

Abstract
Huntingtin-associated protein 1 (HAP1) was initially established as a neuronal binding partner of huntingtin, mutations in which underlie Huntington's disease. Subcellular localization and protein interaction data indicate that HAP1 may be important in vesicle trafficking and cell signalling. In this study, we establish that HAP1 is important in several steps of exocytosis in adrenal chromaffin cells. Using carbon-fibre amperometry, we measured single vesicle exocytosis in chromaffin cells obtained from HAP1−/− and HAP1+/+ littermate mice. Numbers of Ca2+-dependent and Ca2+-independent full fusion events in HAP1−/− cells are significantly decreased compared with those in HAP1+/+ cells. We observed no change in the frequency of ‘kiss-and-run’ fusion events or in Ca2+ entry. Whereas release per full fusion event is unchanged in HAP1−/− cells, early fusion pore duration is prolonged, as indicated by the increased duration of pre-spike foot signals. Kiss-and-run events have a shorter duration, indicating opposing roles for HAP1 in the stabilization of the fusion pore during full fusion and transient fusion, respectively. We use electron microscopy to demonstrate a reduction in the number of vesicles docked at the plasma membrane of HAP1−/− cells, where membrane capacitance measurements reveal the readily releasable pool of vesicles to be reduced in size. Our study therefore illustrates that HAP1 regulates exocytosis by influencing the morphological docking of vesicles at the plasma membrane, the ability of vesicles to be released rapidly upon stimulation, and the early stages of fusion pore formation.
Introduction
Huntingtin-associated protein 1 (HAP1) has been identified as the first interacting partner of huntingtin (Htt), the protein product of the Huntington's disease (HD) gene (Li et al. 1995). Increased binding occurs with mutant Htt and the degree of binding correlates with polyglutamine repeat length (Li et al. 1995). HAP1 has five N-terminal myristoylation sites enabling membrane targeting (Li & Li, 2005). It is primarily localized to synaptic vesicles (Li et al. 2000) and large dense core vesicles (LDCVs) (Wu et al. 2010) in neurons (Li et al. 1995, 1996; Bertaux et al. 1998; Gutekunst et al. 1998; Page et al. 1998) and endocrine cells, including the pituitary gland, pancreatic islets and adrenal medulla (Dragatsis et al. 2000; Liao et al. 2005).
HAP1−/− mice die soon after birth as a result of the degeneration of the hypothalamus, an area of strong HAP1 expression in rodent brain, which causes a loss in feeding drive (Li et al. 2003). HAP1 interacts with several vesicle-trafficking proteins including DUO (Colomer et al. 1997), the dynactin p150Glued subunit (Li et al. 1998) and kinesin light chain (McGuire et al. 2006). HAP1 is transported anterogradely and retrogradely in axons (Block-Galarza et al. 1997) and controls neurite extension (McGuire et al. 2006), a process in which regulated exocytosis is essential (Martinez-Arca et al. 2001; Delgado-Martinez et al. 2007), and insulin release is decreased in pancreatic β cells lacking HAP1 (Cape et al. 2012). These observations infer a role for HAP1 in cytoskeletal function, vesicle trafficking and possibly exocytosis.
During exocytosis, vesicle fusion is tightly coupled to the intracellular Ca2+ signal. Vesicles first attach to the plasma membrane (docking) and then undergo maturation step(s) to become fusion-competent (priming). A fusion pore then forms between a primed vesicle and the plasma membrane primarily under the control of the SNARE proteins and an array of other regulatory proteins. In chromaffin cells, this fusion pore can initially be unstable (‘flickering’) and allow the release of only a small amount of vesicle content (Zhou et al. 1996; Zhang & Jackson, 2010). If an unstable early pore closes and does not progress to a full fusion pore, an occurrence known as transient or ‘kiss-and-run’ fusion, only a proportion of vesicle contents are released. If the fusion pore stabilizes into a full fusion pore, the full release of vesicle contents is thought to follow. All stages of the vesicle fusion process can be measured in adrenal chromaffin cells by using amperometry to detect released catecholamines. The prominent expression of HAP1 in the adrenal medulla (Liao et al. 2005) and on vesicles (Li et al. 2000; Wu et al. 2010), as well as the previously reported interactions of HAP1 with vesicle-trafficking proteins (Colomer et al. 1997; Li et al. 1998; McGuire et al. 2006), led us to investigate whether HAP1 might regulate vesicle fusion and exocytosis. We explored the regulation of exocytosis by HAP1 using adrenal chromaffin cells from HAP1−/− mice (Li et al. 2003). We confirm that loss of HAP1 reduces full fusion exocytosis in chromaffin cells and that this may occur via two potentially interlinked mechanisms: control of vesicle docking, and regulation of fusion pore stabilization.
Methods
Animals
All procedures involving animals were approved by the Animal Welfare Committee of Flinders University. All animals were kept under standardized barrier breeding conditions (12 : 12 h light/dark cycle) with free access to water and food. HAP1−/− neonatal mice were bred from transgenic breeding pairs heterozygous for the HAP1 knockout allele. (C57/Black6 genetic background) (Li et al. 2003a). Briefly, the mouse HAP1 gene was disrupted through homologous recombination (Li et al. 2003a). PCR genotyping of HAP1 knockout mice was carried out using primers 5′-GGG TTT TGG AGG TCT GGT CTC GCT CTG-3′/5′-CTT CAT GTG GAT GCT AGG GAT CC-3′ for wild-type and 5′-GGG TTT TGG AGG TCT GGT CTC GCT CTG-3′/5′-GGG TAC CCT ACC CGG TAG AAT TCG-3′ for knockout animals.
Primary murine chromaffin cell culture
Transgenic pups at P0 were killed by decapitation. The adrenal glands were dissected out and immediately placed in tubes containing ice-cold Locke's solution (154 mm NaCl, 5.6 mm KCl, 3.6 mm NaHCO3, 5.6 mm glucose, 5 mm Hepes; pH 7.4). All tissue from transgenic mouse litters was kept separately for each animal to ensure no integration of tissue from different genotypes. The glands were digested with 5 ml of collagenase A (Roche Diagnostics Australia Pty Ltd, Castle Hill, NSW, Australia) in Locke's buffer (3 mg ml−1). The tubes were incubated in a shaking water bath at 37°C for 10 min and the tissue gently triturated every 5 min. Tubes were then centrifuged at 400 g for 10 min at 4°C. The tissue pellet was then resuspended in 5 ml of supplemented Dulbecco's modified Eagle's medium (DMEM) (Life Technologies Australia Pty Ltd, Sydney, NSW, Australia). The cell suspension was filtered through a 280 μm metal mesh (Sigma-Aldrich Pty Ltd, Castle Hill, NSW, Australia) and centrifuged at 400 g for 10 min at 4°C. The supernatant was discarded and the pellet was resuspended in 200 μl of supplemented DMEM. Cells were plated onto sterile polystyrene-coated 35 mm plastic Petri dishes (100 μl per plate) and plates were left in a 37°C, 5% CO2 incubator for ∼1.5 h to adhere. Quantities of 2 ml of supplemented DMEM containing 10% insulin-transferrin-selenium-ethanolamine (ITS-X) (Sorensen et al. 2003) were carefully added into each plate and the plates were incubated at 37°C in 5% CO2. Media were changed every 3 days and cells were used for experiments 4–7 days post-isolation to maximize secretory capacity.
