

Abstract
Considerable electrophysiological and pharmacological evidence has long suggested an important role for acetylcholine in the regulation of rapid-eye-movement (REM) sleep. For example, injection of the cholinergic agonist carbachol into the dorsomedial pons produces an REM sleep-like state with muscle atonia and cortical activation, both of which are cardinal features of REM sleep. Located within this region of the pons is the sublaterodorsal nucleus (SLD), a structure thought to be both necessary and sufficient for generating REM sleep muscle atonia. Subsets of glutamatergic SLD neurons potently contribute to motor inhibition during REM sleep through descending projections to motor-related glycinergic/GABAergic neurons in the spinal cord and ventromedial medulla. Prior electrophysiological and pharmacological studies examining the effects of acetylcholine on SLD neurons have, however, produced conflicting results. In the present study, we sought to clarify how acetylcholine influences the activity of spinally projecting SLD (SLDsp) neurons. We used retrograde tracing in combination with patch-clamp recordings and recorded pre-and postsynaptic effects of carbachol on SLDsp neurons. Carbachol acted presynaptically by increasing the frequency of glutamatergic miniature excitatory postsynaptic currents. We also found that carbachol directly excited SLDsp neurons by activating an Na+–Ca2+ exchanger. Both pre-and postsynaptic effects were mediated by co-activation of M1 and M3 muscarinic receptors. These observations suggest that acetylcholine produces synergistic, excitatory pre-and postsynaptic responses on SLDsp neurons that, in turn, probably serve to promote muscle atonia during REM sleep.
Introduction
During rapid-eye-movement (REM) sleep, skeletal muscle tone, with the exception of those muscles related to breathing and eye movements, is dramatically reduced. Thus, skeletal muscle atonia is a defining feature of REM sleep. Dysfunction or damage to the supraspinal circuitry regulating REM sleep motor atonia, such as occurs in REM sleep behaviour disorder, results in the appearance of dream-enactment behaviour (Gagnon et al. 2006). Conversely, inappropriate activation of the supraspinal circuitry regulating REM sleep motor atonia, such as occurs in narcolepsy with cataplexy, results in sudden episodes of muscle atonia during wakefulness (Scammell, 2003). A more detailed understanding of the supraspinal circuitry regulating REM muscle atonia would provide not only important details of the neurobiology of REM sleep but probably also the pathophysiological bases of REM sleep behaviour disorder and cataplexy.
The sublaterodorsal nucleus (SLD) contains neurons that are crucial for the generation of atonia during REM sleep (Fuller et al. 2007; Luppi et al. 2012). In rats and mice, the SLD is located in the dorsal pons immediately ventral to the caudal laterodorsal tegmental nucleus and the locus coeruleus (LC; Franklin & Paxinos, 1997; Clement et al. 2011). This region is also known as the subcoeruleus area and is likely to be the homologue of the cat peri-locus coeruleus-α (peri-LCα; Sakai et al. 2001). Considerable evidence suggests that the SLD is both necessary and sufficient for driving muscle atonia during REM sleep (Sakai et al. 2001; Boissard et al. 2002; Lu et al. 2006; Fuller et al. 2007; Luppi et al. 2011; Vetrivelan et al. 2011). Pharmacological activation of putative SLD neurons in cats and rats rapidly produces an REM sleep-like state that is characterized by muscle atonia and cortical activation, with prominent EEG theta activity (Lai & Siegel, 1991; Onoe & Sakai, 1995; Boissard et al. 2002). Electrical stimulation of the SLD region in rats produces bilateral or contralateral suppression of muscle tone, depending on the site of the stimulation (Hajnik et al. 2000). Furthermore, small lesions of this area in cats or rats or focal disruption of glutamatergic transmission in these neurons in mice produces REM sleep without atonia, which is phenotypically very similar to that seen in humans with REM sleep behaviour disorder (Morrison, 1988; Lu et al. 2006; Krenzer et al. 2011). Interestingly, degeneration of the SLD neurons in humans is hypothesized to be a major component of the pathogenesis of human REM sleep behaviour disorder (Boeve et al. 2007; Mathis et al. 2007).
Sublaterodorsal nucleus neurons are glutamatergic, active during REM sleep (Lu et al. 2006; Clement et al. 2011; Krenzer et al. 2011), and are thought to promote atonia through descending projections that couple synaptically with glycinergic/GABAergic premotor neurons in the spinal cord or ventromedial medulla (Soja et al. 1991; Boissard et al. 2002; Lu et al. 2006; Fuller et al. 2007; Lai et al. 2010; Krenzer et al. 2011; Vetrivelan et al. 2011; Chase, 2013). During REM sleep, acetylcholine is thought to participate in the activation of these descending atonia pathways (Jones, 1991; McCarley, 2007). For example, when the cholinergic agonist carbachol is injected in the vicinity of the SLD in rats and, to a lesser extent, in mice, an REM sleep-like state, including muscle atonia, EEG desynchronization, rapid eye movements and ponto-geniculate waves, is induced (Baghdoyan, 1997; Kubin, 2001). The cat SLD has also been shown to contain REM sleep-on neurons that are activated by carbachol (Sakai & Koyama, 1996; Sakai et al. 2001). Although researchers have identified several sites in the medial pontine reticular formation at which carbachol can elicit these REM sleep-like phenomena, the SLD region is the most effective site (Kubin, 2001). Accordingly, the cat SLD has been shown to contain REM sleep-on neurons that are activated by carbachol (Sakai & Koyama, 1996; Sakai et al. 2001).
It has been a major challenge to identify SLD neurons that are active during atonia in an in vitro brain slice preparation. A particular challenge derives from the fact that the SLD region is heterogeneous and includes neurons with no established relationship to REM sleep atonia. This heterogeneity may explain why previous electrophysiological studies of SLD region neurons have found both excitatory and inhibitory responses to carbachol (Sakai & Koyama, 1996; Brown et al. 2006; Garcia-Rill et al. 2007; Heister et al. 2009). In an effort to reconcile these conflicting reports, we developed a novel approach to help clarify the following two key issues. (i) How does carbachol affect SLD neurons that subserve REM sleep atonia? (ii) Does carbachol act directly on these neurons or indirectly through local or distant circuitry? Our experimental approach comprised a combination of retrograde tracing and in vitro electrophysiology techniques that facilitated the identification of spinally projecting SLD (SLDsp) neurons and subsequent characterization of the specific cellular mechanisms by which carbachol excites SLDsp neurons (Lu et al. 2006; Krenzer et al. 2011).
Methods
Animal care and ethical approval
We used 103 C57BL/6 male and female mice, between the ages of 9 and 25 days (The Jackson Laboratory, Bar Harbor, ME, USA). The mice were housed in a pathogen-free barrier animal research facility maintained on a 12 h–12 h light–dark cycle (lights on at 07.00 h) at 22°C ambient temperature and with ad libitum access to food and water. Care of the mice met the National Institutes of Health standards, as set forth in the Guide for the Care and Use of Laboratory Animals, published by the US National Institutes of Health (NIH publication no. 85-23, revised 1996), and all protocols were approved by the Beth Israel Deaconess Medical Center Institutional Animal Care and Use Committee.
Fluorescent retrograde labelling of SLD neurons
We injected rhodamine-labelled fluorescent latex beads (∼0.1 μm diameter; Lumafluor Inc., Naples, FL, USA) into the ventral horn of the spinal cord. These beads are taken up by nerve terminals and retrogradely transported. We initially anaesthetized the mice in an induction chamber with isoflurane (4–5% in oxygen, using an anaesthetic vapourizer). We then fixed the mouse in a stereotaxic frame and reduced the dose of isoflurane to 1–2%. We incised the skin over the spine and, after appropriate dissection and laminectomy, we lowered a glass micropipette vertically into the spinal cord to the designated co-ordinate. To minimize injury to the spinal cord, we slowly injected 150 nl of beads over 5 min using a silane-coated glass micropipette (25–30 μm tip diameter) and an air-pressure injection system. The injections were targeted at laminae VII and VIII at the T1 level (mediolateral, +0.4 mm and dorsoventral, −0.5 mm; Franklin & Paxinos, 1997). In these experiments, we used only mice in which the injection was placed in the ventral horn. Injections into the dorsal horn retrogradely labelled the LC but not the SLD, so we excluded mice with labelling restricted to the LC and mice in which the injection was restricted to the dorsal horn. Two to five days after the surgery, we used the injected mice to prepare brain slices for in vitro recordings. We found many retrogradely labelled SLD neurons ipsilateral and contralateral to the injection, and we recorded neurons from either side of the pons.
Slice preparation and whole-cell patch-clamp recordings
To prepare brain slices, we anaesthetized mice with isoflurane to the point of respiratory arrest, followed by decapitation. Using a vibrating microtome (VT1000; Leica, Bannockburn, IL, USA), we cut coronal brain slices (300 μm thick) in ice-cold artificial cerebral spinal fluid (ACSF; Hepes-buffered solution) oxygenated with 100% O2. We recorded the slices submerged and perfused (2 ml min−1) with ACSF. We recorded only retrogradely labelled neurons (containing the fluorescent beads) in the SLD region in two or three coronal slices (−4.9 to −5.5 mm from bregma; Franklin & Paxinos, 1997; Figs 1 and 2). To guide our recordings, we used fluorescence and infrared differential interference contrast video microscopy using a fixed stage upright microscope (BX51WI; Olympus America Inc.) equipped with a Nomarski water immersion lens (×40/0.8 W) and infrared-sensitive CCD cameras (300T-RC; DAGE MTI, Michigan City, IN, USA; or ORCA-ER; Hamamatsu, Bridgewater, NJ, USA). We recorded in whole-cell configuration at room temperature using a Multiclamp 700B amplifier (Molecular Devices, Foster City, CA, USA), a Digidata 1322A interface and Clampex 9.0 software (Molecular Devices). We monitored the series resistance at regular intervals, and we discarded the data if neurons showed an unstable resting membrane potential or if the series resistance changed by more than 25%.
