

Abstract
Transcription of eukaryotic genes is an exceedingly sophisticated and complicated process, orchestrated by layers of control mechanisms involving a myriad of transcription factors and DNA control sequences, with both groups subject to multiple modifications. The availability of various recent genomic approaches has provided previously unforeseen opportunities to examine the cis-regulatory landscape of the entire genome, resulting in the identification of a potentially overwhelming number of enhancers and novel enhancer functions. In this review, we will focus on the activities of enhancers in metazoans and discuss how they serve to regulate gene expression during early development.
Introduction
The genomes of multicellular organisms are essentially identical among different cell types, and biological differences between cells arise in large part due to differences in gene expression. Enhancers, a type of cis-regulatory sequence distinct from promoters, are frequently located near the transcriptional initiation site of a given gene and are critical for the regulation of gene transcription. Genes often have multiple enhancers, and each enhancer can provide diverse transcriptional regulatory instructions to modulate the levels of gene expression depending on differing biological conditions. Enhancers are thus critical in meeting the demand of normal developmental, environmental and physiological conditions and responding to changes. Failure of normal function of enhancers is likely to cause developmental defects and the occurrence of disease states.
Enhancers were originally identified 30 years ago during study of the regulation of SV40 virus early gene transcription1, 2. A short segment of DNA derived from the SV40 promoter proximal region was placed at varying distances to the basal promoter, and resulted in stimulation of transcription from the promoter regardless of the distance or orientation of the fragment from the promoter and this became the operational definition of enhancer elements. Enhancers have since been found in the genomes of essentially all living organisms (and the viruses that parasitize them), and are recognized as providing a fundamental mechanism for the control of gene expression. A gene having multiple enhancers can display complex expression patterns at different times, and in different regions, of embryos or adult animals utilizing different combinations of transcriptional factors acting on each enhancer. A group of genes sharing similar enhancer sequences acquire the ability to respond to the same transcription factor simultaneously if coordinated gene expression is required. These arrangements of enhancers provide a great flexibility to orchestrate the expression of a cohort of gene activities under varying environmental conditions.
In recent years, significant advances have been made to uncover the role of enhancers in developmental processes. While we have quite a way to go to thoroughly comprehend the complexity of enhancer networks hardwired across the entire genome, a number of important molecular activities of enhancers have been revealed. This review will focus on the fundamental activities of enhancers in metazoans and discuss how they serve to regulate gene expression. Emphasis will be placed on the common methodology to identify and locate enhancers in the genome, and the structural constraints and arrangements of enhancers to direct gene expression in specific cell types. A number of other excellent recent reviews should also be consulted3–7.
Identification of enhancers
The human genome encodes approximately 1700–1900 sequence-specific transcription factors8, and many of them directly bind to enhancers to control gene expression. The current high-side estimate of the total number of enhancers in mammalian genomes is as high as 1 million9. Assuming the average enhancer size to be 100bp, this would translate to a total combined length of 108 bp for all enhancers, or over 3% of the haploid human genome (2.9 × 109 bp). This number exceeds the size of the coding region, which is only about 1.5% of the human genome10, raising the interesting possibility that there may be many, many undiscovered mutations within the bodies of enhancers, with consequent impacts on differential gene regulation. If this is the case, enhancer mutations or enhancer sequence polymorphism within a population likely contribute to rates of occurrence of many heritable disorders and developmental abnormalities. It is therefore important to identify and assign biological function to these enhancers. Several different approaches have been developed to uncover the function of enhancers in metazoans. These approaches include biological assays to identify in vivo function of enhancers, bioinformatics approaches to identify evolutionarily conserve cis-regulatory motifs, and biochemical approaches to identify chromatin signatures.
1) Biological approaches to identify enhancers
Experimentally, enhancers are typically identified by placing a fragment of presumed regulatory DNA near to a promoter, typically from a different gene, and then the output from the promoter is evaluated to determine whether the cis-regulatory element increases transcription over promoter-only constructs. The enhancer and promoter are usually cloned upstream of an easily assayed reporter gene such as luciferase or β-galactosidase in order to readily reveal changes in transcriptional output. A transient transfection assay using established cell lines is the most frequently used approach to perform such an experiment. This type of experiment is rapid and easily carried out, but has potential drawbacks involving the capability of specific cell lines to respond to the particular enhancer in question under the experimental conditions of the cell culture dish, with the result that many enhancers may escape undetected.
A more labor-intensive approach, but potentially more powerful, for uncovering a wide range of enhancers and subtle effects they can exert is to use a transgenesis for examining the function of enhancers in living animals. In a typical transgenic assay, enhancers are again fused to minimal promoters, then introduced into animals via microinjection or electroporation, after which the resultant expression patterns can be examined using a reporter gene11–14. Conversely, to demonstrate the necessity of a putative enhancer and the impact of its loss on gene expression, one can remove (or more subtly mutate) the prospective element from its normal context of a gene regulatory region, then compare transcription levels to the expression of a transgene with the wild type construct. The most sophisticated versions of this approach to identifying enhancers are available in mice, in which site-specific manipulation of the genome and the generation of “knock-ins” are well established15, 16. Unfortunately, the high cost of generating such mouse lines means that a genome-wide approach to identify tens of thousands of mouse enhancers is not feasible, even when the analysis can be performed in the F0 generation animals.