Carbon fibre amperometry
Primary HAP1+/+ and HAP1−/− chromaffin cell cultures were used for amperometric experiments. Cells were prepared for an experiment by removing the culture medium and rinsing several times with Krebs solution (140 mm NaCl, 5 mm KCl, 2 mm CaCl2, 1 mm MgCl2, 10 mm glucose, 10 mm Hepes; pH 7.4). Cells were then viewed using an inverted microscope (Olympus IX71; Olympus Medical Systems Corp., Tokyo, Japan). A 5 μm diameter carbon fibre microelectrode (ProCFE; Dagen Corp., Minneapolis, MN, USA) was positioned with a micromanipulator (MP-285; Sutter Instrument Co., Novarto, CA, USA) in contact with the membrane of an isolated chromaffin cell and +800 mV applied to the electrode under voltage clamp conditions. The microelectrode was kept in place during the stimulation and throughout the secretion process. Current signals attributable to catecholamine oxidation were relayed to a patch clamp amplifier (EPC-7; List Medical GmbH, Darmstadt, Germany) controlled by a DELL computer with Pulse software (V8.78; HEKA Electronik GmbH, Lambrecht/Pfalz, Germany), sampled at 10 kHz and low-pass filtered at 1 kHz. All cells were visually inspected after recording to verify the absence of damage. Solutions used were applied to cells using a gravity perfusion system. All experiments were performed at 35–37°C using a temperature control unit (Warner Instruments, Inc., Hamden, CT, USA). Exocytosis was stimulated with a 70 mm high K+ Krebs solution for 1 min. This increased amount of K+ replaced an equimolar amount of NaCl. After each amperometric measurement, cells were bathed in control Krebs buffer and allowed to recover for at least 5 min before being restimulated for exocytosis. The number of vesicles released via Ca2+-independent exocytosis was evaluated by exposing the cells to a hypertonic external environment with Krebs buffer containing 500 mm sucrose (Rosenmund & Stevens, 1996) for 10 s, followed by control Krebs solution for 1 min.
Data analysis and statistics
Recorded Pulse files were converted to Axon Binary Files using ABF Utility Version 2.1 (Synaptosoft, Inc., Decatur, GA, USA) and secretory events were analysed using Mini Analysis Version 6.0.1 (Synaptosoft, Inc.). The recordings were analysed for 60 s from the start of stimulated secretion. The root-mean-squared (RMS) noise was 0.7–1.9 pA. Only spikes with amplitudes greater than four times the RMS noise threshold and foot signals higher than four times the RMS noise were included in the analysis. The level of noise did not differ between genotypes. All peaks identified by the program were inspected visually and poorly fitted peaks were either manually recalculated or excluded from datasets by the presence of complex traits (exhibiting multiple peak maxima) or noise interference. Such exclusions were not common or more prevalent in any particular genotype. Amperometric traces were not included for analysis if they contained fewer than 15 spikes or over 250 spikes within the 60 s stimulation period. Overlapping spikes were not included for foot or spike analysis. For each amperometric trace, the average spike amplitude, half-width, area, rise time and decay time were determined. The charge of a secretory event is the area under each spike (in picocoulombs, pC). Pre-spike foot (PSF) signal onset was defined when the signal exceeded the peak-to-peak noise of a 5 ms time segment. The end of the PSF was defined as the inflection point between the PSF signal and the spike. All analysed PSF signals had a height greater than four times the RMS noise. Stand-alone foot (SAF) signals were identified as foot signals which did not display a proceeding current spike.
Pooling of spike kinetics data can lead to biased results because they include a large number of events and cells vary in spike parameters (Colliver et al. 2001). To circumvent this potential bias, we determined a single statistic (the cell median of each spike parameter) for each cell and averaged this statistic for all cells of a particular genotype to aid in making comparisons between genotypes. Statistical analysis of amperometric kinetic data was carried out using the Mann–Whitney test, which is a distribution-free test that makes no underlying assumptions about the distribution of the data. With the loss of HAP1 no significant changes were observed during the separate analysis of spike kinetics for spikes without a PSF (stand-alone spikes) and with a PSF; therefore pooled data from both groups of spikes are presented. All data are expressed as the mean ± s.e.m. P-values of < 0.05 are considered to indicate statistical significance.
Capacitance measurements
Whole-cell patch clamp recording was performed using an EPC-10 patch clamp amplifier and PatchMaster software (HEKA Electronik GmbH). Patch pipettes were pulled from borosilicate glass and fire polished, with resistance of 3–5 MΩ. Patch clamping was performed in the perforated patch configuration for capacitance measurements, with internal solution containing 135 mm CsCl, 10 mm NaCl and 10 mm Hepes, adjusted to pH 7.2, and with 500 μg ml−1 amphotericin B. External solution contained 150 mm NaCl, 2.8 mm KCl, 10 mm Hepes, 2 mm MgCl2, 10 mm CaCl2 and 10 mm glucose, adjusted to pH 7.4 with NaOH. Capacitance measurements utilized the Lock-in module of the PatchMaster software, with capacitance change measured in response to a pulse of 200 ms duration to 10 mV from a resting membrane potential of −80 mV. Voltages shown are not adjusted for liquid junction potentials. All experiments were carried out at room temperature (22–24°C). Readily releasable pool (RRP) measurements utilized dual-pulse protocols, with dual 100 ms stimulations to 0 mV and 5 mV, respectively, separated by 100 ms at resting membrane potential (Gillis et al. 1996).
Electron microscopy
The adrenals were dissected out from the pups, cut in half and immediately fixed for 6 h at 4°C in 0.1 m phosphate buffer (PB) pH 7.2, containing 3% glutaraldehyde. The tissue was then transferred to 0.1 m PB, pH 7.2, and kept overnight at 4°C. The samples were washed three times in 0.1 m PB before being post-fixed in 1% osmium tetroxide in 0.1 m PB, pH 7.2, for 1 h at room temperature. After multiple washes the tissue was stained with 2% aqueous uranyl acetate for 30 min. Following a series of dehydration steps with ethanol solutions (50–100%) and propylene oxide, the tissue was incubated for 1 h in a 1 : 1 mixture of propylene oxide and Durcupan resin (Sigma-Aldrich Pty Ltd). The samples were then embedded in pure resin in capsules and polymerization was carried out for 48 h at 60°C. An ultramicrotome (RMC Mechanical Advance Ultramicrotome; Boekeler Instruments, Inc., Tucson, AZ, USA) was used to cut thick sections (1 μm), which were stained with 1% toluidine blue in 1% borax for light microscopic examinations. Ultrathin sections (80–100 nm) were cut using a diamond knife (Diatome AG, Biel, Switzerland) and mounted on single-slot copper grids coated with 0.6–0.8% Butvar solution in chloroform. The sections were then stained with Reynolds’ lead citrate solution (Reynolds, 1963) and allowed to dry overnight. The ultra-thin sections were observed under transmission electron microscopy (1200-EX transmission electron microscope; JEOL Ltd, Tokyo, Japan), and images were taken at ×10,000 and ×30,000 magnification. Ultrastructural images were captured with a MegaView3 camera (Olympus Soft Imaging Solutions GmbH, Münster, Germany) using the item interface program. Higher-resolution montages of selected cells were compiled using the Multiple Image Alignment module in item.
Chromaffin granule counting
Images were analysed using ImageJ software (National Institutes of Health, Bethesda, MD, USA). The number and spatial distribution of LDCVs containing catecholamines were evaluated on micrographs covering the cytoplasmic region of the entire cell. LDCVs were identified by their electron-dense core and an approximate circular membrane. The LDCVs were counted and placed in 100 nm bins according to their distance from the plasma membrane. Docked vesicles were defined as those vesicles making direct contact with the plasma membrane.