Figure 1.
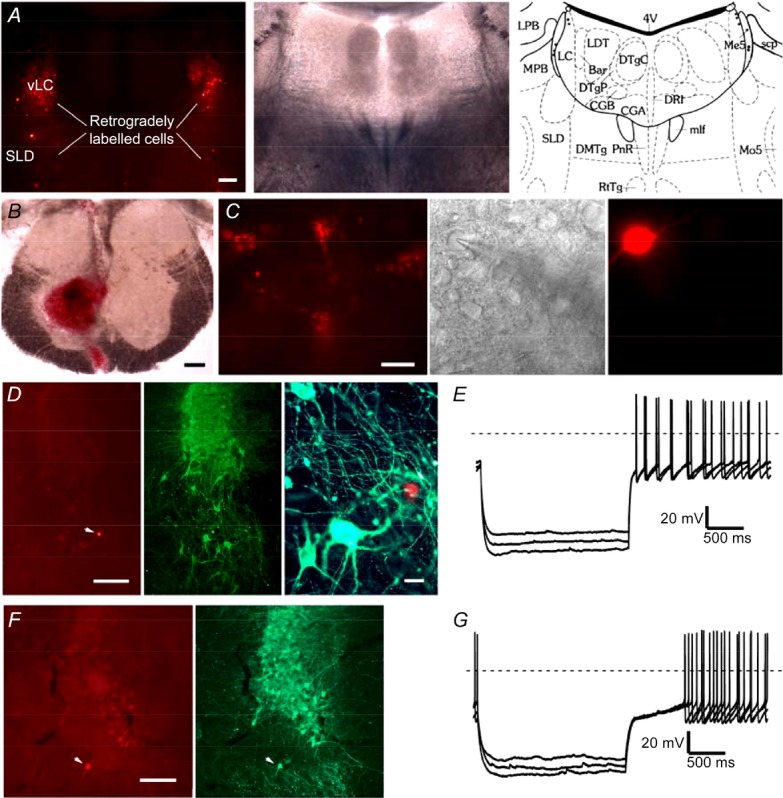
A, photographs show the distribution of fluorescent retrogradely labelled neurons in the ventral locus coeruleus (vLC) and sublaterodorsal nucleus (SLD; left), the same section visualized under bright field illumination (centre) and represented as a schematic drawing of the nuclei, 4th ventricle (4V); Barrington's nucleus (Bar); central gray, alpha part (CGA); central gray, beta part (CGB); dorsomedial tegmental area (DMTg); dorsal raphe nucleus, interfascicular (DRI); dorsal tegmental nucleus central (DTgC); dorsal tegmental nucleus pericentral (DTgP); locus coeruleus (LC); laterodorsal tegmental nucleus (LDT); lateral parabrachial nucleus (LPB); mesencephalic 5 nucleus (Me5); medial longitudinal fasciculus (mlf); medial parabrachial nucleus (MPB); motor trigeminal nucleus (Mo5); pontine raphe nucleus (PnR); reticulotegmental nucleus of pons (RtTg); superior cerebellar peduncle (scp); sublaterodorsal nucleus (SLD); (adapted from Franklin & Paxinos, 1997; scale bar = 200 μm). B, the injection site in the T1 ventral horn (scale bar = 200 μm). C, an example of recordings of SLDsp neurons visualized under fluorescent and infrared differential interference contrast systems and after being filled with Alexa Fluor 555 from the recording pipette (scale bar = 20 μm). D, an example of a recorded SLDsp neuron, filled with Alexa Fluor 555 (left), located immediately ventral to the locus coeruleus (LC), and negative for tyrosine hydroxylase (TH) immunoreactivity (in green; centre panel, scale bar = 200 μm; right panel, scale bar = 20 μm). E, the firing of SLDsp neurons is characterized by a small delay or no delay in rebound firing during repolarization from hyperpolarizing potentials, no low-threshold Ca2+ spikes, and little or no hyperpolarization-activated current (Ih)-mediated depolarizing sag. F and G, an example of a recorded neuron that was retrogradely labelled from the spinal cord, that was located ventral of the LC but was positive for TH immunoreactivity (Alexa Fluor 555 in red and TH immunoreactivity in green; scale bar = 200 μm). This SLD TH-positive spinally projecting neuron has the electrophysiological properties of a typical LC neuron, including delayed rebound firing on recovery from hyperpolarizing current pulses due to activation of an A-type current and no Ih-mediated depolarizing sag.
Figure 2.
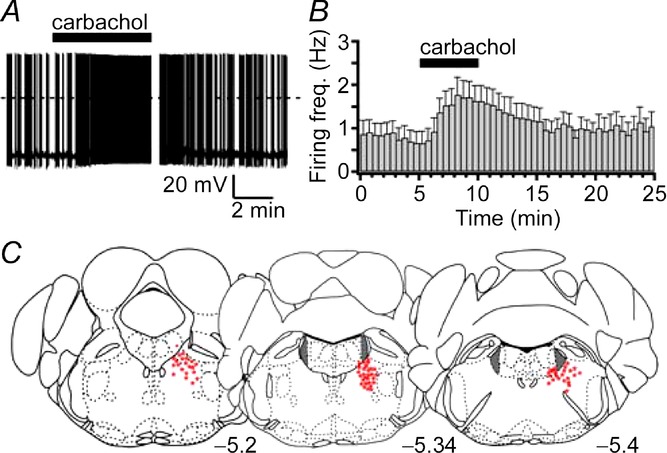
A, the response of a typical SLDsp neuron to carbachol (15 μm). B, a 5 min application of carbachol increases the firing frequency of eight SLDsp neurons. C, the distribution of 72 recorded SLDsp neurons. The LC is highlighted in grey. The numbers at the bottom indicate the distance (in millimetres) posterior to bregma.
Data analysis and statistics
We analysed the current-clamp and voltage-clamp recordings using Clampfit 9.0 (Molecular Devices) and IGOR Pro 6 (WaveMetrics, Lake Oswego, OR, USA). We measured the resting membrane potential in control ACSF and in SLDsp neurons that exhibited spontaneous firing below 2.5 Hz. We calculated input resistance before and during carbachol application using 500 ms hyperpolarizing current pulses in tetrodotoxin (TTX) and at resting membrane potential. We calculated the action potential amplitude as the voltage between the threshold and the action potential peak. We calculated the action potential duration as the width at the voltage halfway between the action potential threshold and the action potential peak. We determined the action potential threshold as the voltage at which the slope of the action potential reached ≥20 V s−1. We calculated the predicted Na+–Ca2+ exchanger reversal potential (ENCX) for a 3:1 stoichiometry as ENCX = 3ENa − 2ECa (Weber et al. 2002). We analysed the spontaneous and miniature excitatory postsynaptic currents (sEPSCs and mEPSCs) offline using Mini Analysis 6 (Synaptosoft, Leonia, NJ, USA). We ranked the synaptic events (5 min in control conditions, the last 5 min of 10 min carbachol application, and the last 5 min of a 15 min washout) by amplitude and interevent interval to prepare cumulative probability distributions. We compared the results using the non-parametric Kolmogorov–Smirnov test. For all the statistical analyses, we used StatView (SAS Institute, Cary, NC, USA). We normalized all the values by dividing the values of control and treatment samples by the mean of the control sample. Such normalization conserves the distribution and the relative variance of the samples, allowing the subsequent use of Student's t test. We used Student's paired t tests or one-way ANOVA (with repeated measures) followed by Fisher's PLSD tests for statistical analysis. A value of α < 0.05 was considered significant. Results are expressed as means ± SEM.
Reagents and solutions
The composition of the ACSF was as follows (mm): 140 NaCl, 3 KCl, 1.3 MgSO4, 2.4 CaCl2, 1.4 NaH2PO4, 11 glucose and 5 Hepes (pH adjusted to 7.2 with NaOH, 315–320 mosmol l−1). The pipette solution was as follows (mm): 120 potassium gluconate, 10 KCl, 3 MgCl2, 10 Hepes, 2.5 potassium ATP, 0.5 sodium GTP and 0.1 Alexa Fluor 555 hydrazide fluorescence dye (Invitrogen Co., Carlsbad, CA, USA; pH adjusted to 7.2 with KOH; 280 mosmol l−1). We recorded mEPSCs in TTX (1 μm). For recordings in extracellular low Na+, we used a choline-based solution in which 80% of the NaCl was replaced with choline chloride and a pipette solution in which we replaced 10 mm potassium gluconate with 10 mm NaCl (final concentration 110 mm potassium gluconate). We purchased TTX, KB-R7943 mesylate, dihydro-β-erythroidine hydrobromide (DHβE), 4-DAMP, AQ-RA-741, PD102807, VU0255035 and J104129 from Tocris Bioscience (Ellisville, MO, USA). We purchased all the other reagents from Sigma-Aldrich (St Louis, MO, USA). We dissolved KB-R7943 mesylate, 4-DAMP, AQ-RA-741, PD102807, VU0255035 and J104129 in dimethyl sulfoxide. The final concentration of dimethyl sulfoxide in the ACSF was <0.1%.
