectively. Fluorescent density was measured by using ImageJ software. The hydrogen peroxide levels were measured as the ratio of oxidized to reduced HyPer intensity.
ATP measurements
Approximately 100 worms of indicated genotype and developmental stages were collected, washed and boiled in M9 buffer for 15 minutes. After centrifugation, the supernatant was used for protein concentration determination (Bradford assay) and ATP levels measured (ENLITEN® ATP Assay, Promega). ATP levels were normalized to total protein content.
qRT-PCR
Approximately 200 worms of indicated genotype and developmental stages were collected, washed in M9 buffer and then homogenized in Trizol reagent (Invitrogen). RNA was extracted according to manufacturer’s protocol. DNA contamination was digested with DNase I (New England Biolabs) and subsequently RNA was reverse-transcribed to cDNA by using the SuperScript® III First-Strand Synthesis System (Life Technologies). Quantitative PCR was performed by using SYBR Green (BioRad). The expression of snb-1 was used to normalize samples.
Statistics
Data are presented as mean ± SEM. Lifespan data were analyzed by using log-rank (Mantel-Cox) test. P < 0.05 was considered as significant. Other data (non lifespan) were analyzed by using unpaired student t test.
Supplementary Material
Highlights.
A conserved proline metabolic gene alh-6 delays aging in a diet type-dependent manner.
alh-6 mutation leads to mitochondrial dysfunction induced by certain diets.
NMUR-1 signaling is essential for diet-induced premature aging in alh-6 mutants.
Dietary restriction induced longevity requires alh-6.
Acknowledgments
We thank T. Nguyen and E. Roh for technical assistance; Dr. Caleb Finch and members of the Curran laboratory for helpful discussions; J. Lo, H. Dalton and D. Lynn for critical reading of the manuscript. The HyPer expression strain was a gift from Dr. B. Braeckman. The muscle mitochondria targeted RFP strain was a gift from Dr. Paul Sternberg. Some strains were provided by the CGC, which is funded by NIH Office of Research Infrastructure Programs (P40 OD010440). S.P.C. is Ellison Medical Foundation New Scholar in Aging; this work was funded by the NIH (AG032308) and the Ellison Medical Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahmad T, Aggarwal K, Pattnaik B, Mukherjee S, Sethi T, Tiwari BK, Kumar M, Micheal a, Mabalirajan U, Ghosh B, et al. Computational classification of mitochondrial shapes reflects stress and redox state. Cell Death Dis. 2013;4:e461. doi: 10.1038/cddis.2012.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An JH, Blackwell TK. SKN-1 links C. elegans mesendodermal specification to a conserved oxidative stress response. Genes Dev. 2003;17:1882–1893. doi: 10.1101/gad.1107803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arantes-Oliveira N, Apfeld J, Dillin A, Kenyon C. Regulation of life-span by germ-line stem cells in Caenorhabditis elegans. Science. 2002;295:502–505. doi: 10.1126/science.1065768. [DOI] [PubMed] [Google Scholar]
- Back P, De Vos WH, Depuydt GG, Matthijssens F, Vanfleteren JR, Braeckman BP. Exploring real-time in vivo redox biology of developing and aging Caenorhabditis elegans. Free Radic Biol Med. 2012;52:850–859. doi: 10.1016/j.freeradbiomed.2011.11.037. [DOI] [PubMed] [Google Scholar]
- Bishop Na, Guarente L. Two neurons mediate diet-restriction-induced longevity in C. elegans. Nature. 2007;447:545–549. doi: 10.1038/nature05904. [DOI] [PubMed] [Google Scholar]