Calcium imaging
Chromaffin cells were loaded with the Ca2+ indicator Fluo-4 AM (5 μm) in Krebs buffer at 37°C for 45 min. Before each recording, cells were allowed to equilibrate in the Krebs bath solution for at least 10 min. Each recording included a 1 min baseline reading, 1 min stimulation with high K+ solution, washing with Krebs for 5 min, and stimulation with 100 μm dimethylphenylpiperazinium (DMPP) followed by washing with Krebs. Images were captured at an exposure of 0.03 s every 2 s. Results were analysed using Imaging Workbench Version 6.0.22 (INDEC Systems, Inc., Santa Clara, CA, USA), exported to GraphPad Prism Version 4.0 (GraphPad Software, Inc., La Jolla, CA, USA) and the average area under the curve calculated in each group. All experiments were carried out at 34–37°C.
Results
Loss of HAP1 reduces catecholamine secretion in chromaffin cells independent of Ca2+ entry
To gauge whether HAP1 regulates catecholamine release in chromaffin cells, we measured the release of catecholamines from single vesicles in HAP1+/+ and HAP1−/− mouse chromaffin cells using carbon fibre amperometry. We stimulated cells with a solution containing 70 mm K+ for 1 min, which triggered exocytosis in both HAP1+/+ (Fig. 1A) and HAP1−/− (Fig. 1B) chromaffin cells. The average number of vesicles undergoing release was significantly lower in HAP1−/− cells than in cells from HAP1+/+ littermates over this stimulation period (Fig. 1C), as was the total amount released per cell (Fig. 1D). To identify whether this reduction in exocytosis in HAP1−/− cells is caused by upstream reductions in Ca2+ entry, we performed single-cell Ca2+ imaging measurements in HAP1+/+ and HAP1−/− cells. In these experiments we repeated the stimulation protocol applied in the amperometry experiments using 70 mm K+ for 1 min. This caused an increase in intracellular Ca2+ levels in both HAP1+/+ (Fig. 2A) and HAP1−/− (Fig. 2B) cells measured as increases in Fluo-4 fluorescence. The total increase in cell fluorescence caused by this stimulation was similar in both groups (Fig. 2C), indicating that HAP1 affects exocytosis downstream of Ca2+ entry. We also observed a small but significant increase in the peak fluorescence change in HAP1−/− cells (Fig. 2D), but no change in the time taken to reach peak Ca2+ levels (Fig. 2E). It is also worth noting that the multiple components observed in the decay phase of the HAP1+/+, but not HAP1−/−, Ca2+ imaging trace provided are not indicative of differences between groups. Thus, a reduction in Ca2+ entry does not underlie the secretory defects we see in HAP1−/− cells.
Figure 1.
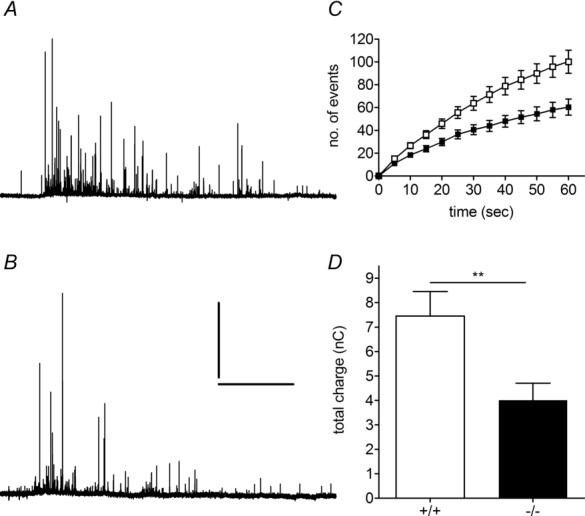
A, B, example amperometric traces from HAP1+/+ (A) and HAP1−/− (B) chromaffin cells. Scale bar in (B) = 20 s/100 pA for (A) and (B). C, rate of HAP1+/+ (white squares, n = 29 cells) and HAP1−/− (black squares, n = 35 cells) exocytosis in chromaffin cells over 1 min. D, average total charge released per cell after 1 min of stimulation in each group (**P < 0.01).
Figure 2.
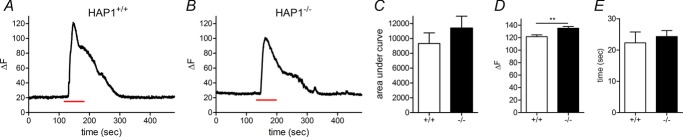
A, B, cells were stimulated with 70 mm K+ solution for 1 min (red lines below traces) causing a reversible increase in Fluo-4 fluorescence in single HAP1+/+ (A) and HAP1−/− (B) cells. C–E, the average area under the curve (C), peak fluorescence change (D) and time to peak fluorescence (E) were calculated for each genotype (n = 13 cells in each genotype; **P < 0.01).
Total release per vesicle is unaffected by HAP1
Although the mean number of exocytotic spikes per cell is reduced in HAP1−/− cells (Fig. 3A), individual spike parameters including spike amplitude, area, rise time, decay time and half-width are unchanged (Fig. 3B–F). We have combined data from all spikes as we observed no differences between genotypes in these spike parameters for events that either do or do not contain a PSF (Table 1). Similarly, when we compared the distribution of these values, including amplitude, area and rise time (Fig. 3G–I), we found no difference between HAP1+/+ and HAP1−/− cells. These data clearly demonstrate that HAP1 does not affect the amount of catecholamine released from vesicles during full fusion.
Figure 3.
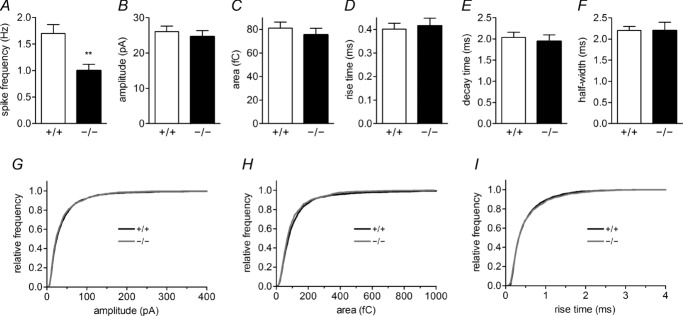
A–F, the number of amperometric spikes per recording is reduced in HAP1−/− cells (A), but no changes are observed in mean spike amplitude (B), area (C), rise time (D), decay time (E) or half-width (F). G–I, between-groups comparisons for these parameters show no difference in amplitude (G), area (H) or rise time (I). (**P < 0.01.)
table 1.
Events were characterized with respect to their peak kinetic parameters (amplitude, 60–80% rise time, area, half-width and 37–80% decay time) and pre-spike foot (PSF) duration, height and area
| PSF parameters |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Events, n | Amplitude, pA | Rise time, ms | Area, fC | Half-width, ms | Decay time, ms | Duration, ms | Height, pA | Area, fC | Events with PSF, % | |
| HAP1+/+ | 102.2 ± 10.2 | 26.1 ± 1.6 | 0.4 ± 0.03 | 81.2 ± 5.1 | 2.2 ± 0.1 | 2.0 ± 0.1 | 2.3 ± 0.1 | 12.2 ± 0.4 | 13.3 ± 0.6 | 30.3 ± 2.5 |
| HAP1−/− | 60.4 ± 7.1* | 24.6 ± 1.7 | 0.4 ± 0.03 | 75.5 ± 5.6 | 2.2 ± 0.2 | 1.9 ± 0.2 | 3.04 ± 0.1† | 11.2 ± 0.4 | 17.1 ± 0.9† | 26.1 ± 3.0 |
The parameter ‘Events with PSF, %’ indicates the mean percentage of amperometric spikes preceded by a PSF signal. Data are given as mean ± s.e.m. Total numbers of events analysed per genotype for spike parameters are HAP1+/+ 1792 (29 cells) and HAP1−/− 1323 (35 cells). Total numbers of events analysed per genotype for PSF parameters are HAP1+/+ 523 and HAP1−/− 326.
P < 0.01;
P < 0.001.