Non-mammalian model organisms can also be utilized to detect enhancers at the genomic scale. In Drosophila, a clever approach known as the “enhancer-trap” was developed17, 18. The principle behind the assay is to use a transgene that will only be expressed if it integrates into a genomic location that is under the influence of endogenous enhancer(s) capable of activating the promoter. The experimental approach takes advantage of the powerful P-element transposon system, such that the enhancer trap construct (a reporter gene driven by an enhancer-less promoter) is allowed to “hop” into different Drosophila chromosomal loci. If the reporter construct hops into the locus near an enhancer that directs a specific spatial or temporal expression pattern, the enhancer’s effects can be easily visualized by specific reporter gene (β-galactosidase or GFP) activity. The major advantage of this transposon tagging system is that unknown enhancer functions may be revealed by examining the appearance of novel reporter gene expression in developing embryos. In other systems in which transposon tagging is not available, alternative enhancer detection techniques have been developed to determine the response of promoters to enhancers via the activity of reporter genes, which include streamlined microinjection approaches13, 19, 20, electroporation into embryos21, and the use of modified transposition systems22, 23.
2) Comparative genomic approaches to identify enhancers
Availability of detailed genomic information from numerous organisms has created opportunities for the application of comparative genomics-based approaches to identify evolutionarily-conserved enhancers. The underlying principle is that natural mutations accumulate much faster in non-functional DNA (because these are not selected against) than at functionally important base positions (such as enhancers) across many millions of years of evolution. By comparing non-coding sequences from orthologs of appropriate evolutionarily-distant animal species, we can identify highly-conserved intragenic regions, which then serve as barometers of functional importance. Such systematic analyses have been conducted in various systems, with the general finding being that evolutionarily-conserved non-coding DNA regions not only directed spatiotemporal expression within a given species, but were also sometimes able to direct expression in heterologous model organisms23, 24. A striking demonstration of this principle was the performance of such “phylogenetic footprinting” analysis between pufferfish (Class Osteichthyes) and human (Class Mammalia)14. After comparing the two genomes, ultra-conserved intragenic regions were identified and their functions tested in vivo using transgenic mice. Among 167 ultra-conserved regions of the human genome tested, 45% displayed tissue-specific expression patterns in the developing mouse embryo at E11.5. This experiment suggests the tendency for highly conserved non-coding sequences to behave as transcriptional enhancers in vivo, but also illustrates a way to test for enhancers present in humans, an animal not easily subjected to experimental manipulations.
Particularly useful model organisms for similar comparative analyses are Xenopus (frog) and zebrafish, in which the evolutionary separation from human is sufficiently distant (350M and 450M years, respectively) to allow effective comparisons. These two species share many fundamentally similar developmental processes and body patterns with mammals, and robust transgenic approaches are possible20, 22, yet these are far more cost effectively carried out than in mice. As the research progresses, there will soon be a large collection of enhancers capable of directing spatiotemporal gene expression patterns in developing animals. These enhancers may serve as important biomarkers of disease, as mutations of such are expected to cause abnormal gene expression and may be the cause of various disorders. Additionally, analysis of evolutionarily-conserved enhancers will lead to an improved understanding of the structural constraints and arrangements of enhancers, which enable us to design more effective synthetic enhancers that can accurately deliver gene expression in specific cell types for the treatment of diseases.
While phylogenetic footprinting analysis is useful in identifying conserved enhancers, this approach has limitations. If the conserved sequences within an enhancer are too short or are too dispersed within the stretch of sequences that are compared, identification of such conservation can be computationally challenging as the chance likelihood of any short sequence to be present increases with the large vertebrate genome sizes. Furthermore, this type of analysis will be limited to the enhancers maintaining an evolutionarily conserved structural arrangement. Recent evidence suggest that a large proportion of enhancers may not be evolutionarily conserved and are highly species specific. For instance, enhancers of even-skipped and pax2 that display identical expression patterns in different Drosophila species fail to reveal any significant sequence conservation25, 26. Furthermore, among enhancers identified within the ENCODE genome (a portion of the genome equal to 1% of the total human complement), only about half of the enhancers were mapped to the evolutionarily-constrained regions27. These findings suggest that enhancers can turn over rapidly over a relatively short evolutionary time scale and that animals may co-opt and use different enhancer machinery to direct gene expression in similar tissues. Thus, an approach solely based on evolutionary constraints as a criterion is only partially useful, and other criteria should be applied to uncover as yet undetected enhancers.
3. Chromatin signatures of enhancers
The formation of transcriptionally-active enhancer complexes involves the dynamic interplay of transcriptional apparatus such as transcription factors, RNA polymerase and chromatin. It is generally accepted that in the area of compacted chromatin enhancer sequences are typically not accessible to components of the transcriptional apparatus, and therefore such enhancers are protected from the activity of nucleases like DNAse I. Conversely, transcriptionally active regions correlate with the presence of DNAse I hypersensitive sites in the genome because DNA binding sites in these areas are more accessible to transcription factors and accessory proteins28–30. Traditional methods for identifying DNAse I hypersensitive sites were based on low throughput methods using Southern blot analysis. However, this approach has recently been transformed by a high-throughput method that identifies DNAse I hypersensitive sites across the entire genome using a deep sequencing approach31. While DNAse-seq will never replace ChIP-seq analysis, the former approach is extremely valuable in identifying a broad variety of active enhancers and promoters, independent of a priori knowledge of the identity of bound transcriptional factors32. Moreover, DNAse-seq is capable of identifying individual protein-DNA footprints throughout the genome with base-pair resolution33, thus providing an unprecedented opportunity to examine how individual transcriptional factors bind DNA and interact with neighboring transcription factors to control gene expression at the whole genome level.