- Brenner S. The genetics of Caenorhabditis elegnns. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks KK, Liang B, Watts JL. The influence of bacterial diet on fat storage in C. elegans. PLoS One. 2009;4:e7545. doi: 10.1371/journal.pone.0007545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabreiro F, Au C, Leung KY, Vergara-Irigaray N, Cochemé HM, Noori T, Weinkove D, Schuster E, Greene NDE, Gems D. Metformin retards aging in C. elegans by altering microbial folate and methionine metabolism. Cell. 2013;153:228–239. doi: 10.1016/j.cell.2013.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coolon JD, Jones KL, Todd TC, Carr BC, Herman MA. Caenorhabditis elegans genomic response to soil bacteria predicts environment-specific genetic effects on life history traits. PLoS Genet. 2009;5:e1000503. doi: 10.1371/journal.pgen.1000503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curran SP, Ruvkun G. Lifespan regulation by evolutionarily conserved genes essential for viability. PLoS Genet. 2007;3:e56. doi: 10.1371/journal.pgen.0030056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillin A, Crawford DK, Kenyon C. Timing requirements for insulin/IGF-1 signaling in C. elegans. Science. 2002a;298:830–834. doi: 10.1126/science.1074240. [DOI] [PubMed] [Google Scholar]
- Dillin A, Hsu A, Arantes-Oliveira N, Lehrer-Graiwer J, Hsin H, Fraser AG, Kamath RS, Ahringer J, Kenyon C. Rates of Behavior and Aging Specified by Mitochondrial Function During Development. Science. 2002b;298:2398–2401. doi: 10.1126/science.1077780. [DOI] [PubMed] [Google Scholar]
- Fontana L, Partridge L, Longo VD. Extending healthy life span--from yeast to humans. Science. 2010;328:321–326. doi: 10.1126/science.1172539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Der Graaff E, Kunze R, Frommer WB. The Role of D 1 -Pyrroline-5-Carboxylate Dehydrogenase in Proline Degradation. Plant Cell. 2004;16:3413–3425. doi: 10.1105/tpc.104.023622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gracida X, Eckmann CR. Fertility and Germline Stem Cell Maintenance under Different Diets Requires nhr-114/HNF4 in C. elegans. Curr Biol. 2013;23:607–613. doi: 10.1016/j.cub.2013.02.034. [DOI] [PubMed] [Google Scholar]
- Grandison RC, Piper MDW, Partridge L. Amino-acid imbalance explains extension of lifespan by dietary restriction in Drosophila. Nature. 2009;462:1061–1064. doi: 10.1038/nature08619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer EL, Dowlatshahi D, Banko MR, Villen J, Hoang K, Blanchard D, Gygi SP, Brunet A. An AMPK-FOXO pathway mediates longevity induced by a novel method of dietary restriction in C. elegans. Curr Biol. 2007;17:1646–1656. doi: 10.1016/j.cub.2007.08.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanada R, Teranishi H, Pearson JT, Kurokawa M, Hosoda H, Fukushima N, Fukue Y, Serino R, Fujihara H, Ueta Y, et al. Neuromedin U has a novel anorexigenic effect independent of the leptin signaling pathway. Nat Med. 2004;10:1067–1073. doi: 10.1038/nm1106. [DOI] [PubMed] [Google Scholar]
- Harman D. Aging: a theory based on free radical and radiation chemistry. J Gerontol. 1956;11:298–300. doi: 10.1093/geronj/11.3.298. [DOI] [PubMed] [Google Scholar]
- Hekimi S, Lapointe J, Wen Y. Taking a “good” look at free radicals in the aging process. Trends Cell Biol. 2011;21:569–576. doi: 10.1016/j.tcb.2011.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard AD, Wang R, Pong S, Mellin TN, Strack A, Guan X, Zeng Z, Williams DL, Feighner SD, Nunes CN, et al. Identification of receptors for neuromedin U and its role in feeding. Nature. 2000;406:4–8. doi: 10.1038/35017610. [DOI] [PubMed] [Google Scholar]
- Hughes SE, Evason K, Xiong C, Kornfeld K. Genetic and Pharmacological Factors That Influence Reproductive Aging in Nematodes. PLoS Genet. 2007;3:e25. doi: 10.1371/journal.pgen.0030025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenyon CJ. The genetics of ageing. Nature. 2010;464:504–512. doi: 10.1038/nature08980. [DOI] [PubMed] [Google Scholar]