HAP1 differentially regulates fusion pore formation during full fusion or kiss-and-run exocytosis
We also analysed the PSF signal in these recordings as this represents the amount of catecholamine flux through the unstable fusion pore before it stabilizes into a full fusion pore (Zhou et al. 1996; Fang et al. 2008; Zhang & Jackson, 2010). The PSF is visible before many amperometric spikes (Fig. 4A) and SAF signals represent kiss-and-run exocytosis when an unstable fusion pore closes prior to full fusion occurring (Fig. 4B). We hypothesized that if HAP1 affects fusion pore stability, then the incidence of kiss-and-run fusion may be altered in the absence of HAP1. We observed SAF signals in our recordings and found the average number of SAF signals observed during stimulation to be similar in HAP1+/+ and HAP1−/− cells (Fig. 4C). Thus the relative number of kiss-and-run events to full fusion events increases in HAP1−/− cells and we therefore found, on average, a SAF signal for every 12 full spikes in HAP1−/− cells compared to every 24 full spikes in HAP1+/+ cells (Fig. 4D). We observed obvious differences when we compared the effect of HAP1 on the kinetics associated with the early fusion pore when it either does (PSF) or does not (SAF) transition into a full fusion event. HAP1−/− cells display no change in PSF amplitude (Fig. 4E) but a larger SAF amplitude (Fig. 4F), an increased PSF duration (Fig. 4G) but decreased SAF duration (Fig. 4H), and an increased PSF area (Fig. 4I) but unchanged SAF area (Fig. 4J). These data suggest HAP1 is involved in the earliest stages of fusion pore development, but differentially regulates the stabilization of the early fusion pore according to whether full fusion or kiss-and-run fusion occur.
Figure 4.
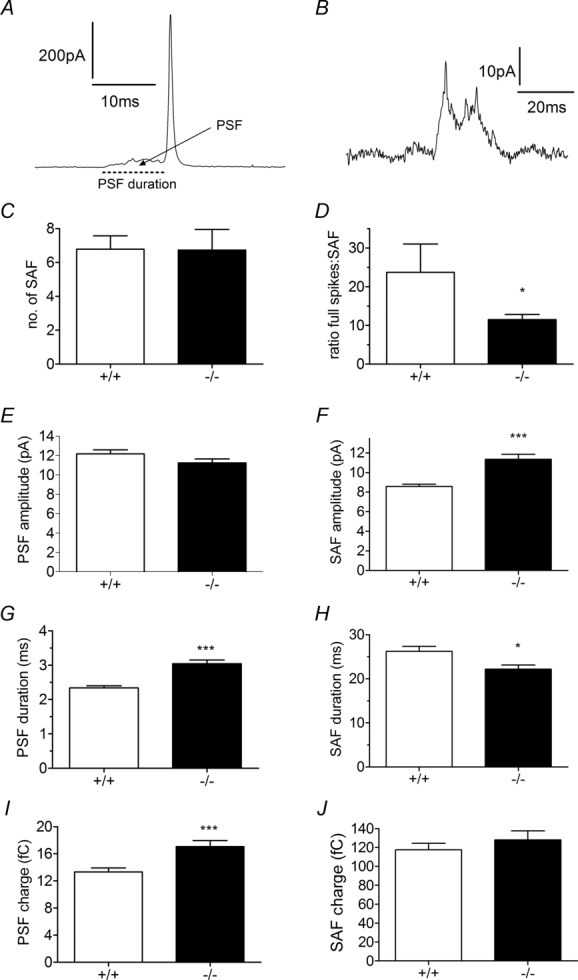
A, a full fusion event preceded by a pre-spike foot (PSF) signal (arrow, dotted line indicates PSF duration) representing catecholamine release during fusion pore formation and stabilization. B, when a fusion pore does not stabilize into a full fusion pore but instead reverses and closes, only a stand-alone foot (SAF) signal is observed. C, D, there is no change in the average number of SAF events (C), and SAF events are more frequent in relation to full fusion events in HAP1−/− cells (D). E, F, in HAP1−/− cells, PSF amplitude is unchanged (E), but SAF amplitude is increased (F). G, H, PSF duration is longer in HAP1−/− cells (G), but SAF duration is shorter (H). I, J, in HAP1−/− cells, PSF area is larger (I), but SAF charge is unchanged (J). (*P < 0.05, ***P < 0.001; n = 523 PSF and n = 186 SAF in HAP1+/+ cells; n = 326 PSF and n = 192 SAF in HAP1−/− cells.)
HAP1 reduces the fraction released during early pore flickering
Given that we observed an increased PSF duration and area in HAP1−/− cells, we measured the fraction of total catecholamine released from the PSF compared with the PSF plus full spike. Unsurprisingly, the fractional release from the PSF is larger in HAP1−/− cells in terms of both the mean value (Fig. 5A) and at any particular range of Q⅓ (Fig. 5B).
Figure 5.
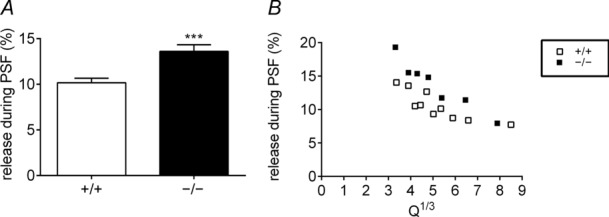
A, B, the fractional release from the PSF is larger in HAP1−/− cells, as illustrated by the average of the cell mean values (A) and plots of specific kinetic parameters at different values of Q⅓ (B).
Ca2+-independent exocytosis is also regulated by HAP1
We tested whether Ca2+-independent exocytosis is reduced in HAP1−/− cells by exposing cells to a hypertonic solution containing 500 mm sucrose. Brief (10 s) exposure of cells to such a solution followed by their rapid return to isotonic solution causes Ca2+-independent release of vesicles. In these experiments, we observed the release of vesicles in HAP1+/+ cells (Fig. 6A) and in HAP1−/− cells (Fig. 6B). However, significantly less release is observed with this stimulus in HAP1−/− cells (Fig. 6C).
Figure 6.
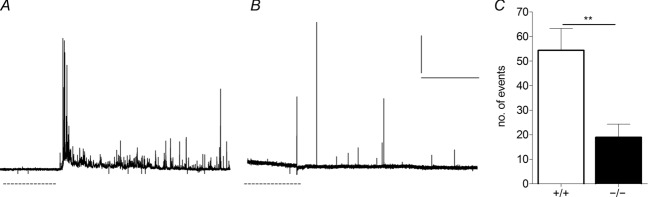
A, B, HAP1+/+ (A) and HAP1−/− (B) cells were exposed for 10 s to a hyperosmotic solution (dashed line) and rapidly returned to isotonic solution to measure the size of the readily releasable pool (RRP). C, the mean number of vesicles in the RRP is lower in HAP1−/− cells than in HAP1+/+ cells. (**P < 0.01; n = 7 HAP1+/+ cells, n = 9 HAP1−/− cells.)
HAP1 controls vesicle docking and the size of the RRP
As HAP1 is associated with vesicle trafficking and as recent work illustrates a potential vesicle-docking defect in the absence of HAP1 (Cape et al. 2012), we hypothesized that the earliest phase of release may be negatively affected in these cells. To examine this further, we undertook electron microscopy analysis of the average vesicle number and vesicle localization in HAP1+/+ and HAP1−/− chromaffin cells. We distinguished vesicle localization to the resolution of those vesicles in direct contact (docked) or not in contact (undocked) with the plasma membrane. We have demonstrated this with a diagram (Fig. 7Aa) and with an electron micrograph image (Fig. 7Ab). Electron micrographs clearly illustrate the presence of LDCVs in both HAP1+/+ (Fig. 7B) and HAP1−/− (Fig. 7C) cells. We did not observe any difference between these groups in terms of the number of vesicles per unit area (Fig. 7D). In addition, little difference in the localization of vesicles emerges when vesicles are pooled into 100 nm bins according to their distance from the plasma membrane (Fig. 7E). However, when we assessed the number of vesicles in contact with the plasma membrane, we found significantly fewer LDCVs on the membrane of HAP1−/− cells (Fig. 7F).