Histology
Immediately after the preparation of brain slices, we removed the spinal cords, fixed them overnight in 10% buffered formalin (Fisher Scientific, Pittsburg, PA, USA), then cut them into 80 μm axial sections on a freezing microtome and examined them with fluorescence microscopy to validate the location of the injected beads.
After the slice recordings, we fixed the brain slices (300 μm) overnight in 10% buffered formalin. We photographed the brain slices under fluorescence microscopy to document the location of retrogradely labelled neurons. To identify the recorded neurons filled with the red/orange fluorescent dye (Alexa Fluor 555) we dissolved the beads in xylene. We dehydrated the recorded slices in a series of graded ethanols (50, 70, 95 and 100%; 10 min for each solution) and then placed the slices in xylene for 10 min. This technique thoroughly dissolved the beads, and we then photographed the slice. By digitally superimposing the two images, we mapped the location of the recorded neurons in relationship to the cluster of retrogradely labelled cells in the SLD.
Next, we resectioned the recorded slices into 60 μm sections on a freezing microtome and then processed them for tyrosine hydroxylase (TH) immunoreactivity. We incubated the sections overnight in rabbit TH primary antibody (1:1000 dilution; AB152; lot JC1634159; Chemicon International/Millipore, Temecula, CA, USA) in PBS containing 0.3% Triton X-100 and then the next day in goat Cy2-anti-rabbit IgG secondary antibody (1:500 dilution in PBS containing 0.3% Triton X-100; Jackson ImmunoResearch, West Grove, PA, USA) for 2 h. This rabbit polyclonal antibody is raised against purified rat pheochromacytoma TH. It recognizes a single band of 62 kDa molecular weight on Western blots of rat brain (manufacturer) and stains a pattern of neurons in the rat pons consistent with previous reports (Bruinstroop et al. 2011). Omission of the TH primary antisera showed no immunoreactivity above background. We mounted the sections on gelatin-coated slices, dehydrated them in ascending concentrations of ethanol, delipidated in xylene and coverslipped them. We examined the sections under fluorescence to determine the location of the recorded neurons with respect to the TH-labelled neurons of the LC and whether or not they were positive for TH immunoreactivity.
Results
Recordings from REM-atonia neurons of the SLD in brainstem in vitro slices
To facilitate the identification of SLDsp neurons in our in vitro brainstem slice preparation, we injected fluorescent latex microspheres (which are physiologically inert) into the ventral horn of the spinal cord in vivo (Fig. 1). Two to five days later, the mice were killed for in vitro recordings. We recorded only from retrogradely labelled SLD neurons containing the fluorescent microspheres (Figs 1 and 2). The LC, which is immediately dorsal to the SLD, also projects to the spinal cord (VanderHorst & Ulfhake, 2006; Bruinstroop et al. 2011). To avoid any spinally projecting LC neurons, we focused our analysis on neurons that were located ventral to the LC and dorsal to the trigeminal motor nucleus, positive for microspheres but lacking TH immunoreactivity (determined post hoc).
We found that the SLDsp neurons were a relatively homogeneous population with electrophysiological properties distinct from the neighbouring noradrenergic LC neurons. The SLDsp neurons had an average membrane potential of −41.2 ± 0.8 mV and an average input resistance of 597 ± 51 MΩ (n = 32). Seventy-seven per cent of SLDsp neurons were spontaneously active (n = 65 in which spontaneous firing was assessed). These spontaneously active SLDsp neurons had a mean firing frequency of 3.0 ± 0.3 Hz (n = 50) and a mean firing threshold of 47.3 ± 0.6 mV (n = 10). The SLDsp neurons had a relatively narrow action potential (1.91 ± 0.09 ms, n = 50) compared with LC neurons (2.80 ± 0.19 ms, n = 9; P < 0.01; Student's unpaired t test) and had an after-hyperpolarization amplitude of −15.2 ± 0.8 mV (n = 13). In contrast to LC neurons, SLDsp neurons showed little or no delay in rebound firing during repolarization from hyperpolarizing potentials (SLDsp neurons, 151.6 ± 23.3 ms, n = 24 vs. LC neurons, 685.5 ± 108.7 ms, n = 9; P < 0.01; Student's unpaired t test). In addition, they lacked low-threshold Ca2+ spikes and had an inwardly rectifying K+ current and little or no hyperpolarization-activated current (Ih). These firing characteristics are similar to those of a previously described population in this region (Brown et al. 2006), although their spinal projections were not known at the time.
Carbachol excites SLDsp neurons through M1 and M3 muscarinic receptors
We found that carbachol (15 μm) excites 83% of SLDsp neurons. Of 54 SLDsp neurons tested, only six were inhibited and three showed no response. In current-clamp recordings of SLDsp neurons that were excited, carbachol induced membrane depolarization (+6.1 ± 0.4 mV; n = 7) and increased the spontaneous firing rate (by 100.8 ± 25.3%; n = 8; P < 0.01, Student's paired t test, carbachol vs. control conditions; Fig. 2). These effects were not associated with a change in input resistance (control conditions, 525 ± 61 MΩ and carbachol, 507 ± 61 MΩ; n = 10; P = 0.54, Student's paired t test, carbachol vs. control conditions). In addition, carbachol did not change the action potential amplitude (control conditions, 58.0 ± 4.0 mV and carbachol, 57.1 ± 3.9 mV; n = 9; P > 0.05, Student's paired t test, carbachol vs. control conditions), duration (control conditions 1.78 ± 0.16 ms and carbachol, 1.81 ± 0.16 ms; n = 9; P > 0.05, Student's paired t test, carbachol vs. control conditions) and action potential threshold (control conditions, −30.1 ± 1.8 mV and carbachol, −29.2 ± 1.9 mV; n = 9; P > 0.05, Student's paired t test, carbachol vs. control conditions).
In voltage-clamp recordings [holding potential (Vh) = −60 mV] from SLDsp neurons excited by carbachol, carbachol evoked an inward current (−20.1 ± 5.2 pA; n = 12; P < 0.01, Student's paired t test, carbachol vs. control conditions) accompanied by an increase in membrane current noise. These effects were maintained in the presence of 1 μm TTX (−20.6 ± 2.2 pA; n = 22; P < 0.01, Student's paired t test, carbachol vs. control conditions). The carbachol-mediated current was unaffected by a cocktail of nicotinic receptor antagonists, i.e. hexamethonium chloride (C6; 100 μm) plus methyllycaconitine citrate (MLA; 10 nm) plus mecamylamine (MEC; 1 μm) plus dihydro-β-erythroidine hydrobromide (DHβE; 500 nm), but was abolished by the muscarinic receptor antagonist scopolamine (10 μm), indicating that carbachol directly excites SLDsp neurons through muscarinic receptors (Fig. 3). In the presence of the nicotinic receptor antagonists, carbachol still evoked an inward current of comparable amplitude to that in control ACSF (−19.8 ± 3.6 pA; n = 7; P < 0.01, Student's paired t test, carbachol + C6 + MLA + MEC + DHβE vs. C6 + MLA + MEC + DHβE), but not in the presence of scopolamine (−2.9 ± 0.8 pA; n = 9; P > 0.05, Student's paired t test, carbachol + scopolamine vs. scopolamine). Application of pirenzepine, an preferential M1 receptor antagonist, or 1,1-Dimethyl-4-diphenylacetoxypiperidinium iodide (4-DAMP), a preferential M3/M1 receptor antagonist, both reduced the response of carbachol. Pirenzepine (10 μm) reduced the carbachol-mediated current to 23.3 ± 7.6% (n = 8), whereas 4-DAMP (100 nm) reduced it to 43.8 ± 9.1% (n = 12). It is known that pirenzepine also binds with relative high affinity to M3 and M4 receptors, although it has much lower affinity for M2 (Buckley et al. 1989). It is unclear whether 10 μm pirenzepine can antagonize the carbachol response at M3 receptors. For instance, it antagonizes ligand-induced responses at M3 receptors in submaxillary gland membranes but not in subthalamic neurons (Buckley et al. 1989; Shen & Johnson, 2000). Therefore, based on our results with pirenzepine and 4-DAMP, we concluded that M1 receptors are likely to mediate arbachol excitation of SLDsp neurons, but the involvement of the M3 subtype was still uncertain. Combined application of pirenzepine and 4-DAMP (10 μm pirenzepine + 100 nm 4-DAMP) abolished the carbachol-evoked inward current (−1.3 ± 0.6 pA; n = 6; P > 0.05, Student's paired t test, carbachol + pirenzepine + 4-DAMP vs. pirenzepine + 4-DAMP), whereas combined application of the preferential M2/M4 receptor antagonist AQ-RA-741 and the M4-selective receptor antagonist PD102807 (AQ-RA-741 200 nm + PD102807 1 μm) was ineffective in antagonizing the carbachol-evoked inward current (−22.3 ± 3.1 pA; n = 7; P < 0.01, Student's paired t test, carbachol + AQ-RA-741 + PD102807 vs. AQ-RA-741 + PD102807). Therefore, and in agreement with the excitatory response on SLDsp neurons, we concluded that carbachol acts through M1 and/or M3 receptor subtypes (Gq-coupled receptors) but not through M2 and M4 subtypes (Gi-coupled receptors). We next used two selective antagonists for M1 and M3 receptors, namely VU0255035 (M1– selective; Sheffler et al. 2009) and J104129 (M3– selective; Mitsuya et al. 1999). We found that both reduced the response of carbachol but did not abolish it. VU0255035 (1 μm) reduced the carbachol-mediated current to 56.1 ± 17.8% (n = 7) and J104129 (50 nm) reduced it to 53.1 ± 15.0% (n = 7). Combined application of VU0255035 and J104129 (VU0255035 1 μm + J104129 50 nm) abolished carbachol-mediated current (−3.9 ± 1.6 pA; n = 5; P > 0.05, Student's paired t test, carbachol + VU0255035 + J104129 vs. VU0255035 + J104129). Altogether, these results strongly suggest that the carbachol-mediated excitation of the SLDsp neurons occurs by co-activation of M1 and M3 receptor subtypes (Fig. 3).