- Kenyon CJ, Chang J, Gensch E, Runder A, Tabtlang R. A C. elegans mutant that lives twice as long as wild type. Nature. 1993;366:461–464. doi: 10.1038/366461a0. [DOI] [PubMed] [Google Scholar]
- Kimura KD, Tissenbaum Ha, Liu Y, Ruvkun G. daf-2, an Insulin Receptor-Like Gene That Regulates Longevity and Diapause in Caenorhabditis elegans. Science. 1997;277:942–946. doi: 10.1126/science.277.5328.942. [DOI] [PubMed] [Google Scholar]
- Lakowski B, Hekimi S. The genetics of caloric restriction in Caenorhabditis elegans. Proc Natl Acad Sci U S A. 1998;95:13091–13096. doi: 10.1073/pnas.95.22.13091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S-J, Murphy CT, Kenyon C. Glucose shortens the life span of C. elegans by downregulating DAF-16/FOXO activity and aquaporin gene expression. Cell Metab. 2009;10:379–391. doi: 10.1016/j.cmet.2009.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S-J, Hwang AB, Kenyon C. Inhibition of respiration extends C. elegans life span via reactive oxygen species that increase HIF-1 activity. Curr Biol. 2010;20:2131–2136. doi: 10.1016/j.cub.2010.10.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SS, Lee RYN, Fraser AG, Kamath RS, Ahringer J, Ruvkun G. A systematic RNAi screen identifies a critical role for mitochondria in C. elegans longevity. Nat Genet. 2003;33:40–48. doi: 10.1038/ng1056. [DOI] [PubMed] [Google Scholar]
- Li X, Matilainen O, Jin C, Glover-Cutter KM, Holmberg CI, Blackwell TK. Specific SKN-1/Nrf Stress Responses to Perturbations in Translation Elongation and Proteasome Activity. PLoS Genet. 2011;7:e1002119. doi: 10.1371/journal.pgen.1002119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacNeil LT, Watson E, Arda HE, Zhu LJ, Walhout AJM. Diet-Induced Developmental Acceleration Independent of TOR and Insulin in C. elegans. Cell. 2013;153:240–252. doi: 10.1016/j.cell.2013.02.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier W, Adilov B, Regenass M, Alcedo J. A neuromedin U receptor acts with the sensory system to modulate food type-dependent effects on C. elegans lifespan. PLoS Biol. 2010;8:e1000376. doi: 10.1371/journal.pbio.1000376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mair W, Goymer P, Pletcher SD, Partridge L. Demography of dietary restriction and death in Drosophila. Science. 2003;301:1731–1733. doi: 10.1126/science.1086016. [DOI] [PubMed] [Google Scholar]
- Melcher C, Pankratz MJ. Candidate Gustatory Interneurons Modulating Feeding Behavior in the Drosophila Brain. PLoS Biol. 2005;3:e305. doi: 10.1371/journal.pbio.0030305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melov S, Ravenscroft J, Malik S, Gill MS, Walker DW, Clayton PE, Wallace DC, Malfroy B, Doctrow SR, Lithgow GJ. Extension of Life-Span with Superoxide Dismutase/Catalase Mimetics. Science. 2000;289:1567–1569. doi: 10.1126/science.289.5484.1567. [DOI] [PubMed] [Google Scholar]
- Miller G, Honig A, Stein H, Suzuki N, Mittler R, Zilberstein A. Unraveling delta1-pyrroline-5-carboxylate-proline cycle in plants by uncoupled expression of proline oxidation enzymes. J Biol Chem. 2009;284:26482–26492. doi: 10.1074/jbc.M109.009340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomura M, Takagi H. Role of the yeast acetyltransferase Mpr1 in oxidative stress: regulation of oxygen reactive species caused by a toxic proline catabolism intermediate. Proc Natl Acad Sci U S A. 2004;101:12616–12621. doi: 10.1073/pnas.0403349101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ocampo A, Liu J, Schroeder EA, Shadel GS, Barrientos A. Mitochondrial Respiratory Thresholds Regulate Yeast Chronological Life Span and its Extension by Caloric Restriction. Cell Metab. 2012;16:55–67. doi: 10.1016/j.cmet.2012.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paek J, Lo JY, Narasimhan SD, Nguyen TN, Glover-Cutter K, Robida-Stubbs S, Suzuki T, Yamamoto M, Blackwell TK, Curran SP. Mitochondrial SKN-1/Nrf mediates a conserved starvation response. Cell Metab. 2012;16:526–537. doi: 10.1016/j.cmet.2012.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panowski SH, Wolff S, Aguilaniu H, Durieux J, Dillin A. PHA-4/Foxa mediates diet-restriction-induced longevity of C. elegans. Nature. 2007;447:550–555. doi: 10.1038/nature05837. [DOI] [PubMed] [Google Scholar]