Genome-wide studies examining histone modification patterns among different cell lines have revealed that the location of enhancers is often correlated with the binding of protein p300 and the presence of specific histone H3 modifications. Particularly, H3K4me1 and H3K4me3 (mono- and tri-methylation of histone H3 on lysine 4) modifications are extensively studied27. The presence of p300 binding and the H3K4me1 modification are signs of active enhancers, and colocalization of H3K4me1 and H3K4me3 modifications are often correlated with the promoter sites9, 34. Based on the H3K4me1 marking and p300 binding, the total number of enhancers in the mammalian genome was estimated to be in the range of 105–106 9. This number represents an average of one enhancer every 3–30kb in the genome, which is in agreement with the occurrence of H3K4me1-marked enhancers identified by the ENCODE project27. It is also interesting to note that when hundreds of randomly selected intragenic regions of sea urchin genome were used in a transient transgenic approach to identify enhancer activities capable of directing spatiotemporal gene expression patterns in sea urchin embryos, an enhancer was identified in an average of every 30kb13.
Long distance effect of enhancers
In simple organisms (yeast and bacteria), enhancers are located relatively close to the basal promoter, but metazoan enhancers are known to function at considerable genomic distances from the promoters they act upon. This intriguing behavior of enhancers raises a number of questions. What is the underlying mechanism for this flexibility and the source of their apparent autonomy? What is the 3-dimensional structure of the genome allowing such interactions to occur within the physically constrained space of the nucleus? Evidence accumulated thus far indicates that insulators, which can block the action of nearby enhancers, are in fact scattered across the chromosomes of metazoans, thus creating isolated gene regulatory units between enhancers and their cognate promoters35, 36. This arrangement prevents inappropriate cross-regulation of gene expression from neighboring gene’s enhancers. The most popular model explaining the autonomy of enhancers is the DNA looping model, wherein the enhancer and promoter complexes forms direct physical interactions and the intervening DNA between the enhancer and promoter complexes form a loop37, 38. Major support for this model comes from a study utilizing the chromosome conformation capture assay39. In this technique, proteins and DNA in complexes at the bases of the loops are first covalently cross-linked, digested with restriction enzymes, then subjected to intra-complex ligation. This permits covalently linking DNA fragments from different genomic regions bound within the complex to be linked to one another. After reversing protein-DNA cross-links, the pool of resulting chimeric DNA fragments containing re-ligated DNA can be analyzed by PCR, using oligonucleotide primers flanking regions of suspected looping. A more recent version of this method, ChIA-PET (Chromatin Interaction Analysis by Paired-End Tag) sequencing, involves chromatin immunoprecipitation, intra-complex ligation, and high-throughput sequencing of the ligation products from both ends to obtain genome-wide information on DNA looping40. Use of these methods has provided confirmatory evidence of direct physical interactions between promoter and enhancer complexes. If enhancers can function at distance, how far can they act? How do different enhancers “decide” when to interact with specific promoters without interfering the function of other enhancers? The answers to these questions are likely to be complex, involving both the state of local chromatin modifications/conformations and the availability of transcriptional factors in a given cell. However, with the advent of new high-throughput sequencing technology, we will have the opportunity to address these questions in the near future.
Arrangement of enhancers and development
One of fundamental, unanswered questions in developmental biology today is the mechanism by which enhancers interact with transcriptional machinery to produce quantitatively precise and spatiotemporally-restricted gene expression patterns during development. In the following section, some specific examples of how animals utilize enhancers to coordinate gene expression are discussed.
An enhancer can be composed of a modular arrangement of short (6–8bp) conserved transcriptional factor binding sequences, to control spatial expression patterns. This example can be illustrated using a case from the amphibian gastrula stage embryo. In the frog Xenopus, initiation of gastrulation is controlled by a small region of the early embryo known as Spemann’s organizer, occupying about 60 degrees of horizontal arc in the dorsal equator of amphibian late blastula-stage embryos. The organizer is induced by two overlapping growth factor signaling inputs and contains the future dorsal anterior endodermal and mesodermal tissues. When this region is isolated and transplanted into the ventral equatorial region of a host embryo, the organizer induces a secondary, “twinned”, embryonic axis containing anterior-posterior and dorsal-ventral patterned tissues. The mechanism of the organizer’s induction has been extensively studied for over 85 years41–43. The two growth factor signaling pathways implicated in establishing the organizer are those of Wnt and Nodal signaling (Figure 1). Wnt signaling is activated on the dorsal side of embryos after fertilization, leading to the activation of the homeobox genes twin and siamois via a short enhancer, composed of multiple copies of Lef/Tcf-binding sequences, located within the promoter regions of these genes44, 45. Once twin and siamois are activated, their homeobox gene products bind to the short proximal enhancer (50bp) of the organizer homeobox gene, goosecoid (gsc). However, this binding alone is not sufficient to fully activate gsc transcription, so another input is required. This input is provided by the vegetal region, where the Nodal signaling cascade activates its key signal transducers, various Smad proteins, to bind the gsc promoter’s distal enhancer (19bp), together with another transcriptional factor 46. Thus, the organizer region is formed in the dorsal equatorial region of the embryo where dorsal expression of Wnt-induced twin and siamois overlaps with a zone of active Nodal signaling. This synergistic interaction, mediated by two short distal and proximal enhancers located within 226bp of the transcriptional start site, is required to express gsc at the appropriate location and time of development.
Figure 1.

Spatial regulation of goosecoid by short enhancers. (A) Wnt and Nodal signaling areas are shaded in red and in green. The overlapping region (right panel) corresponds to Spemann’s organizer where gsc is activated. (B) Upon Wnt signaling activation, siamois and twin are transcribed via β-catenin and Tcf/Lef. Siamois and Twin homeodomain proteins bind to the proximal enhancer of gsc. A Smad2/4 complex is also formed in the distal enhancer of gsc where nodal signaling is active. These complexes interact synergistically and gsc expression becomes spatially defined in the Spemann’s organizer region.