Figure 3.

A–C, carbachol (15 μm) activates an inward current (A) that is unaffected by the following cocktail of nicotinic receptor antagonists: hexamethonium chloride (C6; 100 μm) + methyllycaconitine citrate (MLA; 10 nm) + mecamylamine (MEC; 1 μm) + dihydro-β-erythroidine hydrobromide (DHβE; 500 nm; B), but is blocked by the muscarinic receptor antagonist, scopolamine (10 μm; C). D–F, the preferential M1 receptor antagonist pirenzepine (10 μm; D) and the preferential M3/M1 receptor antagonist 1,1-Dimethyl-4-diphenylacetoxypiperidinium iodide (4-DAMP; 100 nm; E) only produce a partial block of the carbachol-mediated current, whereas, when co-applied (pirenzepine + 4-DAMP) they abolish the effects of carbachol (F). G, combined application of the preferential M2/M4 receptor antagonist AQ-RA-741 (200 nm) and the M4-selective receptor antagonist PD102807 (1 μm) does not block carbachol-evoked inward current. H–J, both the M1-selective antagonist VU0255035 (1 μm; H) and the M3-selective antagonist J104129 (50 nm; I) only produce a partial block of the response of carbachol, but when co-applied (VU0255035 + J104129) they abolish the effects of carbachol (J). K, summary histogram of carbachol-mediated current recorded in control artificial cerebrospinal fluid (ACSF; con; n = 22) and in the presence of C6 + MLA + MEC + DHβE (n = 7), of scopolamine (scop; n = 9), of pirenzepine (pirenz; n = 8), of 4-DAMP (n = 12), of pirenzepine + 4-DAMP (n = 6), of AQ-RA-741 + PD102807 (n = 7), of U0255035 (n = 7), of J104129 (n = 7) and of VU0255035 + J104129 (n = 5). *P < 0.05 and **P < 0.01, Student's paired t test, comparing holding currents before and during carbachol applications in the different conditions. †P < 0.05, ††P < 0.01, F(9,80) = 9.068, P < 0.001, one-way ANOVA, Fisher's PLSD, comparing carbachol-mediated current in control ACSF (con) vs. the carbachol-mediated current of the pharmacologically treated groups (C6 + MLA + MEC + DHβE, scop, pirenz, 4DAMP, pirenz + 4DAMP, AQRA741 + PD102807, VU0255035, J104129 and VU0255035 + J104129). All recordings were conducted in TTX (1 μm) and at a holding potential (Vh) of −60 mV.
Carbachol excites SLDsp neurons through the activation of an Na+–Ca2+ exchanger
We next examined the ionic mechanisms mediating carbachol excitation of SLDsp neurons. We first determined the current–voltage (I–V) relationship of the carbachol response using a voltage ramp protocol (from −100 to −40 mV, at 6 mV s−1). We then tested whether cations that can block K+ conductances could block the response of carbachol (Fig. 4). Barium chloride (1 mm) did not affect the carbachol-evoked inward current (−23.50 ± 3.42 pA; n = 6; P < 0.01, Student's paired t test, carbachol + BaCl2 vs. BaCl2; and P = 0.38, one-way ANOVA, carbachol in BaCl2 vs. carbachol in control ACSF), indicating that the effect of carbachol was not mediated through a K+ conductance. Carbachol produced an inward current across the entire voltage ramp, resulting in a parallel shift of the I–V relationship. This suggested that the reversal potential of the carbachol-mediated current is at a potential far from the range of potentials of our ramp protocol. It has been previously reported that carbachol excites basal forebrain neurons through activation of the Na+–Ca2+ exchanger (Xu et al. 2006), which based on the composition of our extracellular and pipette solutions and a 3 Na+:1 Ca2+ stoichiometry, has a predicted reversal potential of approximately +175 mV (Weber et al. 2002). To test whether the effect of carbachol was mediated through an Na+–Ca2+ exchanger, we used the following three approaches: (i) we lowered the Na+ gradient across the membrane; (iii) we buffered intracellular Ca2+; and (iii) we blocked the Na+–Ca2+ exchanger with KB-R7943. We first replaced 80% of extracellular Na+ with choline chloride (low-Na+ ACSF) and increased the concentration of Na+ in the recording pipette to 10 mm. This shifted the predicted reversal potential of the Na+–Ca2+ exchanger to approximately −55 mV and abolished carbachol-evoked inward current (−0.85 ± 0.57 pA; n = 8; P > 0.05, Student's paired t test, carbachol in low-Na+ ACSF vs. control low-Na+ ACSF). The carbachol response was also sensitive to intracellular free Ca2+. We found that the response to carbachol was greatly reduced when we buffered intracellular Ca2+ with 1,2-bis-(o-aminophen-oxy)ethane-N,N,N′,N′-tetraacetic acid (BAPTA; 10 mM in the recording pipette; −7.53 ± 1.43 pA; n = 8; P < 0.01, Student's paired t test, carbachol with BAPTA vs. control conditions with BAPTA) and it was blocked by EGTA (10 mm in the recording pipette; −2.20 ± 1.38 pA; n = 7; P > 0.05, Student's paired t test, carbachol with EGTA vs. control conditions with EGTA). In addition, application of KB-R7943 (35 μm), which at this concentration inhibits the Na+–Ca2+ exchanger in forward mode (Iwamoto et al. 1996), completely blocked the carbachol-mediated inward current (−0.79 ± 1.75 pA; n = 12; P > 0.05, Student's paired t test, carbachol + KB-R7943 vs. KB-R7943). Altogether, these results strongly suggest that carbachol directly excites SLDsp neurons by activating an Na+–Ca2+ exchanger (Fig. 4).
Figure 4.

A, carbachol (15 μm) activates an inward current across the entire voltage ramp, resulting in a parallel shift of the current–voltage relationship. B, blocking K+ conductances with BaCl2 (1 mm) does not block the effect of carbachol. C–F, the effect of carbachol is instead blocked by replacing 80% of extracellular Na+ with choline chloride (low-Na+ ACSF; C), is reduced by BAPTA (10 mm in the recording pipette; D) and is blocked by EGTA (10 mm in the recording pipette; E) or by the Na+–Ca2+ exchanger blocker, KB-R7943 (KB-R; 35 μm; F). All the current–voltage relationships were obtained using voltage ramps (from −100 to −40 mV; 6 mV s−1). G, examples of voltage-clamp recordings (Vh = −60 mV) before and during carbachol applications in BaCl2 (1 mm), low-Na+ ACSF, with BAPTA (10 mm) or EGTA (10 mm) in the recording pipette, and in KB-R7943. H, summary histogram of carbachol-mediated current recorded in control ACSF (con; n = 22; data also represented in Fig. 3H), in BaCl2 (BaCl2; n = 6), in low-Na+ ACSF (low Na+; n = 8), with BAPTA or EGTA in the recording pipette (BAPTA, n = 8; and EGTA, n = 7) and in KB-R7943 (KB-R; n = 12). **P < 0.01, Student's paired t test, comparing holding currents before and during carbachol applications. ††P < 0.01, F(5,57) = 18.04, P < 0.001, one-way ANOVA, Fisher's PLSD, comparing carbachol-mediated current in control ACSF (con) vs. the carbachol-mediated current of the pharmacologically treated groups (BaCl2, low Na+, BAPTA, EGTA and KB-R). All recordings were conducted in TTX (1 μm).
Carbachol increases glutamatergic input to SLDsp neurons through presynaptic M1 and M3 muscarinic receptors
We next examined the effects of carbachol on glutamatergic input to SLDsp neurons. Carbachol (15 μm) administration nearly doubled the frequency of glutamatergic sEPSCs (control conditions, 3.15 ± 1.00 Hz and carbachol, 6.12 ± 1.32 Hz; n = 8; P < 0.01, Student's paired t test; Fig. 5). This effect was reversed after 20 min if washout, and the sEPSCs were completely abolished by the AMPA receptor antagonist DNQX (20 μm; n = 3; recordings in 10 μm bicuculline at Vh = −60 mV).
Figure 5.