Figure 7.

Aa, undocked and morphologically docked large dense core vesicles (LDCVs) at the plasma membrane (PM). Ab, an electron micrograph shows a primed vesicle undergoing fusion. B, C, electron micrographs from HAP1+/+ (B) and HAP1−/− (C) chromaffin cells clearly identify LDCVs; red arrows indicate morphologically docked LDCVs. D, E, there are no changes in the total number of vesicles per unit area (D) or the distance of vesicles from the plasma membrane (in 100 nm bins) except in those 300–500 nm from the PM (E). F, the number of vesicles morphologically docked at the plasma membrane is significantly lower in HAP1−/− cells. (*P < 0.05; n = 9 HAP1+/+ cells, n = 7 HAP1−/− cells. Scale bars: Ab, 100 nm; B, C, 500 nm. Data represent the mean ± s.e.m.)
To further examine vesicle release from the RRP immediately following stimulation, we undertook membrane capacitance measurements. Cells were stimulated with a dual-pulse depolarization protocol (Fig. 8A), and membrane capacitance was monitored before and after stimulation. Depolarization induced a robust increase in membrane capacitance in HAP1+/+ cells (black lines, Fig. 8A), reflecting the exocytosis of secretory granules (Smith et al. 1998; Xu et al. 1999). The secretory response was clearly reduced in HAP1−/− cells (grey lines, Fig. 8A), whereas the Ca2+ currents were identical to those in HAP1+/+ cells (Fig. 8B and C). Data from this dual-pulse protocol were used to calculate the size of the RRP of vesicles (Gillis et al. 1996; Smith et al. 1998; Xu et al. 1999), a kinetically defined parameter considered to reflect vesicles already docked to the plasma membrane in a fusion-competent state. When the two depolarizations are given in rapid succession, in this case with a 100-ms delay, the maximal size of the RRP, Bmax, can be derived from the equation: Bmax = S/(1 – R2), where S = the sum of the capacitance responses to the first (ΔC1) and second (ΔC2) depolarizations, and R = the ratio of ΔC2/ΔC1 to reflect the fraction of LDCVs in the RRP mobilized by stimulation. Our analysis demonstrates that the size of the initial capacitance jump is reduced in HAP1−/− cells (P < 0.05) (Fig. 8D) and that the RRP in HAP1−/− cells is only 45% of that in wild-type cells (P < 0.05) (Fig. 8E). R-values were similar for HAP1+/+ and HAP1−/− chromaffin cells (Fig. 8F), indicating that HAP1 depletion does not change the release rate. We also elicited secretion in these experiments using a single voltage pulse from −80 mV to 10 mV for 200 ms. This triggered a significant increase in membrane capacitance in both HAP1+/+ and HAP1−/− cells (Fig. 8G). Such a stimulation protocol triggered two distinct rates of exocytosis: a fast component over the first ∼3 s, followed by a slower component. When we compared the average changes in membrane capacitance before and after this 3 s time-point, we found fast exocytosis immediately after the pulse to be significantly decreased in HAP1−/− cells, but the slower component to be unchanged (Fig. 8H). Again, this change in exocytosis does not reflect smaller Ca2+ currents in HAP1−/− cells (Fig. 8I).
Figure 8.
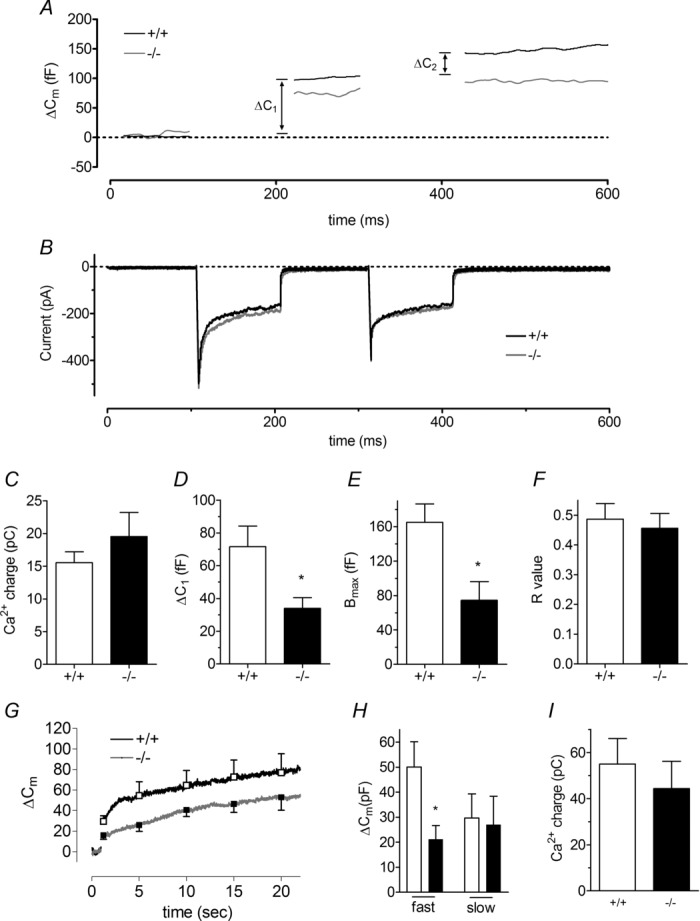
A–C, in the absence of HAP1, exocytosis is decreased during a dual-pulse stimulation (A), whereas the size of the integrated Ca2+ current is unchanged (B, C). D–F, there is a significant difference in both ΔC1 (D), and the maximal size of the readily releasable pool (RRP) (Bmax) (E), whereas the ratio of ΔC1 to ΔC2 (R) is unchanged (F) (n = 17 HAP1+/+ cells, n = 4 HAP1−/− cells). G, fast (<3 s) and slow (>3 s) phases of exocytosis are triggered by a single 200 ms voltage pulse to 0 mV. H, the fast but not the slow component of exocytosis is significantly reduced in HAP1−/− cells. I, these changes do not reflect reduced Ca2+ current size in HAP1−/− cells. (*P < 0.05; n = 6 HAP1+/+ cells, n = 9 HAP1−/− cells.)
Discussion
This study illustrates novel roles for HAP1 in the control of cell signalling. We demonstrate that loss of HAP1 negatively affects the number of vesicles undergoing exocytosis in adrenal chromaffin cells and that this effect is not attributable to changes in Ca2+ entry or the number of LDCVs. HAP1 regulates exocytosis via two potentially interlinked mechanisms: by regulating the docking of vesicles on the plasma membrane and the size of the RRP, and by influencing the stability of the fusion pore as it forms.