Instead of short enhancers, longer enhancer “modules” can be used to independently regulate complex spatial expression patterns of a gene (Figure 2). In Drosophila, maternal gene products Bicoid and Caudal control the expression of gap genes such as hunchback, kruppel, knirps, and giant. Shortly after the activation of these gap genes, activation of pair-rule genes occurs. Transcription of pair rule genes is regulated by the combined inputs of the maternal Bicoid and Caudal products acting together with the gap gene products. Even-skipped (eve) is a pair-rule gene, with expression characterized by a distinctive seven stripe pattern. During Drosophila blastoderm formation, an initially less well-defined seven stripe pattern becomes sharper, with distinct anterior and posterior expression borders. The formation of even-skipped borders is regulated by five separate enhancer modules dispersed around the eve exons5. Each of the five enhancers functions independently to direct the expression of each eve stripe in the fly embryo. This can be readily demonstrated by placing one of the five enhancers upstream of a β-galactosidase reporter gene. Each enhancer drives the expression of β-galactosidase in a specific stripe or stripes. One of these enhancers, eve stripe 2 (Eve2), is a 480-bp enhancer that has been studied extensively47. The Eve2 enhancer contains 12 transcription factor binding sites. Maternally-expressed transcription factors Bicoid and Hunchback, bind six of the sites to positively regulate the expression of eve in forming the anterior expression border of eve stripe 2. Gap proteins Giant and Kruppel bind the other six sites of Eve2 to repress stripe 2 enhancer activity, thus regulating the borders of eve stripe 2 expression48. Other enhancer modules in eve use similar logic, but each utilizes a different combination of maternal and gap proteins. Thus, in the case of eve, each long enhancer module has a specific role to establish proper anterior and posterior boundaries in specific cell types when appropriate transcriptional factors are present. Collectively, after integrating the activities of all five independent-enhancer modules, a characteristic seven stripe eve pattern is generated.
Figure 2.

Arrangement of Even-skipped enhancer modules. (A) The seven stripe of Eve expression in a Drosophila embryo. Eve stripe 2 domain is highlighted. (B) Five different enhancer modules control the expression of Eve. (C) Stripe 2 enhancer module consists of multiple transcriptional factor binding sites. When the Bicoid and Hunchback transcription factors are present, eve stripe 2 is activated. When Giant and Kruppel proteins are available, eve strip 2 element is repressed.
In vertebrates, complex spatially and temporally defined expression patterns of a gene can be similarly attained by incorporating multiple enhancers that act at a distance. For instance, the mouse Sonic hedgehog (Shh) gene is expressed in many different tissues including limbs, central nervous system, and gastrointestinal tract during development. This complex expression pattern is the result of the combined action of multiple enhancers, each of which acts independently to control Shh transcription in different tissues at different developmental times (Figure 3). In order to generate the characteristic CNS expression patterns of Shh, the SFPE1 enhancer located at −8 kb and enhancer SFPE2 located within intron 2 of Shh, each acts independently to control expression in the floor plate of the hindbrain and spinal cord, respectively 49. The Shh brain enhancer 1 (SBE1) is located in intron 2, directing Shh expression to the ventral midbrain and caudal diencephalon. The more distal enhancer elements, SBE2, SBE3, and SBE4, which are located greater than 400 kb upstream of the Shh transcription start site, drive reporter expression in the ventral forebrain. It is the combined activity of these enhancers that gives rise to the detailed and specific Shh expression patterns observed in the developing central nervous system of mouse embryos50. Shn expression patterns in limbs and gastrointestinal tract are similarly regulated by multiple, independently acting enhancers51, 52.
Figure 3.
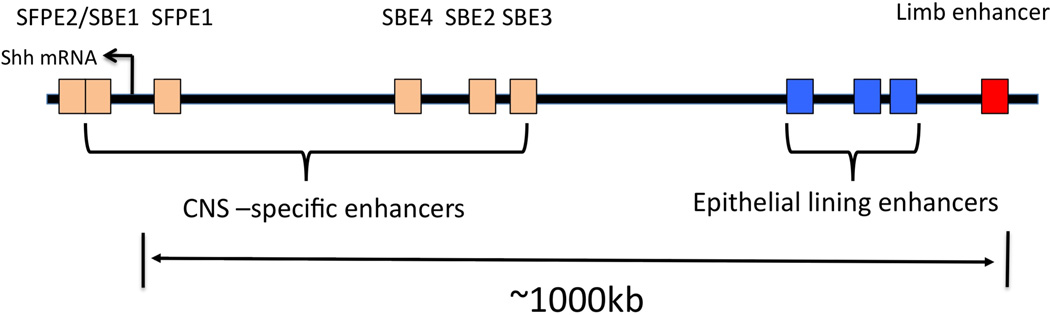
Arrangement of Shh enhancers. CNS specific enhancers and epithelial lining enhancers are shown. Limb enhancer is located approximately 1MB away from the Shh promoter region.
Enhancers and Evolution
A change in the activity of a transcription factor can bring a profound effect on developmental processes as the expression of batteries of target genes in every cell type where the transcription factor functions, might be altered significantly. It is easy to imagine that many point mutations within the coding sequence of a transcriptional factor can alter the proteins architecture or ability to interact with DNA or other proteins, resulting in inactivation or hypo- or hyper-activation of the transcription factor, bringing a significant and sometimes catastrophic impact on developmental processes. On the other hand, mutations within cis-regulatory elements may be more subtle and specific. A sequence change within an individual enhancer recognized by a specific transcriptional factor, or physical replacement or removal an enhancer near a target gene of the transcriptional factor can exert limited phenotypic outcomes, without influencing other normal developmental processes mediated by the transcriptional factor. Such enhancer mutations may prove to be more important for evolutionary innovation than mutation of transcription factor coding sequence. Incremental changes in phenotype that can be accomplished by mutating cis-regulatory sequences are more in line with gradualistic models for evolutionary adaptation and survival in response to environmental challenges.