A, carbachol (15 μm) increases spontaneous excitatory postsynaptic current (sEPSC) frequency without affecting amplitude. Current traces are shown for a representative SLDsp neuron in control, carbachol and washout (wash) conditions. B, mean effect of carbachol on sEPSC frequency (n = 8; left panel) and sEPSC amplitude (F(2,19) = 0.641, P = 0.547, one-way ANOVA; right panel). C, effects of carbachol on the miniature excitatory postsynaptic currents (mEPSCs) shown in a representative SLDsp neuron. The current traces are represented in the left panel and the cumulative distribution plots of mEPSC interevent interval and amplitude are represented in the centre and right panels, respectively. Carbachol significantly decreases the interevent interval without affecting the mEPSC amplitude (P values from Kolmogorov–Smirnov test). D, the effect of carbachol is completely abolished in the presence of scopolamine (10 μm), indicating that carbachol acts on presynaptic muscarinic receptors (current traces for scopolamine and scopolamine + carbachol are represented in the left panel; cumulative distribution plots of the mEPSC interevent interval and amplitude are represented in the centre and right panels, respectively; P values from Kolmogorov–Smirnov test). Spontaneous EPSCs and mEPSCs were recorded at Vh = −60 mV, and mEPSCs were recorded in TTX (1 μm). In the cumulative distributions, results are represented before carbachol application as dotted lines, during carbachol as bold lines and after carbachol application as thinner lines.
To determine whether carbachol acts on presynaptic glutamatergic terminals, we tested its effects on glutamatergic mEPSCs. Carbachol increased the frequency of glutamatergic mEPSCs by 66% (control conditions, 1.12 ± 0.18 Hz and carbachol, 1.87 ± 0.37 Hz; n = 10; P < 0.01, Student's paired t test; and washout, 1.07 ± 0.21 Hz). Carbachol had no effect on mEPSC amplitude (control conditions, 12.65 ± 1.13 pA and carbachol, 12.44 ± 1.17 pA; n = 10; P = 0.59, Student's paired t test; washout, 11.97 ± 1.36 pA), consistent with a presynaptic effect. Application of scopolamine (10 μm) completely blocked the effects of carbachol on mEPSC frequency (scopolamine, 0.80 ± 0.14 Hz and scopolamine + carbachol, 0.78 ± 0.14 Hz; n = 8; P = 0.83, Student's paired t test), indicating that carbachol acts presynaptically through muscarinic receptors (Fig. 5).
Application of pirenzepine (preferential M1 receptor antagonist) or 4-DAMP (preferential M3/M1 receptor antagonist) reduced but did not block the effect of carbachol on mEPSC frequency (Fig. 6). In the presence of pirenzepine (10 μm), carbachol still increased the frequency of glutamatergic mEPSCs by 44% (pirenzepine, 1.21 ± 0.12 Hz and pirenzepine + carbachol, 1.74 ± 0.15 Hz; n = 8; P = 0.007, Student's paired t test), whereas in 4-DAMP (100 nm), carbachol increased mEPSC frequency by only 14% (4-DAMP, 0.74 ± 0.07 Hz and 4-DAMP + carbachol, 0.85 ± 0.09 Hz; n = 10; P = 0.013, Student's paired t test). Combined application of pirenzepine and 4-DAMP (10 μm pirenzepine + 100 nm 4-DAMP) completely abolished the effects of carbachol on the mEPSC frequency (pirenzepine + 4-DAMP, 1.70 ± 0.21 Hz and pirenzepine + 4-DAMP + carbachol, 1.69 ± 0.25 Hz; n = 6; P = 0.95, Student's paired t test), whereas combined application of the preferential M2/M4 receptor antagonist AQ-RA-741 and the M4-selective receptor antagonist PD102807 (200 nm AQ-RA-741 + 1 μm PD102807) was ineffective (AQ-RA-741 + PD102807, 1.26 ± 0.19 Hz and AQ-RA-741 + PD102807 + carbachol, 2.23 ± 0.29 Hz, n = 7; P = 0.026, Student's paired t test). This suggests that carbachol increases the glutamatergic input to the SLDsp neurons through M1 and/or M3, but not through M2 and M4 presynaptic receptors (Fig. 6).
Figure 6.

A and B, the preferential M1 receptor antagonist pirenzepine (10 μm; A) or the preferential M3/M1 receptor antagonist 4-DAMP (100 nm; B) lead to only a partial block of the effects of carbachol on the mEPSC frequency. C, co-application of pirenzepine + 4-DAMP abolishes the effects of carbachol. D, co-application of the preferential M2/M4 receptor antagonist AQ-RA-741 (200 nm) and the M4-selective receptor antagonist PD102807 (1 μm) are ineffective in reversing the effects of carbachol on mEPSC frequency. Effects of carbachol on the mEPSCs in the presence of pirenzepine, 4-DAMP, pirenzepine + 4-DAMP and AQ-RA-741 + PD102807 are shown in four representative SLDsp neurons. The current traces are represented in the left panels and the cumulative distribution plots of mEPSC interevent interval and amplitude are represented in the centre and right panels, respectively. Miniature EPSCs were recorded in TTX (1 μm) at Vh = −60 mV. In the cumulative distributions, results are represented before carbachol application as dotted lines, during carbachol as bold lines and after carbachol application as thinner lines, and P values refer to Kolmogorov–Smirnov tests.
To test this possibility further, we used VU0255035 (M1-selective antagonist; Sheffler et al. 2009) and J104129 (M3-selective antagonist; Mitsuya et al. 1999). Both these antagonists reduced the effect of carbachol on mEPSC frequency but did not abolish it (Fig. 7). In the presence of VU0255035 (1 μm), carbachol increased the mEPSC frequency by 46% (VU0255035, 1.13 ± 0.08 Hz and VU0255035 + carbachol, 1.64 ± 0.16 Hz; n = 7; P = 0.007, Student's paired t test), whereas in J104129 (50 nm), carbachol increased the glutamatergic mEPSC frequency by 25% (J104129, 1.31 ± 0.16 Hz and J104129 + carbachol, 1.64 ± 0.24 Hz; n = 7; P = 0.024, Student's paired t test). Combined application of VU0255035 and J104129 (1 μm VU0255035 + 50 nm J104129) completely abolished the effects of carbachol on the mEPSC frequency (VU0255035 + J104129, 1.63 ± 0.34 Hz and VU0255035 + J104129 + carbachol, 1.56 ± 0.37 Hz; n = 5; P = 0.58, Student's paired t test). These results indicate that carbachol increases the glutamatergic input to the SLDsp neurons through both M1 and M3 presynaptic receptors, with the predominant effect occurring through the M3 subtype.
Figure 7.

A and B, the M1-selective and the M3-selective muscarinic receptor antagonists, VU0255035 (1 μm) and J104129 (50 nm), produce only a partial block of the effects of carbachol on the mEPSC frequency. C, co-application VU0255035 (1 μm) + J104129 (50 nm) abolishes the effects of carbachol. The effects of carbachol on the mEPSCs in the presence of VU0255035, J104129 and VU0255035 + J104129 are shown in three representative SLDsp neurons. The current traces are represented in the left panels and the cumulative distribution plots of mEPSC interevent interval and amplitude are represented in the centre and right panels, respectively. D and E, summary bar graphs compare the effects on mEPSC frequency (D) and mEPSC amplitude (E) of carbachol applied alone (n = 10) and in the presence of scopolamine (10 μm, n = 8), pirenzepine (10 μm, n = 8), 4-DAMP (100 nm, n = 10), 4-DAMP + pirezepine (n = 6), AQ-RA-741 (200 nm) + PD102807 (1 μm, n = 7), VU0255035 (1 μm, n = 7), J104129 (50 nm, n = 7), and VU0255035 + J104129 (n = 5). *P < 0.05 and **P < 0.01, Student's paired t test comparing the effects on mEPSC frequency and mEPSC amplitude before and during carbachol applications (carb vs. con; scop + carb vs. scop; pirenz + carb vs. pirenz; 4-DAMP + carb vs. 4-DAMP; pirenz + 4-DAMP + carb vs. pirenz + 4-DAMP; AQ-RA-741 + PD102807 + carb vs. AQ-RA-741 + PD102807; VU0255035 + carb vs. VU0255035; J104129 + carb vs. J104129; and VU0255035 + J104129 + carb vs. VU0255035 + J104129). Effects of carbachol on mEPSC frequency (F(8,59) = 2.635, P = 0.015, one-way ANOVA) and mEPSC amplitude (F(8,59) = 0.153, P = 0.996, one-way ANOVA), †P < 0.05 and ††P < 0.01, Fisher's PLSD comparing the effects of carbachol in control ACSF and in the presence of muscarinic receptor antagonists (carb vs. carb in scop; carb vs. carb in pirenz; carb vs. carb in 4-DAMP; carb vs. carb in pirenz + 4-DAMP; carb vs. carb in AQ-RA-741 + PD102807; carb vs. carb in VU0255035; carb vs. carb in J104129; and carb vs. carb + VU0255035 + J104129). Miniature EPSCs were recorded in TTX (1 μm) at Vh = −60 mV. In the cumulative distributions, results are represented before carbachol application as dotted lines, during carbachol application as bold lines and after carbachol application as thinner lines, and P values refer to Kolmogorov–Smirnov tests.
Discussion
We found that carbachol excites SLDsp neurons both through a direct postsynaptic effect and by increasing glutamatergic synaptic inputs. Both of these effects were mediated through M1 and M3 receptors and strongly suggest cellular mechanisms through which acetylcholine can contribute to the generation of atonia during REM sleep.
Carbachol directly excites the SLDsp neurons through M1 and M3 muscarinic receptors
Previous in vivo single-unit recording studies in cats reported that iontophoretic application of carbachol activates REM sleep-active neurons in the SLD (Sakai et al. 2001). Identifying and studying these neurons in vitro has been a challenge because the SLD contains a great variety of cell types, including glutamatergic, noradrenergic and GABAergic neurons (Lu et al. 2006; VanderHorst & Ulfhake, 2006; Brown et al. 2008; Bruinstroop et al. 2011) and lacks markers specific for REM sleep-active neurons. It is therefore not surprising that previous in vitro studies have found both excitatory and inhibitory responses to carbachol in neurons of the SLD region in rats and mice (Brown et al. 2006; Garcia-Rill et al. 2007; Heister et al. 2009). Our findings, which derive from a selective interrogation of spinally projecting SLD neurons, reveal that carbachol directly and consistently excites SLDsp neurons through postsynaptic muscarinic receptors.