HAP1 is localized to synaptic vesicles (Li et al. 2000) and LDCVs (Wu et al. 2010) and a lack of HAP1 reduces insulin secretion in pancreatic β cells (Cape et al. 2012). HAP1 is abundantly expressed in hypothalamic orexin neurons (Lin et al. 2010), which play an important role in regulating feeding and behaviour. Ablation of HAP1 expression in these neurons causes a loss of feeding, impairs neuronal process length and reduces the distribution of trafficking protein complexes and cargo proteins to synapses (Lin et al. 2010). Our data illustrating that catecholamine secretion is reduced in HAP1−/− chromaffin cells suggest that HAP1 may be an important protein controlling exocytosis in endocrine cells. Such a role is supported by the discrete expression pattern of HAP1 in neurons and endocrine cells but not in non-endocrine tissues (Liao et al. 2005). The reduced RRP size and vesicle docking we observe in HAP1−/− chromaffin cells is in agreement with the altered vesicle localization reported in HAP1−/− β cells (Cape et al. 2012) and suggests that reduced trafficking of vesicles to the plasma membrane and priming for release may be a major mechanism by which HAP1 regulates exocytosis. HAP1 has previously been associated with vesicle trafficking in neurons as a result of its association with the cytoskeletal protein DUO (Colomer et al. 1997), the p150Glued subunit of the motor protein dynactin (Li et al. 1998) and the trafficking protein kinesin light chain (McGuire et al. 2006). Whether the altered function of these or other unidentified HAP1 binding partners in HAP1−/− chromaffin cells underlies this docking defect is currently unknown.
Our amperometry experiments demonstrate that HAP1 does not affect the amount of catecholamine released per vesicle during full fusion events. This indicates that HAP1 does not regulate the loading of catecholamines into vesicles and that once full fusion occurs, HAP1 has no further influence on exocytosis. The PSF represents the release of catecholamines through the developing fusion pore as it undergoes the transition from a small unstable, flickering pore to a stable, irreversible full fusion state (Zhou et al. 1996; Zhang & Jackson, 2010). Similarly, the SAF represents this flickering, unstable fusion pore that transitions to a closed state rather than full pore opening. The duration of the PSF or SAF signal reflects the transient pore lifetime, and PSF or SAF signal height relates to pore conductance during this transient state (Zhou et al. 1996; Fang et al. 2008; Zhang & Jackson, 2010). Thus, the changes in PSF and SAF signal duration in HAP1−/− cells indicate that HAP1 plays a role in stabilization and/or conductance in the fusion pore as it transitions to either a full fusion state or pore re-closure. It is curious that HAP1 appears to affect the early fusion pore lifetime differently depending on the fate of that fusion event. We do not as yet understand how HAP1 would accelerate pore transition from early fusion to full fusion but prolong fusion pore flickering during kiss-and-run fusion. These data do, however, provide further evidence that full fusion and kiss-and-run fusion may be controlled by different means, as has been suggested previously (Zanin et al. 2011).
The observation that HAP1 has no effect on the incidence of kiss-and-run exocytosis per cell is unexpected given the significant drop in full fusion events in HAP1−/− cells. Thus, there is an increase in kiss-and-run events relative to the total number of fusion events occurring in HAP1−/− cells. There are two possible explanations for this. Firstly, different mechanisms may regulate whether a vesicle will undergo full or transient fusion, as we have previously postulated (Zanin et al. 2011), and HAP1 may not be involved in regulating the incidence of kiss-and-run fusion. Alternatively, altered fusion pore development and stabilization attributable to the lack of HAP1 may underlie the occurrence of relatively more kiss-and-run events in HAP1−/− cells.
Exocytosis consists of a series of interlinked steps. Several studies illustrate that docking and priming are molecularly intertwined and that the functional disruption of priming and fusion proteins significantly affects docking (de Wit et al. 2006; Hammarlund et al. 2007; Yizhar & Ashery, 2008). As electron microscopy cannot distinguish between morphologically docked vesicles and ‘functionally docked’ vesicles that are primed and fusion competent, our data do not exclude a role of HAP1 in vesicle priming. By affecting priming, HAP1 may alter both vesicle docking and fusion, thereby decreasing the size of the RRP and total number of exocytotic events. Other vesicle trafficking-related proteins such as myosin II and F-actin also regulate fusion pore stability and lifetime (Berberian et al. 2009; Doreian et al. 2009). As fusion pore opening can only occur once a vesicle is docked and primed for release, the HAP1-dependent mechanism regulating docking may be the same as that regulating early fusion pore stability and lifetime.
HAP1 is a binding partner of the trafficking protein, huntingtin (Htt), mutations in which underlie HD. The binding of HAP1 to mutant Htt is stronger than to normal Htt (Li et al. 1995), potentially altering some of the functions of HAP1. Reductions in chromaffin cell (Johnson et al. 2007) and β cell (Bjorkqvist et al. 2005) secretion, and in presynaptic release (Johnson et al. 2006), are observed in mouse models of HD. The role of HAP1 as a positive regulator of chromaffin cell exocytosis has implications in the release of adrenaline and noradrenaline during the fight-or-flight response. Similarities in the mechanisms controlling hormone release exist in other HAP1-expressing endocrine tissues (e.g. pituitary, thyroid and pancreas) (Dragatsis et al. 2000; Liao et al. 2005) and, as such, HAP1 may be a key regulator of cell signalling in these endocrine tissues also. Consistent with this hypothesis is the reduced vesicle docking and the release of insulin when HAP1 expression is ablated specifically in pancreatic β cells (Cape et al. 2012). The reduced catecholamine and insulin secretion (Bjorkqvist et al. 2005; Johnson et al. 2007), as well as the hypothalamic degeneration (Petersen et al. 2005) observed in HD mouse models parallel the reduced plasma adrenaline levels (Aminoff et al. 1974) and increased incidence of diabetes (Podolsky et al. 1972) in HD individuals, and the degeneration of up to 90% of the lateral hypothalamic neurons occurs in late stages of HD (Kremer et al. 1990, 1991). Given these similarities, the fact that HAP1 binds Htt, that this binding increases in HD (Li et al. 1995) and that polymorphisms in HAP1 alter the age of onset in HD individuals (Metzger et al. 2008), our data support the proposition that HAP1 may play a role in several neurochemical and endocrine changes occurring in HD.
The molecular mechanisms underlying impaired neurotransmission in HD are still largely unknown and are likely to involve multiple pathogenic mechanisms. Both Htt and HAP1 are anterogradely and retrogradely transported (Block-Galarza et al. 1997) and mutant Htt and HAP1 colocalize in axonal terminals in the brains of HD transgenic mice, but tighter binding to synaptic vesicles by mutant Htt decreases HAP1 synaptic vesicle localization (Li et al. 2003b). As our data indicate that HAP1 regulates exocytosis, it is conceivable that mutant Htt disrupts the normal role of HAP1 in exocytosis. Defective synaptic vesicle exocytosis has been directly linked to neurodegeneration: for example, a complete loss of neurotransmitter release in Munc18–1 null mice was found to lead to widespread neuronal death (Verhage et al. 2000) and a reduced number of synaptic connections (Bouwman et al. 2004). Such examples illustrate a direct relationship between defects in synaptic exocytosis and the onset of neurodegeneration. In the brain, HAP1 is present in the highest concentration in the hypothalamus and the lethal phenotype of HAP1−/− animals is linked to disturbed hypothalamic function. Thus, future work regarding the potential roles of HAP1 in the context of HD and cell signalling may provide new insights into cellular dysfunction in this complex neurological disorder.
Key points
Huntingtin-associated protein 1 (HAP1) is expressed in neurons and endocrine cells, in which it is thought to regulate vesicle trafficking.
HAP1 is a binding partner of the Huntington's disease (HD)-causing protein huntingtin, and binding is stronger in HD.
Whether HAP1 regulates a significant end-point of vesicle transport, exocytosis, and what stage of exocytosis HAP1 may regulate, is unknown.
We use mouse chromaffin cells to demonstrate that HAP1 regulates exocytosis via two potentially interlinked mechanisms: control of vesicle docking and the readily releasable vesicle pool, and regulation of fusion pore stabilization.
These results establish HAP1 as a significant player in exocytosis control with potential relevance for HD and for a number of neuronal and homeostatic pathways.
Acknowledgments
All experiments were performed in the Molecular and Cellular Neuroscience Laboratory, Centre for Neuroscience, Flinders University.