A striking evolutionary example of mutation of cis-regulatory information is found in the stickleback fish’s pelvic fin development. Sticklebacks are commonly oceanic, but some species were trapped in freshwater lakes during recession of ice-age glaciers. Geographic isolation has permitted independent evolution of these populations segregated from any significant mixing with the oceanic varieties for some 22,000 years ago53. When the genetic nature of differences in pelvic fins between freshwater and oceanic sticklebacks was examined, a mutation was mapped to a single location, in an enhancer for the gene Pitx154. In sticklebacks that have a pronounced pelvic fin, this enhancer is active and specifically drives Pitx1 gene expression in the pelvic fin during development. On the other hand, in freshwater sticklebacks lacking the pelvic fin, the enhancer regulating Pitx1 expression in the pelvic fin is lost. This example illustrates how enhancers can be rapidly altered to deliver a major evolutionary consequence without altering other developmental processes of the gene product they control. Importantly, a reverse evolution experiment was recently performed whereby the pelvic fin-controlling Pitx1 enhancer element was reinstated, in populations with only rudimentary pelvic fins. When this transgene was introduced back into this stickleback population, the transgene was sufficient to restore the pelvic fin to these fish even though they had lost the fin since their divergence from pelvic-finned stickleback populations55.
One well-documented case of an enhancer operating over long distance is the limb bud enhancer that controls Sonic hedgehog (Shh) gene expression51, 56. This 800bp enhancer is evolutionarily conserved between fish and mammals. It is located within the intron of another genes, Lmbr1, which is over 1Mb away from the Shh gene itself, and regulates Shh expression in the zone of polarizing activity, or ZPA, of the limb bud51 (Figure 3). Mice with certain mutations in this enhancer fail to form the ulna and digits 2 to 5, while other mutations to this enhancer, in humans, cause congenital limb malformations56. When, the genomic sequences of the equivalent regions of limbless lizards and newts, and some snakes were examined, this conserved enhancer was absent, suggesting the possibility that loss of this region might be causally linked to the evolutionary loss of limbs in these animals 57. Taking the analogy of pelvic fin development in stickleback fish, it is tempting to speculate that these limbless reptiles might grow limbs and “revert” to the phenotype of their last common ancestors with limbed reptiles, if the limb enhancer in these limbless animals could be replaced. Unfortunately, this experiment is technically challenging and cannot be performed at present time.
While some enhancer mutations do display striking and even drastic phenotypes, there is a new study indicating that evolution may have selected for organisms with built in safeguards to resist such drastic changes by duplicating enhancer regions. Recent work done in Drosophila indicates that one-third to one-half of all Dorsal (a transcription factor involved in embryonic dorsal-ventral axis specification) target genes may be regulated by secondary enhancers. Such secondary enhancers were termed “shadow enhancers”, reflecting the notion that the secondary enhancer mediates activity that overlaps the function of the primary enhancer. Placement of either a primary enhancer or a shadow enhancer upstream of a reporter gene shows that both are capable of driving nearly identical patterns of gene expression 58. It has been hypothesized that these shadow enhancers help ensure precise and reproducible patterns of gene expression during embryogenesis in case the primary enhancer fails for any reason, reminiscient of what Hans Spemann once described as the concept of “double-assurance”, explaining the robustness of developmental systems against perturbation41. It is possible that shadow enhancers are employed to protect the organism against critical mutations or environmental changes that could have deleterious affects on development. Consistent with this notion, it has been demonstrated experimentally that a single enhancer sometimes fails to drive the complete pattern of expression under temperature stress59, whereas the presence of both enhancers (primary and shadow) permits normal gene expression under these conditions. Thus, while the name of “shadow enhancer” implies a very passive role, these cis-regulatory elements may play a more active role in the robustness of metazoan development, the maintenance of which has been favored by evolution.
New surprises
Enhancer’s classical definition includes their function as distal cis-acting regulatory elements that regulate transcription from a promoter. However, recent genome-wide transcriptome analyses have revealed that enhancers are themselves also transcribed by RNA polymerase II and therefore might exert functions through a long noncoding RNA (ncRNA)4. Previously, many enhancers including globin locus control region were shown to be transcribed into ncRNAs in the cells60, and these enhancer-derived RNAs were considered as by-products of transcription or transcriptional anomality. However, recent reports suggest that two distinct types of ncRNAs can modulate the activity of enhancers. These are long polyadenylated ncRNAs that are directionally expressed, and enhancer RNAs (eRNAs) that are non-polyadenylated and transcribed bidirectionally61–64. Both types of ncRNAs are shown to actively engage with transcription complexes to regulate transcriptional activity as revealed by the loss-of-function analyses of some of these RNAs. While the exact mechanisms of their actions are unclear, it has been speculated that ncRNAs play a role in stabilizing local configurations of DNA tertiary structure to allow the enhancer and transcription complexes to continue to interact or strengthen that interaction. In fact, ncRNAs such as lincRNAs (long intergenic noncoding RNAs) has been shown to mediate epigenetic changes by recruiting chromatin remodeling complexes to specific genomic loci64. If the activation of gene expression by ncRNA or eRNA is a general function of these classes of RNA, certainly this will provide an additional layer of complexity to the already exceedingly sophisticated and complex process of enhancer functions. At the same time, we may also find a new opportunity to control enhancer activity via a way of modulating the expression of ncRNA or eRNA, which may be useful for the development of novel enhancer-based therapies for the treatments of various diseases.
Acknowledgement
I thank Ira Blitz for critically reviewing this manuscript. Work in the Cho lab is supported by the National Institutes of Health.