Of the five known muscarinic receptor subtypes, the M1, M2 and M3 subtypes are expressed in the SLD region (Baghdoyan et al. 1994; Mallios et al. 1995). We selected a combination of widely used antagonists that have poor receptor subtype specificity, namely pirenzepine (M1 preferring), 4-DAMP (M3/M1 preferring) and AQ-RA-741 (M2/M4 preferring), as well as three recently developed subtype-selective antagonists, namely VU0255035 (M1 selective; Sheffler et al. 2009), J104129 (M3 selective; Mitsuya et al. 1999) and PD102807 (M4 selective; Kitaichi et al. 1999). We found that the effect of carbachol on SLDsp neurons was mediated by the M1 and M3 receptor subtypes, but not M2 and M4. The M2 receptor is thought to be the predominant subtype expressed in GABAergic neurons of the pontine reticular formation, but not in glutamatergic SLDsp neurons (Coleman et al. 2004; Brischoux et al. 2008). Our results are generally consistent with this finding. The M1 subtype activates SLD neurons in vitro (Heister et al. 2009), but it has been reported that manipulation of the M1 receptors in the SLD failed to produce effects on REM sleep (Velazquez-Moctezuma et al. 1991; Coleman et al. 2004). It was therefore surprising to find in the present study that M1 receptors contribute to the carbachol-mediated excitation of SLDsp neurons. In contrast, and in agreement with our findings, the M3 subtype is thought to activate the pontine REM generator neurons directly (Sakai & Onoe 1997).
In conclusion, our combined electrophysiological and pharmacological approach demonstrates that SLDsp neurons are excited by carbachol and that this response is mediated by the combined activation of M3 as well as M1 postsyanaptic receptors.
Our experiments also revealed that the postsynaptic effect of carbachol on SLDsp neurons is mediated by activation of an Na+–Ca2+ exchanger. This conclusion is supported by the sensitivity of the carbachol-mediated inward current to extracellular Na+ and intracellular Ca2+ concentrations, as well as to KB-R7943, a selective inhibitor of the Na+–Ca2+ exchanger. The Na+–Ca2+ exchangers are Ca2+ transporters that extrude Ca2+ from the cytoplasm with an exchange ratio of three Na+ ions brought in for every extruded Ca2+ ion (i.e. forward mode). This activity generates a net influx of positive charges that results in a steady-state inward current and can be accompanied by an increase in membrane current noise (Xu et al. 2006). All three isoforms of the Na+–Ca2+ exchangers (NCX1, NCX2 and NCX3) are expressed in the brain (Lytton, 2007). In addition to acetylcholine, other neurotransmitters, such as orexin-A and thyrotrophin-releasing hormone, excite target neurons through Gq-coupled signalling and activation of Na+–Ca2+ exchangers (Eriksson et al. 2001; Burdakov et al. 2003; Xu et al. 2006; Parmentier et al. 2009). A debate continues, however, over whether these neurotransmitters directly affect the Na+–Ca2+ exchangers or, rather, that their increased activity is a consequence of increased intracellular Ca2+ levels. In addition, a carbachol-mediated increase in intracellular Ca2+ levels could also produce an increase in the voltage-gated Na+ currents (Bulatko & Greeff, 1995). Still, the lack of changes in the action potential amplitude, duration and threshold in response to carbachol suggests that it is unlikely that modulation of the voltage-gated Na+ current contributes to the carbachol-mediated excitation of SLDsp neurons. In further support of a mechanism for the activation of the Na+–Ca2+ exchanger through intracellular Ca2+ elevation are the observations that Gq signalling pathways can activate intracellular Ca2+ signalling and that Na+–Ca2+ exchangers become maximally activated when Ca2+ rises above resting levels (Lytton, 2007). Future studies using Ca2+ imaging might be able to provide a definitive answer concerning this mechanism of action. Nevertheless, it is clear that, directly or indirectly, the activation of the Na+–Ca2+ exchangers depolarizes the membrane potential and promotes neuronal Firing.
Carbachol acts through presynaptic M1 and M3 muscarinic receptors to increase excitatory inputs to SLDsp neurons
Previous microdialysis studies have suggested that glutamatergic input to SLD neurons is important in REM sleep regulation. Injection of glutamate receptor agonists into the SLD region of cats and rats increased the firing of SLD REM-active neurons and induced a REM-like state with continuous muscle atonia (Lai & Siegel, 1991; Onoe & Sakai, 1995; Hajnik et al. 2000). Kynurenate, a glutamate antagonist, rapidly reversed the muscle atonia induced by local application of bicuculline in rats, but did not alter the cortical REM-like state, suggesting that the glutamatergic input to the SLD is required for the generation of atonia during REM sleep (Boissard et al. 2002). We found that carbachol increases glutamatergic synaptic drive to SLDsp neurons, which may serve as an additional excitatory mechanism for cholinergic activation of atonia pathways. This observation runs counter to a previous electrophysiological in vitro study, which found that carbachol mostly reduced the glutamatergic input to neurons in the SLD region of rats (Heister et al. 2009). However, that study did not identify the phenotype of the recorded neurons, and it is not clear to what extent the recordings included SLDsp neurons. We also found that carbachol increased the frequency of mEPSCs without affecting their amplitude, indicating a presynaptic effect with no change in postsynaptic efficacy. Using the same pharmacological approach that we used to identify the muscarinic receptor subtypes responsible for the postsynaptic response to carbachol, we found that the effect of carbachol on the mEPSC frequency was abolished by scopolamine (non-subtype selective) or by co-application of an M1-and an M3-selective antagonist, namely VU0255035 (M1 selective; Sheffler et al. 2009) and J104129 (M3 selective; Mitsuya et al. 1999), which is consistent with a muscarinic presynaptic response mediated by M1 and M3 receptor subtypes. Our pharmacological study also shows that the M3 receptors are the larger contributor of the presynaptic response to carbachol.
Methods to detect glutamatergic neurons definitively by the presence of vesicular transporters are relatively new. Previous studies on glutamatergic inputs to the SLD used glutamate antibodies, which are neither sensitive nor specific. Hence, the source of glutamatergic input to the SLD remains unknown. The lateral hypothalamus, ventrolateral periaqueductal grey matter, lateral pontine tegmentum and ventrolateral medulla all contain glutamatergic neurons and project to the SLD (Shammah-Lagnado et al. 1987; Lai et al. 1993; Semba, 1993; Boissard et al. 2003), although it is not known whether the neurons projecting to the SLD from these regions are glutamatergic. A small number of cells in the contralateral SLD region and a larger number of cells in the ipsi-and contralateral pontine reticular formation were found to project to the SLD (Lai et al. 1993; Boissard et al. 2003), but their phenotype, too, remains unidentified. In addition, it is still unclear whether glutamatergic input to the SLD REM-atonia neurons is stable across all behavioural states or whether it increases at the onset of REM sleep. Our results suggest that a muscarinic presynaptic mechanism increases glutamatergic input to SLD neurons during REM sleep when the levels of acetylcholine in the dorsal pons are highest (Leonard & Lydic, 1997). Defining which of these glutamatergic inputs contribute to the development of atonia should be achievable using optogenetic stimulation or other techniques.
Physiological significance and neuronal circuitry controlling REM muscle atonia
Based on present and prior results, it appears most likely that acetylcholine promotes REM sleep in the dorsomedial pons by activating two separate sets of REM-generating neurons; the first set promotes cortical activation through ascending inputs to the thalamus, the posterior hypothalamus and the basal forebrain, whereas the second set generates muscle atonia through descending inputs to glycinergic/GABAergic premotor neurons in the spinal cord and ventral medulla (Fuller et al. 2007; Luppi et al. 2012; Chase, 2013). The existence of separate pathways mediating the cortical and motor components of REM sleep also provides a possible basis for the occasional dissociation of atonia and cortical activation during pathological states such as cataplexy, sleep paralysis and REM sleep behaviour disorder (Lu et al. 2006; Vetrivelan et al. 2009, 2011; Luppi et al. 2011). There is general consensus that descending projections for REM atonia originate from glutamatergic REM sleep-active neurons of the SLD, and that during REM sleep the motorneurons are actively inhibited by glycinergic and GABAergic inputs and through ionotrophic glycine and GABAA receptors and metabotropic GABAB receptors (Chase et al. 1989; Brooks & Peever, 2012; Chase, 2013). There remains, however, an open debate over the respective contribution of premotor neurons of the ventromedial medulla vs. spinal interneurons in mediating REM atonia (Chase, 2013). Moreover, it is possible that premotor neurons in the medulla and spinal ventral horn are activated by distinct groups of SLD neurons. Along these lines, a previous single-unit recording study in cats found that spinally projecting neurons in the peri-LCα were inactive during REM sleep (Sakai et al. 1981). Importantly, however, only a relatively small number of neurons were sampled, and many of these may have been noradrenergic neurons, which are known to be silent during REM sleep (Aston-Jones & Bloom, 1981; Bruinstroop et al. 2011). Indeed, in cats, glutamatergic subcoeruleus neurons are intermingled with noradrenergic neurons of the LC complex. Therefore, while the medullary pathway may contribute to atonia in intact animals, the available data continue to suggest that the SLD–spinal cord pathway is necessary to produce muscle atonia during REM sleep; hence, it was the focus of the present study.