- Van Raamsdonk JM, Hekimi S. Superoxide dismutase is dispensable for normal animal lifespan. Proc Natl Acad Sci U S A. 2012;109:5785–5790. doi: 10.1073/pnas.1116158109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rea SL, Ventura N, Johnson TE. Relationship between mitochondrial electron transport chain dysfunction, development, and life extension in Caenorhabditis elegans. PLoS Biol. 2007;5:e259. doi: 10.1371/journal.pbio.0050259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz TJ, Zarse K, Voigt A, Urban N, Birringer M, Ristow M. Glucose restriction extends Caenorhabditis elegans life span by inducing mitochondrial respiration and increasing oxidative stress. Cell Metab. 2007;6:280–293. doi: 10.1016/j.cmet.2007.08.011. [DOI] [PubMed] [Google Scholar]
- Shtonda BB, Avery L. Dietary choice behavior in Caenorhabditis elegans. J Exp Biol. 2006;209:89–102. doi: 10.1242/jeb.01955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soukas AA, Kane EA, Carr CE, Melo JA, Ruvkun G. Rictor/TORC2 regulates fat metabolism, feeding, growth, and life span in Caenorhabditis elegans. Genes Dev. 2009;23:496–511. doi: 10.1101/gad.1775409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmons L, Court DL, Fire A. Ingestion of bacterially expressed dsRNAs can produce specific and potent genetic interference in Caenorhabditis elegans. Gene. 2001;263:103–112. doi: 10.1016/s0378-1119(00)00579-5. [DOI] [PubMed] [Google Scholar]
- Tullet JMa, Hertweck M, An JH, Baker J, Hwang JY, Liu S, Oliveira RP, Baumeister R, Blackwell TK. Direct inhibition of the longevity-promoting factor SKN-1 by insulin-like signaling in C. elegans. Cell. 2008;132:1025–1038. doi: 10.1016/j.cell.2008.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Robida-Stubbs S, Tullet J, Ma Rual J-F, Vidal M, Blackwell TK. RNAi screening implicates a SKN-1-dependent transcriptional response in stress resistance and longevity deriving from translation inhibition. PLoS Genet. 2010;6:e1001048. doi: 10.1371/journal.pgen.1001048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang MC, O’Rourke EJ, Ruvkun G. Fat metabolism links germline stem cells and longevity in C. elegans. Science. 2008;322:957–960. doi: 10.1126/science.1162011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson E, MacNeil LT, Arda HE, Zhu LJ, Walhout AJM. Integration of metabolic and gene regulatory networks modulates the C. elegans dietary response. Cell. 2013;153:253–266. doi: 10.1016/j.cell.2013.02.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, Hekimi S. A mitochondrial superoxide signal triggers increased longevity in Caenorhabditis elegans. PLoS Biol. 2010;8:e1000556. doi: 10.1371/journal.pbio.1000556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon K-A, Nakamura Y, Arakawa H. Identification of ALDH4 as a p53-inducible gene and its protective role in cellular stresses. J Hum Genet. 2004;49:134–140. doi: 10.1007/s10038-003-0122-3. [DOI] [PubMed] [Google Scholar]
- Zarse K, Schmeisser S, Groth M, Priebe S, Beuster G, Kuhlow D, Guthke R, Platzer M, Kahn CR, Ristow M. Impaired insulin/IGF1 signaling extends life span by promoting mitochondrial L-proline catabolism to induce a transient ROS signal. Cell Metab. 2012;15:451–465. doi: 10.1016/j.cmet.2012.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.