References
- 1.Banerji J, Rusconi S, Schaffner W. Expression of a b-globin gene is enhanced by remote SV40 DNA sequences. Cell. 1981;27:299–308. doi: 10.1016/0092-8674(81)90413-x. [DOI] [PubMed] [Google Scholar]
- 2.Moreau P, Hen R, Wasylyk B, Everrett R, Gaub M, Chambon P. The SV40 72-bp repeat has a striking effect on gene expression both in SV 40 and other chimeric recombinants. Nucleic Acids Res. 1981;9:6047–6069. doi: 10.1093/nar/9.22.6047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Visel A, Rubin EM, Pennacchio LA. Genomic views of distant-acting enhancers. Nature. 2009;461:199–205. doi: 10.1038/nature08451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mercer TR, Dinger ME, Mattick JS. Long non-coding RNAs: insights into functions. Nature Reviews Genetics. 2009;10:155–159. doi: 10.1038/nrg2521. [DOI] [PubMed] [Google Scholar]
- 5.Levine M. Transcriptional enhancers in animal development and evolution. Curr Biol. 2010;20:754–763. doi: 10.1016/j.cub.2010.06.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bulger M, Groudine M. Enhancers: the abundance and function of regulatory sequences beyond promoters. Dev Biol. 2010;339:250–257. doi: 10.1016/j.ydbio.2009.11.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bulger M, Groudine M. Functional and mechanistic diversity of distal transcription enhancers. Cell. 2011;144:327–339. doi: 10.1016/j.cell.2011.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vaquerizas JM, Kummerfeld SK, Teichmann SA, Luscombe NM. A census of human transcription factors: function, expression and evolution. Nat Rev Genet. 2009;10:252–263. doi: 10.1038/nrg2538. [DOI] [PubMed] [Google Scholar]
- 9.Heintzman ND, Hon GC, Hawkins RD, Kheradpour P, Stark A, Harp LF, Ye Z, Lee LK, Stuart RK, Ching CW, et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature. 2009;459:108–112. doi: 10.1038/nature07829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.International Human Genome Sequencing Consortium. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 11.Watabe T, Kim S, Candia A, Rothbächer U, Hashimoto C, Inoue K, Cho KW. Molecular mechanisms of Spemann's organizer formation: conserved growth factor synergy between Xenopus and mouse. Genes Dev. 1995;9:3038–3050. doi: 10.1101/gad.9.24.3038. [DOI] [PubMed] [Google Scholar]
- 12.Momose T, Tonegawa A, Takeuchi J, Ogawa H, Umesono K, Yasuda K. Efficient targeting of gene expression in chick embryos by microelectroporation. Dev Growth Differ. 1999;41:335–344. doi: 10.1046/j.1440-169x.1999.413437.x. [DOI] [PubMed] [Google Scholar]
- 13.Cameron RA, Oliveri P, Wyllie J, Davidson EH. cis-Regulatory activity of randomly chosen genomic fragments from the sea urchin. Gene Expr Patterns. 2004;4:205–213. doi: 10.1016/j.modgep.2003.08.007. [DOI] [PubMed] [Google Scholar]
- 14.Pennacchio LA, Ahituv N, Moses AM, Prabhakar S, Nobrega MA, Shoukry M, Minovitsky S, Dubchak I, Holt A, Lewis KD, et al. In vivo enhancer analysis of human conserved non-coding sequences. Nature. 2006;444:499–502. doi: 10.1038/nature05295. [DOI] [PubMed] [Google Scholar]
- 15.Palmiter RD, Brinster RL. Germ-line transformation of mice. Annu Rev Genet. 1986;20:465–499. doi: 10.1146/annurev.ge.20.120186.002341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mikkola HK, Orkin SH. Gene targeting and transgenic strategies for the analysis of hematopoietic development in the mouse. Methods Mol Med. 2005;105:3–22. doi: 10.1385/1-59259-826-9:003. [DOI] [PubMed] [Google Scholar]
- 17.O'Kane CJ, Gehring WJ. Detection in situ of genomic regulatory elements in Drosophila. Proc Natl Acad Sci USA. 1987;84:9123–9127. doi: 10.1073/pnas.84.24.9123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bier E, Vaessin H, Shepherd S, Lee K, McCall K, Barbel S, Ackerman L, Carretto R, Uemura T, Grell E, et al. Searching for pattern and mutation in the Drosophila genome with a P-lacZ vector. Genes Dev. 1989;3:1273–1287. doi: 10.1101/gad.3.9.1273. [DOI] [PubMed] [Google Scholar]
- 19.Ogino H, McConnell WB, Grainger RM. High-throughput transgenesis in Xenopus using I-SceI meganuclease. Nat Protoc. 2006;1:1703–1710. doi: 10.1038/nprot.2006.208. [DOI] [PubMed] [Google Scholar]
- 20.Ishibashi S, Kroll KL, Amaya E. A method for generating transgenic frog embryos. Methods Mol Biol. 2008;461:447–466. doi: 10.1007/978-1-60327-483-8_31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Itasaki N, Bel-Vialar S, Krumlauf R. Shocking' developments in chick embryology: electroporation and in ovo gene expression. Nat Cell Biol. 1999;1:203–207. doi: 10.1038/70231. [DOI] [PubMed] [Google Scholar]
- 22.Thermes V, Grabher C, Ristoratore F, Bourrat F, Choulika A, Wittbrodt J, Joly JS. I-SceI meganuclease mediates highly efficient transgenesis in fish. Mech Dev. 2002;118:91–98. doi: 10.1016/s0925-4773(02)00218-6. [DOI] [PubMed] [Google Scholar]
- 23.Navratilova P, Fredman D, Hawkins TA, Turner K, Lenhard B, Becker TS. Systematic human/zebrafish comparative identification of cis-regulatory activity around vertebrate developmental transcription factor genes. Dev Biol. 2009;327:526–540. doi: 10.1016/j.ydbio.2008.10.044. [DOI] [PubMed] [Google Scholar]