Although the amount and onset of REM sleep generated by local administration of carbachol to the pons vary depending on the location and dose of the injections, species or type of preparation (Kubin, 2001), the classical hypothesis that cholinergic neurons participate in the genesis of REM sleep is still widely accepted. Indeed, acetylcholine levels in the dorsal pons are twice as high in REM sleep compared with slow-wave sleep and wakefulness (Leonard & Lydic, 1997), and depletion of acetylcholine inhibits REM sleep, whereas blocking acetylcholine degradation promotes REM sleep (reviewed by Jones, 1991). These findings are also consistent with the report of innervation of the SLD by choline acetyltransferase-immunoreactive fibres that originate in the pedunculuopontine and laterodorsal tegmental nuclei (Jones, 1990; Semba, 1993). Our results provide important insights into the cellular mechanisms by which acetylcholine can activate SLD REM-atonia neurons.
Our findings indicate that acetylcholine produces synergistic, excitatory pre-and postsynaptic effects on spinally projecting SLD neurons. These effects should promote muscle atonia during REM sleep. Simultaneous withdrawal of monoaminergic, orexinergic and GABAergic neurotransmission onto SLD neurons may also contribute to muscle atonia (Jones, 1991; Vetrivelan et al. 2011; Luppi et al. 2012); however, how these neurotransmitters and peptides affect the SLD REM-atonia neurons is still poorly understood and is of considerable interest for future studies.
Key points
Activation of spinally projecting sublaterodorsal nucleus (SLD) neurons inhibits motor activity, in part through spinal inhibitory interneurons, to produce muscle atonia during rapid-eye-movement (REM) sleep.
It has long been hypothesized that acetylcholine released during REM sleep contributes to REM sleep atonia through activation of SLD neurons.
We show, using whole-cell recordings in brainstem slices, that acetylcholine directly excites spinally projecting SLD neurons via M1 and M3 muscarinic receptors, and increases afferent excitatory synaptic input to these neurons.
These results suggest that acetylcholine contributes to REM sleep muscle atonia through excitation of spinally projecting SLD neurons.
Acknowledgments
The authors wish to thank Sofia Z. Iqbal and Sarah A. Keating for assistance with animal surgeries and immunohistochemistry and Dr Patrick M. Fuller for his helpful comments on the manuscript.
Glossary
- 4-DAMP
1,1-Dimethyl-4-diphenylacetoxypiperidinium iodide
- ACSF
artificial cerebrospinal fluid
- BAPTA
1,2-bis-(o-aminophen-oxy)ethane-N,N,N′,N′-tetraacetic acid
- C6
hexamethonium chloride
- DHβE
dihydro-β-erythroidine hydrobromide
- ENCX
Na+–Ca2+ exchanger reversal potential
- Ih
hyperpolarization-activated current
- I–V
current–voltage relationship
- LC
locus coeruleus
- MEC
mecamylamine
- mEPSC
miniature excitatory postsynaptic current
- MLA
methyllycaconitine citrate
- peri-LCα
peri-locus coeruleus-α
- REM
rapid eye movement
- sEPSC
spontaneous excitatory postsynaptic currents
- SLD
sublaterodorsal nucleus
- SLDsp
spinally projecting sublaterodorsal neurons
- TH
tyrosine hydroxylase
- TTX
tetrodotoxin
- Vh
holding potential
Additional Information
Competing interests
None declared.
Author contributions
Spinal cord injections and whole-cell recordings were performed by W.F.J., R.H.W. and J.M.H. Conception and design of experiments: W.F.J., R.H.W., J.L. and E.A. Data analysis and interpretation: W.F.J., R.H.W. and E.A. Manuscript writing: T.E.S., C.B.S. and E.A. All authors have approved the final version of the manuscript.
Funding
This study was supported by NIH grants: 1RO1NS061863, the Administrative Supplement Utilizing Recovery Act Funds 3RO1NS061863 and 1P01HL095491.
References
- Aston-Jones G, Bloom FE. Activity of norepinephrine-containing locus coeruleus neurons in behaving rats anticipates fluctuations in the sleep-waking cycle. J Neurosci. 1981;1:876–886. doi: 10.1523/JNEUROSCI.01-08-00876.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baghdoyan HA. Cholinergic mechanisms regulating REM sleep. In: Schwartz WJ, editor. Sleep Science: IntegratingBasic Research and Clinical practice. Basel: Karger Publishing; 1997. pp. 88–116. [Google Scholar]
- Baghdoyan HA, Mallios VJ, Duckrow RB, Mash DC. Localization of muscarinic receptor subtypes in brain stem areas regulating sleep. Neuroreport. 1994;5:1631–1634. doi: 10.1097/00001756-199408150-00022. [DOI] [PubMed] [Google Scholar]
- Boeve BF, Silber MH, Saper CB, Ferman TJ, Dickson DW, Parisi JE, Benarroch EE, Ahlskog JE, Smith GE, Caselli RC, Tippman-Peikert M, Olson EJ, Lin SC, Young T, Wszolek Z, Schenck CH, Mahowald MW, Castillo PR, Del Tredici K, Braak H. Pathophysiology of REM sleep behaviour disorder and relevance to neurodegenerative disease. Brain. 2007;130:2770–2788. doi: 10.1093/brain/awm056. [DOI] [PubMed] [Google Scholar]
- Boissard R, Fort P, Gervasoni D, Barbagli B, Luppi PH. Localization of the GABAergic and non-GABAergic neurons projecting to the sublaterodorsal nucleus and potentially gating paradoxical sleep onset. Eur J Neurosci. 2003;18:1627–1639. doi: 10.1046/j.1460-9568.2003.02861.x. [DOI] [PubMed] [Google Scholar]
- Boissard R, Gervasoni D, Schmidt MH, Barbagli B, Fort P, Luppi PH. The rat ponto-medullary network responsible for paradoxical sleep onset and maintenance: a combined microinjection and functional neuroanatomical study. Eur J Neurosci. 2002;16:1959–1973. doi: 10.1046/j.1460-9568.2002.02257.x. [DOI] [PubMed] [Google Scholar]
- Brischoux F, Mainville L, Jones BE. Muscarinic-2 and orexin-2 receptors on GABAergic and other neurons in the rat mesopontine tegmentum and their potential role in sleep–wake state control. J Comp Neurol. 2008;510:607–630. doi: 10.1002/cne.21803. [DOI] [PubMed] [Google Scholar]
- Brooks PL, Peever JH. Identification of the transmitter and receptor mechanisms responsible for REM sleep paralysis. J Neurosci. 2012;32:9785–9795. doi: 10.1523/JNEUROSCI.0482-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown RE, McKenna JT, Winston S, Basheer R, Yanagawa Y, Thakkar MM, McCarley RW. Characterization of GABAergic neurons in rapid-eye-movement sleep controlling regions of the brainstem reticular formation in GAD67-green fluorescent protein knock-in mice. Eur J Neurosci. 2008;27:352–363. doi: 10.1111/j.1460-9568.2008.06024.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown RE, Winston S, Basheer R, Thakkar MM, McCarley RW. Electrophysiological characterization of neurons in the dorsolateral pontine rapid-eye-movement sleep induction zone of the rat: intrinsic membrane properties and responses to carbachol and orexins. Neuroscience. 2006;143:739–755. doi: 10.1016/j.neuroscience.2006.08.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruinstroop E, Cano G, Vanderhorst VG, Cavalcante JC, Wirth J, Sena-Esteves M, Saper CB. Spinal projections of the A5, A6 (locus coeruleus), and A7 noradrenergic cell groups in rats. J Comp Neurol. 2011;520:1985–2001. doi: 10.1002/cne.23024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckley NJ, Bonner TI, Buckley CM, Brann MR. Antagonist binding properties of five cloned muscarinic receptors expressed in CHO-K1 cells. Mol Pharmacol. 1989;35:469–476. [PubMed] [Google Scholar]
- Bulatko AK, Greeff NG. Functional availability of sodium channels modulated by cytosolic free Ca2+ in cultured mammalian neurons (N1E-115) J Physiol. 1995;484:307–312. doi: 10.1113/jphysiol.1995.sp020666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burdakov D, Liss B, Ashcroft FM. Orexin excites GABAergic neurons of the arcuate nucleus by activating the sodium–calcium exchanger. J Neurosci. 2003;23:4951–4957. doi: 10.1523/JNEUROSCI.23-12-04951.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chase MH. Motor control during sleep and wakefulness: clarifying controversies and resolving paradoxes. Sleep Med Rev. 2013;17:299–312. doi: 10.1016/j.smrv.2012.09.003. [DOI] [PubMed] [Google Scholar]