Glossary
- HAP1
huntingtin-associated protein 1
- HD
Huntington's disease
- Htt
huntingtin
- LDCVs
large dense core vesicles
- PSF
pre-spike foot
- RRP
readily releasable pool
- SAF
stand-alone foot
Additional information
Competing interests
None declared.
Author contributions
D.J.K., X.-F.Z. and K.D.M. were involved in the conception and design of the experiments. K.D.M., M.D.D., H.P., L.P., M.P.Z., E.H.T. and D.J.K. were involved in the collection, analysis and interpretation of data. D.J.K., X.-F.Z., K.D.M. and M.D.D. drafted the article and revised it critically for important intellectual content. All authors approved the final version of the manuscript.
Funding
This work was funded by the Australian Research Council through a Discovery Grant (D.J.K. and X.-F.Z.) and Future Fellowship (D.J.K.), and by a National Health and Medical Research Council Senior Research Fellowship (X.-F.Z.).
References
- Aminoff MJ, Trenchard A, Turner P, Wood WG, Hills M. Plasma uptake of dopamine and 5-hydroxytryptamine and plasma-catecholamine levels in patients with Huntington's chorea. Lancet. 1974;2:1115–1116. doi: 10.1016/s0140-6736(74)90873-3. [DOI] [PubMed] [Google Scholar]
- Berberian K, Torres AJ, Fang Q, Kisler K, Lindau M. F-actin and myosin II accelerate catecholamine release from chromaffin granules. J Neurosci. 2009;29:863–870. doi: 10.1523/JNEUROSCI.2818-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertaux F, Sharp AH, Ross CA, Lehrach H, Bates GP, Wanker E. HAP1–huntingtin interactions do not contribute to the molecular pathology in Huntington's disease transgenic mice. FEBS Letters. 1998;426:229–232. doi: 10.1016/s0014-5793(98)00352-4. [DOI] [PubMed] [Google Scholar]
- Bjorkqvist M, Fex M, Renstrom E, Wierup N, Petersen A, Gil J, Bacos K, Popovic N, Li JY, Sundler F, Brundin P, Mulder H. The R6/2 transgenic mouse model of Huntington's disease develops diabetes due to deficient β-cell mass and exocytosis. Hum Mol Genet. 2005;14:565–574. doi: 10.1093/hmg/ddi053. [DOI] [PubMed] [Google Scholar]
- Block-Galarza J, Chase KO, Sapp E, Vaughn KT, Vallee RB, DiFiglia M, Aronin N. Fast transport and retrograde movement of huntingtin and HAP 1 in axons. Neuroreport. 1997;8:2247–2251. doi: 10.1097/00001756-199707070-00031. [DOI] [PubMed] [Google Scholar]
- Bouwman J, Maia AS, Camoletto PG, Posthuma G, Roubos EW, Oorschot VM, Klumperman J, Verhage M. Quantification of synapse formation and maintenance in vivo in the absence of synaptic release. Neuroscience. 2004;126:115–126. doi: 10.1016/j.neuroscience.2004.03.027. [DOI] [PubMed] [Google Scholar]
- Cape A, Chen X, Wang CE, O'Neill A, Lin YF, He J, Xu XS, Yi H, Li H, Li S, Li XJ. Loss of huntingtin-associated protein 1 impairs insulin secretion from pancreatic β-cells. Cell Mol Life Sci. 2012;69:1305–1317. doi: 10.1007/s00018-011-0692-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colliver TL, Hess EJ, Ewing AG. Amperometric analysis of exocytosis at chromaffin cells from genetically distinct mice. J Neurosci Methods. 2001;105:95–103. doi: 10.1016/s0165-0270(00)00359-9. [DOI] [PubMed] [Google Scholar]
- Colomer V, Engelender S, Sharp AH, Duan K, Cooper JK, Lanahan A, Lyford G, Worley P, Ross CA. Huntingtin-associated protein 1 (HAP1) binds to a Trio-like polypeptide, with a rac1 guanine nucleotide exchange factor domain. Hum Mol Genet. 1997;6:1519–1525. doi: 10.1093/hmg/6.9.1519. [DOI] [PubMed] [Google Scholar]
- de Wit H, Cornelisse LN, Toonen RF, Verhage M. Docking of secretory vesicles is syntaxin dependent. PLoS One. 2006;1:e126. doi: 10.1371/journal.pone.0000126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delgado-Martinez I, Nehring RB, Sorensen JB. Differential abilities of SNAP-25 homologs to support neuronal function. J Neurosci. 2007;27:9380–9391. doi: 10.1523/JNEUROSCI.5092-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doreian BW, Fulop TG, Meklemburg RL, Smith CB. Cortical F-actin, the exocytic mode, and neuropeptide release in mouse chromaffin cells is regulated by myristoylated alanine-rich C-kinase substrate and myosin II. Mol Biol Cell. 2009;20:3142–3154. doi: 10.1091/mbc.E09-03-0197. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Dragatsis I, Dietrich P, Zeitlin S. Expression of the huntingtin-associated protein 1 gene in the developing and adult mouse. Neurosci Lett. 2000;282:37–40. doi: 10.1016/s0304-3940(00)00872-7. [DOI] [PubMed] [Google Scholar]
- Fang Q, Berberian K, Gong LW, Hafez I, Sorensen JB, Lindau M. The role of the C terminus of the SNARE protein SNAP-25 in fusion pore opening and a model for fusion pore mechanics. Proc Natl Acad Sci U S A. 2008;105:15388–15392. doi: 10.1073/pnas.0805377105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillis KD, Mossner R, Neher E. Protein kinase C enhances exocytosis from chromaffin cells by increasing the size of the readily releasable pool of secretory granules. Neuron. 1996;16:1209–1220. doi: 10.1016/s0896-6273(00)80147-6. [DOI] [PubMed] [Google Scholar]
- Gutekunst CA, Li SH, Yi H, Ferrante RJ, Li XJ, Hersch SM. The cellular and subcellular localization of huntingtin-associated protein 1 (HAP1): comparison with huntingtin in rat and human. J Neurosci. 1998;18:7674–7686. doi: 10.1523/JNEUROSCI.18-19-07674.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammarlund M, Palfreyman MT, Watanabe S, Olsen S, Jorgensen EM. Open syntaxin docks synaptic vesicles. PLoS Biol. 2007;5:e198. doi: 10.1371/journal.pbio.0050198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson MA, Rajan V, Miller CE, Wightman RM. Dopamine release is severely compromised in the R6/2 mouse model of Huntington's disease. J Neurochem. 2006;97:737–746. doi: 10.1111/j.1471-4159.2006.03762.x. [DOI] [PubMed] [Google Scholar]
- Johnson MA, Villanueva M, Haynes CL, Seipel AT, Buhler LA, Wightman RM. Catecholamine exocytosis is diminished in R6/2 Huntington's disease model mice. J Neurochem. 2007;103:2102–2110. doi: 10.1111/j.1471-4159.2007.04908.x. [DOI] [PubMed] [Google Scholar]
- Kremer HP, Roos RA, Dingjan G, Marani E, Bots GT. Atrophy of the hypothalamic lateral tuberal nucleus in Huntington's disease. J Neuropathol Exp Neurol. 1990;49:371–382. doi: 10.1097/00005072-199007000-00002. [DOI] [PubMed] [Google Scholar]
- Kremer HP, Roos RA, Dingjan GM, Bots GT, Bruyn GW, Hofman MA. The hypothalamic lateral tuberal nucleus and the characteristics of neuronal loss in Huntington's disease. Neurosci Lett. 1991;132:101–104. doi: 10.1016/0304-3940(91)90443-w. [DOI] [PubMed] [Google Scholar]