- 24.Kimura-Yoshida C, Kitajima K, Oda-Ishii I, Tian E, Suzuki M, Yamamoto M, Suzuki T, Kobayashi M, Aizawa S, Matsuo I. Characterization of the pufferfish Otx2 cis-regulators reveals evolutionarily conserved genetic mechanisms for vertebrate head specification. Development. 2004;131:57–71. doi: 10.1242/dev.00877. [DOI] [PubMed] [Google Scholar]
- 25.Hare EE, Peterson BK, Eisen MB. A careful look at binding site reorganization in the even-skipped enhancers of Drosophila and sepsids. PLoS Genet. 2008;4 doi: 10.1371/journal.pgen.1000268. e1000268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Swanson CI, Evans NC, Barolo S. Structural rules and complex regulatory circuitry constrain expression of a Notch- and EGFR-regulated eye enhancer. Dev Cell. 2010;18:359–370. doi: 10.1016/j.devcel.2009.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.The ENCODE Project Consortium. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447:799–816. doi: 10.1038/nature05874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu C, Wong YC, Elgin SC. The chromatin structure of specific genes, II: Disruption of chromatin structure during gene activity. Cell. 1979;16:807–814. doi: 10.1016/0092-8674(79)90096-5. [DOI] [PubMed] [Google Scholar]
- 29.Elgin SC. DNAase I-hypersensitive sites of chromatin. Cell. 1981;27:413–415. doi: 10.1016/0092-8674(81)90381-0. [DOI] [PubMed] [Google Scholar]
- 30.Gross DS, Garrard WT. Nuclease hypersensitive sites in chromatin. Annu. Rev. Biochem. 1988;57:159–197. doi: 10.1146/annurev.bi.57.070188.001111. [DOI] [PubMed] [Google Scholar]
- 31.Song L, Crawford GE. DNase-seq: A high-resolution technique for mapping active gene regulatory elements across the genome from mammalian cells. Cold Spring Harbor Protoc. 2010 doi: 10.1101/pdb.prot5384. pdb.prot5384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Boyle AP, Davis S, Shulha HP, Meltzer P, Margulies EH, Weng Z, Furey TS, Crawford GE. High resolution mapping and characterization of open chromatin across the genome. Cell. 2008;132:311–322. doi: 10.1016/j.cell.2007.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Boyle AP, Song L, Lee BK, London D, Keefe D, Birney E, Iyer VR, Crawford GE, Furey TS. High-resolution genome-wide in vivo footprinting of diverse transcription factors in human cells. Genome Res. 2011;21:456–464. doi: 10.1101/gr.112656.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Visel A, Blow MJ, Li Z, Zhang T, Akiyama JA, Holt A, Plajzer-Frick I, Shoukry M, Wright C, Chen F, et al. ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature. 2009;457:854–858. doi: 10.1038/nature07730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wei GH, Liu DP, Liang CC. Chromatin domain boundaries: insulators and beyond. Cell Res. 2005;15:292–300. doi: 10.1038/sj.cr.7290298. [DOI] [PubMed] [Google Scholar]
- 36.Giles KE, Gowher H, Ghirlando R, Jin C, Felsenfeld G. Chromatin boundaries, insulators, and long-range interactions in the nucleus. Cold Spring Harb Symp Quant Biol. 2010;75:79–85. doi: 10.1101/sqb.2010.75.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Blackwood EM, Kadonaga JT. Going the distance: a current view of enhancer action. Science. 1998;281:60–63. doi: 10.1126/science.281.5373.60. [DOI] [PubMed] [Google Scholar]
- 38.Bulger M, Groudine M. Looping versus linking: toward a model for long-distance gene activation. Genes Dev. 1999;13:2465–2477. doi: 10.1101/gad.13.19.2465. [DOI] [PubMed] [Google Scholar]
- 39.Dekker J, Rippe K, Dekker M, Kleckner N. Capturing chromosome conformation. Science. 2002;295:1306–1311. doi: 10.1126/science.1067799. [DOI] [PubMed] [Google Scholar]
- 40.Fullwood MJ, Liu MH, Pan YF, Liu J, Xu H, Mohamed YB, Orlov YL, Velkov S, Ho A, Mei PH, et al. An oestrogen-receptor-alpha-bound human chromatin interactome. Nature. 2009;462:58–64. doi: 10.1038/nature08497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hamburger V. The Heritage of Experimental Embryology: Hans Spemann and the Organizer. New York: Oxford University Press; 1988. [Google Scholar]
- 42.Harland R, Gerhart J. Formation and function of Spemann’s organizer. Annual Review of Cell and Developmental Biology. 1997;13:611–667. doi: 10.1146/annurev.cellbio.13.1.611. [DOI] [PubMed] [Google Scholar]
- 43.De Robertis EM, Larraín J, Oelgeschläger M, Wessely O. The establishment of spemann's organizer and patterning of the vertebrate embryo. Nature Reviews Genetics. 2000;1:171–181. doi: 10.1038/35042039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Laurent M, Hashimoto C, Blitz IL, Rothbacher U, Cho KWY. The Xenopus homeobox gene Twin mediates Wnt induction of goosecoid in establishment of Spemann’s organizer. Development. 1997;124:4905–4916. doi: 10.1242/dev.124.23.4905. [DOI] [PubMed] [Google Scholar]
- 45.Brannon M, Gomperts M, Sumoy L, Moon RT, Kimelman D. A beta-catenin/XTcf-3 complex binds to the siamois promoter to regulate dorsal axis specification in Xenopus. Genes Dev. 1997;11:2359–2370. doi: 10.1101/gad.11.18.2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ring C, Ogata S, Meek L, Song J, Ohta T, Miyazono K, Cho KW. The role of a Williams-Beuren syndrome-associated helix-loop-helix domain-containing transcription factor in activin/nodal signaling. Genes Dev. 2002;16:820–835. doi: 10.1101/gad.963802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Small S, Blair A, Levine M. Regulation of even-skipped stripe 2 in the Drosophila embryo. EMBO J. 1992;11:4047–4057. doi: 10.1002/j.1460-2075.1992.tb05498.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stanojevic D, Small S, Levine M. Regulation of a segmentation stripe by overlapping activators and repressors in the Drosophila embryo. Science. 1991;254:1385–1387. doi: 10.1126/science.1683715. [DOI] [PubMed] [Google Scholar]
- 49.Epstein DJ, McMahon AP, Joyner AL. Regionalization of Sonic hedgehog transcription along the anteroposterior axis of the mouse central nervous system is regulated by Hnf3-dependent and -independent mechanisms. Development. 1999;126:281–292. doi: 10.1242/dev.126.2.281. [DOI] [PubMed] [Google Scholar]
- 50.Jeong Y, El-Jaick K, Roessler E, Muenke M, Epstein DJ. A functional screen for sonic hedgehog regulatory elements across a 1 Mb interval identifies long-range ventral forebrain enhancers. Development. 2006;133:761–772. doi: 10.1242/dev.02239. [DOI] [PubMed] [Google Scholar]
- 51.Sagai T, Hosoya M, Mizushina Y, Tamura M, Shiroishi T. Elimination of a long-range cis-regulatory module causes complete loss of limb-specific Shh expression and truncation of the mouse limb. Development. 2005;132:797–803. doi: 10.1242/dev.01613. [DOI] [PubMed] [Google Scholar]
- 52.Sagai T, Amano T, Tamura M, Mizushina Y, Sumiyama K, Shiroishi T. A cluster of three long-range enhancers directs regional Shh expression in the epithelial linings. Development. 2009;136:1665–1674. doi: 10.1242/dev.032714. [DOI] [PubMed] [Google Scholar]
- 53.Pennisi E. Evolutionary Biology: Changing a fish’s bony armor in the wink of a gene. Science. 2004;304:1736–1739. doi: 10.1126/science.304.5678.1736. [DOI] [PubMed] [Google Scholar]
- 54.Shapiro MD, Marks ME, Peichel CL, Blackman BK, Nereng KS, Jónsson B, Schluter D, Kingsley DM. Genetic and developmental basis of evolutionary pelvic reduction in threespine sticklebacks. Nature. 2004;428:717–723. doi: 10.1038/nature02415. [DOI] [PubMed] [Google Scholar]
- 55.Chan YF, Marks ME, Jones FC, Villarreal G, Jr, Shapiro MD, Brady SD, Southwick AM, Absher DM, Grimwood J, Schmutz J, et al. Adaptive evolution of pelvic reduction in sticklebacks by recurrent deletion of a Pitx1 enhancer. Science. 2010;327:302–305. doi: 10.1126/science.1182213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lettice LA, Heaney SJ, Purdie LA, Li L, de Beer P, Oostra BA, Goode D, Elgar G, Hill RE, de Graaff E. A long-range Shh enhancer regulates expression in the developing limb and fin and is associated with preaxial polydactyly. Hum. Mol. Genet. 2003;12:1725–1735. doi: 10.1093/hmg/ddg180. [DOI] [PubMed] [Google Scholar]
- 57.Sagai T, Masuya H, Tamura M, Shimizu K, Yada Y, Wakana S, Gondo Y, Noda T, Shiroishi T. Phylogenetic conservation of a limb-specific, cis-acting regulator of Sonic hedgehog (Shh) Mamm Genome. 2004;15:23–34. doi: 10.1007/s00335-033-2317-5. [DOI] [PubMed] [Google Scholar]
- 58.Hong JW, Hendrix DA, Levine MS. Shadow enhancers as a source of evolutionary novelty. Science. 2008;321:1314. doi: 10.1126/science.1160631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Perry MW, Boettiger AN, Bothma JP, Levine M. Shadow enhancers foster robustness of Drosophila gastrulation. Curr Biol. 2010;20:1562–1567. doi: 10.1016/j.cub.2010.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ashe HL, Monks J, Wijgerde M, Fraser P, Proudfoot NJ. Intergenic transcription and transinduction of the human beta-globin locus. Genes Dev. 1997;11:2494–2509. doi: 10.1101/gad.11.19.2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kim TK, Hemberg M, Gray JM, Costa AM, Bear DM, Wu J, Harmin DA, Laptewicz M, Barbara-Haley K, Kuersten S, Markenscoff-Papadimitriou E, Kuhl D, Bito H, Worley PF, Kreiman G, Greenberg ME. Widespread transcription at neuronal activity-regulated enhancers. Nature. 2010;465:182–187. doi: 10.1038/nature09033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.De Santa F, Barozzi I, Mietton F, Ghisletti S, Polletti S, Tusi BK, Muller H, Ragoussis J, Wei CL, Natoli G. A large fraction of extragenic RNA pol II transcription sites overlap enhancers. PLoS Biol. 2010;8 doi: 10.1371/journal.pbio.1000384. e1000384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ørom UA, Derrien T, Beringer M, Gumireddy K, Gardini A, Bussotti G, Lai F, Zytnicki M, Notredame C, Huang Q, et al. Cell. 2010;143:46–58. doi: 10.1016/j.cell.2010.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Guttman M, Donaghey J, Carey BW, Garber M, Grenier JK, Munson G, Young G, Lucas AB, Ach R, Bruhn L, et al. lincRNAs act in the circuitry controlling pluripotency and differentiation. Nature. 2011;477:295–300. doi: 10.1038/nature10398. [DOI] [PMC free article] [PubMed] [Google Scholar]