- Li SH, Gutekunst CA, Hersch SM, Li XJ. Interaction of huntingtin-associated protein with dynactin P150Glued. J Neurosci. 1998;18:1261–1269. doi: 10.1523/JNEUROSCI.18-04-01261.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li SH, Li H, Torre ER, Li XJ. Expression of huntingtin-associated protein-1 in neuronal cells implicates a role in neuritic growth. Mol Cell Neurosci. 2000;16:168–183. doi: 10.1006/mcne.2000.0858. [DOI] [PubMed] [Google Scholar]
- Li SH, Yu ZX, Li CL, Nguyen HP, Zhou YX, Deng C, Li XJ. Lack of huntingtin-associated protein-1 causes neuronal death resembling hypothalamic degeneration in Huntington's disease. J Neurosci. 2003a;23:6956–6964. doi: 10.1523/JNEUROSCI.23-17-06956.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Wyman T, Yu ZX, Li SH, Li XJ. Abnormal association of mutant huntingtin with synaptic vesicles inhibits glutamate release. Hum Mol Genet. 2003b;12:2021–2030. doi: 10.1093/hmg/ddg218. [DOI] [PubMed] [Google Scholar]
- Li XJ, Li SH. HAP1 and intracellular trafficking. Trends Pharmacol Sci. 2005;26:1–3. doi: 10.1016/j.tips.2004.11.001. [DOI] [PubMed] [Google Scholar]
- Li XJ, Li SH, Sharp AH, Nucifora FC, Jr, Schilling G, Lanahan A, Worley P, Snyder SH, Ross CA. A huntingtin-associated protein enriched in brain with implications for pathology. Nature. 1995;378:398–402. doi: 10.1038/378398a0. [DOI] [PubMed] [Google Scholar]
- Li XJ, Sharp AH, Li SH, Dawson TM, Snyder SH, Ross CA. Huntingtin-associated protein (HAP1): discrete neuronal localizations in the brain resemble those of neuronal nitric oxide synthase. Proc Natl Acad Sci U S A. 1996;93:4839–4844. doi: 10.1073/pnas.93.10.4839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao M, Shen J, Zhang Y, Li SH, Li XJ, Li H. Immunohistochemical localization of huntingtin-associated protein 1 in endocrine system of the rat. J Histochem Cytochem. 2005;53:1517–1524. doi: 10.1369/jhc.5A6662.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin YF, Xu X, Cape A, Li S, Li XJ. Huntingtin-associated protein-1 deficiency in orexin-producing neurons impairs neuronal process extension and leads to abnormal behaviour in mice. J Biol Chem. 2010;285:15941–15949. doi: 10.1074/jbc.M110.107318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Arca S, Coco S, Mainguy G, Schenk U, Alberts P, Bouille P, Mezzina M, Prochiantz A, Matteoli M, Louvard D, Galli T. A common exocytotic mechanism mediates axonal and dendritic outgrowth. J Neurosci. 2001;21:3830–3838. doi: 10.1523/JNEUROSCI.21-11-03830.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuire JR, Rong J, Li SH, Li XJ. Interaction of huntingtin-associated protein-1 with kinesin light chain: implications in intracellular trafficking in neurons. J Biol Chem. 2006;281:3552–3559. doi: 10.1074/jbc.M509806200. [DOI] [PubMed] [Google Scholar]
- Metzger S, Rong J, Nguyen HP, Cape A, Tomiuk J, Soehn AS, Propping P, Freudenberg-Hua Y, Freudenberg J, Tong L, Li SH, Li XJ, Riess O. Huntingtin-associated protein-1 is a modifier of the age-at-onset of Huntington's disease. Hum Mol Genet. 2008;17:1137–1146. doi: 10.1093/hmg/ddn003. [DOI] [PubMed] [Google Scholar]
- Page KJ, Potter L, Aronni S, Everitt BJ, Dunnett SB. The expression of huntingtin-associated protein (HAP1) mRNA in developing, adult and ageing rat CNS: implications for Huntington's disease neuropathology. Euro J Neurosci. 1998;10:1835–1845. doi: 10.1046/j.1460-9568.1998.00185.x. [DOI] [PubMed] [Google Scholar]
- Petersen A, Gil J, Maat-Schieman ML, Bjorkqvist M, Tanila H, Araujo IM, Smith R, Popovic N, Wierup N, Norlen P, Li JY, Roos RA, Sundler F, Mulder H, Brundin P. Orexin loss in Huntington's disease. Hum Mol Genet. 2005;14:39–47. doi: 10.1093/hmg/ddi004. [DOI] [PubMed] [Google Scholar]
- Podolsky S, Leopold NA, Sax DS. Increased frequency of diabetes mellitus in patients with Huntington's chorea. Lancet. 1972;1:1356–1358. doi: 10.1016/s0140-6736(72)91092-6. [DOI] [PubMed] [Google Scholar]
- Reynolds ES. The use of lead citrate at high pH as an electron-opaque stain in electron microscopy. J Cell Biol. 1963;17:208–212. doi: 10.1083/jcb.17.1.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenmund C, Stevens CF. Definition of the readily releasable pool of vesicles at hippocampal synapses. Neuron. 1996;16:1197–1207. doi: 10.1016/s0896-6273(00)80146-4. [DOI] [PubMed] [Google Scholar]
- Smith C, Moser T, Xu T, Neher E. Cytosolic Ca2+ acts by two separate pathways to modulate the supply of release-competent vesicles in chromaffin cells. Neuron. 1998;20:1243–1253. doi: 10.1016/s0896-6273(00)80504-8. [DOI] [PubMed] [Google Scholar]
- Sorensen JB, Nagy G, Varoqueaux F, Nehring RB, Brose N, Wilson MC, Neher E. Differential control of the releasable vesicle pools by SNAP-25 splice variants and SNAP-23. Cell. 2003;114:75–86. doi: 10.1016/s0092-8674(03)00477-x. [DOI] [PubMed] [Google Scholar]
- Verhage M, Maia AS, Plomp JJ, Brussaard AB, Heeroma JH, Vermeer H, Toonen RF, Hammer RE, van den Berg TK, Missler M, Geuze HJ, Sudhof TC. Synaptic assembly of the brain in the absence of neurotransmitter secretion. Science. 2000;287:864–869. doi: 10.1126/science.287.5454.864. [DOI] [PubMed] [Google Scholar]
- Wu LL, Fan Y, Li S, Li XJ, Zhou XF. Huntingtin-associated protein-1 interacts with pro-brain-derived neurotrophic factor and mediates its transport and release. J Biol Chem. 2010;285:5614–5623. doi: 10.1074/jbc.M109.073197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu T, Ashery U, Burgoyne RD, Neher E. Early requirement for α-SNAP and NSF in the secretory cascade in chromaffin cells. EMBO J. 1999;18:3293–3304. doi: 10.1093/emboj/18.12.3293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yizhar O, Ashery U. Modulating vesicle priming reveals that vesicle immobilization is necessary but not sufficient for fusion-competence. PLoS One. 2008;3:e2694. doi: 10.1371/journal.pone.0002694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanin MP, Phillips L, Mackenzie KD, Keating DJ. Aging differentially affects multiple aspects of vesicle fusion kinetics. PLoS One. 2011;6:e27820. doi: 10.1371/journal.pone.0027820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Jackson MB. Membrane bending energy and fusion pore kinetics in Ca2+-triggered exocytosis. Biophys J. 2010;98:2524–2534. doi: 10.1016/j.bpj.2010.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Misler S, Chow RH. Rapid fluctuations in transmitter release from single vesicles in bovine adrenal chromaffin cells. Biophys J. 1996;70:1543–1552. doi: 10.1016/S0006-3495(96)79718-7. [DOI] [PMC free article] [PubMed] [Google Scholar]