- Chase MH, Soja PJ, Morales FR. Evidence that glycine mediates the postsynaptic potentials that inhibit lumbar motoneurons during the atonia of active sleep. J Neurosci. 1989;9:743–751. doi: 10.1523/JNEUROSCI.09-03-00743.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clement O, Sapin E, Berod A, Fort P, Luppi PH. Evidence that neurons of the sublaterodorsal tegmental nucleus triggering paradoxical (REM) sleep are glutamatergic. Sleep. 2011;34:419–423. doi: 10.1093/sleep/34.4.419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman CG, Lydic R, Baghdoyan HA. M2 muscarinic receptors in pontine reticular formation of C57BL/6J mouse contribute to rapid eye movement sleep generation. Neuroscience. 2004;126:821–830. doi: 10.1016/j.neuroscience.2004.04.029. [DOI] [PubMed] [Google Scholar]
- Eriksson KS, Sergeeva O, Brown RE, Haas HL. Orexin/hypocretin excites the histaminergic neurons of the tuberomammillary nucleus. J Neurosci. 2001;21:9273–9279. doi: 10.1523/JNEUROSCI.21-23-09273.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin FBJ, Paxinos G. The Mouse Brain in Stereotaxic Coordinates. San Diego, CA, USA: Academic Press Inc; 1997. [Google Scholar]
- Fuller PM, Saper CB, Lu J. The pontine REM switch: past and present. J Physiol. 2007;584:735–741. doi: 10.1113/jphysiol.2007.140160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gagnon JF, Postuma RB, Mazza S, Doyon J, Montplaisir J. Rapid-eye-movement sleep behaviour disorder and neurodegenerative diseases. Lancet Neurol. 2006;5:424–432. doi: 10.1016/S1474-4422(06)70441-0. [DOI] [PubMed] [Google Scholar]
- Garcia-Rill E, Heister DS, Ye M, Charlesworth A, Hayar A. Electrical coupling: novel mechanism for sleep-wake control. Sleep. 2007;30:1405–1414. doi: 10.1093/sleep/30.11.1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajnik T, Lai YY, Siegel JM. Atonia-related regions in the rodent pons and medulla. J Neurophysiol. 2000;84:1942–1948. doi: 10.1152/jn.2000.84.4.1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heister DS, Hayar A, Garcia-Rill E. Cholinergic modulation of GABAergic and glutamatergic transmission in the dorsal subcoeruleus: mechanisms for REM sleep control. Sleep. 2009;32:1135–1147. doi: 10.1093/sleep/32.9.1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwamoto T, Watano T, Shigekawa M. A novel isothiourea derivative selectively inhibits the reverse mode of Na+/Ca2+ exchange in cells expressing NCX1. J Biol Chem. 1996;271:22391–22397. doi: 10.1074/jbc.271.37.22391. [DOI] [PubMed] [Google Scholar]
- Jones BE. Immunohistochemical study of choline acetyltransferase-immunoreactive processes and cells innervating the pontomedullary reticular formation in the rat. J Comp Neurol. 1990;295:485–514. doi: 10.1002/cne.902950311. [DOI] [PubMed] [Google Scholar]
- Jones BE. Paradoxical sleep and its chemical/structural substrates in the brain. Neuroscience. 1991;40:637–656. doi: 10.1016/0306-4522(91)90002-6. [DOI] [PubMed] [Google Scholar]
- Kitaichi K, Day JC, Quirion R. A novel muscarinic M4 receptor antagonist provides further evidence of an autoreceptor role for the muscarinic M2 receptor sub-type. Eur J Pharmacol. 1999;383:53–56. doi: 10.1016/s0014-2999(99)00607-x. [DOI] [PubMed] [Google Scholar]
- Krenzer M, Anaclet C, Vetrivelan R, Wang N, Vong L, Lowell BB, Fuller PM, Lu J. Brainstem and spinal cord circuitry regulating REM sleep and muscle atonia. PLoS One. 2011;6:e24998. doi: 10.1371/journal.pone.0024998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubin L. Carbachol models of REM sleep: recent developments and new directions. Arch Ital Biol. 2001;139:147–168. [PubMed] [Google Scholar]
- Lai YY, Clements JR, Siegel JM. Glutamatergic and cholinergic projections to the pontine inhibitory area identified with horseradish peroxidase retrograde transport and immunohistochemistry. J Comp Neurol. 1993;336:321–330. doi: 10.1002/cne.903360302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai YY, Kodama T, Schenkel E, Siegel JM. Behavioral response and transmitter release during atonia elicited by medial medullary stimulation. J Neurophysiol. 2010;104:2024–2033. doi: 10.1152/jn.00528.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai YY, Siegel JM. Pontomedullary glutamate receptors mediating locomotion and muscle tone suppression. J Neurosci. 1991;11:2931–2937. doi: 10.1523/JNEUROSCI.11-09-02931.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard TO, Lydic R. Pontine nitric oxide modulates acetylcholine release, rapid eye movement sleep generation, and respiratory rate. J Neurosci. 1997;17:774–785. doi: 10.1523/JNEUROSCI.17-02-00774.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Sherman D, Devor M, Saper CB. A putative flip-flop switch for control of REM sleep. Nature. 2006;441:589–594. doi: 10.1038/nature04767. [DOI] [PubMed] [Google Scholar]
- Luppi PH, Clement O, Sapin E, Gervasoni D, Peyron C, Leger L, Salvert D, Fort P. The neuronal network responsible for paradoxical sleep and its dysfunctions causing narcolepsy and rapid eye movement (REM) behaviour disorder. Sleep Med Rev. 2011;15:153–163. doi: 10.1016/j.smrv.2010.08.002. [DOI] [PubMed] [Google Scholar]
- Luppi PH, Clement O, Sapin E, Peyron C, Gervasoni D, Leger L, Fort P. Brainstem mechanisms of paradoxical (REM) sleep generation. Pflugers Arch. 2012;463:43–52. doi: 10.1007/s00424-011-1054-y. [DOI] [PubMed] [Google Scholar]
- Lytton J. Na+/Ca2+ exchangers: three mammalian gene families control Ca2+ transport. Biochem J. 2007;406:365–382. doi: 10.1042/BJ20070619. [DOI] [PubMed] [Google Scholar]
- McCarley RW. Neurobiology of REM and NREM sleep. Sleep Med. 2007;8:302–330. doi: 10.1016/j.sleep.2007.03.005. [DOI] [PubMed] [Google Scholar]
- Mallios VJ, Lydic R, Baghdoyan HA. Muscarinic receptor subtypes are differentially distributed across brain stem respiratory nuclei. Am J Physiol Lung Cell Mol Physiol. 1995;268:L941–L949. doi: 10.1152/ajplung.1995.268.6.L941. [DOI] [PubMed] [Google Scholar]
- Mathis J, Hess CW, Bassetti C. Isolated mediotegmental lesion causing narcolepsy and rapid eye movement sleep behaviour disorder: a case evidencing a common pathway in narcolepsy and rapid eye movement sleep behaviour disorder. J Neurol Neurosurg Psychiatry. 2007;78:427–429. doi: 10.1136/jnnp.2006.099515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsuya M, Mase T, Tsuchiya Y, Kawakami K, Hattori H, Kobayashi K, Ogino Y, Fujikawa T, Satoh A, Kimura T, Noguchi K, Ohtake N, Tomimoto K. J-104129, a novel muscarinic M3 receptor antagonist with high selectivity for M3 over M2 receptors. Bioorg Med Chem. 1999;7:2555–2567. doi: 10.1016/s0968-0896(99)00177-7. [DOI] [PubMed] [Google Scholar]
- Morrison AR. Paradoxical sleep without atonia. Arch Ital Biol. 1988;126:275–289. [PubMed] [Google Scholar]
- Onoe H, Sakai K. Kainate receptors: a novel mechanism in paradoxical (REM) sleep generation. Neuroreport. 1995;6:353–356. [PubMed] [Google Scholar]
- Parmentier R, Kolbaev S, Klyuch BP, Vandael D, Lin JS, Selbach O, Haas HL, Sergeeva OA. Excitation of histaminergic tuberomamillary neurons by thyrotropin-releasing hormone. J Neurosci. 2009;29:4471–4483. doi: 10.1523/JNEUROSCI.2976-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai K, Crochet S, Onoe H. Pontine structures and mechanisms involved in the generation of paradoxical (REM) sleep. Arch Ital Biol. 2001;139:93–107. [PubMed] [Google Scholar]
- Sakai K, Koyama Y. Are there cholinergic and non-cholinergic paradoxical sleep-on neurones in the pons. Neuroreport. 1996;7:2449–2453. doi: 10.1097/00001756-199611040-00009. [DOI] [PubMed] [Google Scholar]
- Sakai K, Onoe H. Critical role for M3 muscarinic receptors in paradoxical sleep generation in the cat. Eur J Neurosci. 1997;9:415–423. doi: 10.1111/j.1460-9568.1997.tb01619.x. [DOI] [PubMed] [Google Scholar]
- Sakai K, Sastre JP, Kanamori N, Jouvet M. State-specific neurones in the ponto-medullary reticular formation with specific reference to the postural atonia during paradoxical sleep in cat. In: Pompeiano M, Aimone MarsanC, editors. Brain Mechanisms of Perceptual Awareness and Purposeful Behavior. New York: Raven Press; 1981. pp. 405–429. [Google Scholar]
- Scammell TE. The neurobiology, diagnosis, and treatment of narcolepsy. Ann Neurol. 2003;53:154–166. doi: 10.1002/ana.10444. [DOI] [PubMed] [Google Scholar]
- Semba K. Aminergic and cholinergic afferents to REM sleep induction regions of the pontine reticular formation in the rat. J Comp Neurol. 1993;330:543–556. doi: 10.1002/cne.903300410. [DOI] [PubMed] [Google Scholar]
- Shammah-Lagnado SJ, Negrao N, Silva BA, Ricardo JA. Afferent connections of the nuclei reticularis pontis oralis and caudalis: a horseradish peroxidase study in the rat. Neuroscience. 1987;20:961–989. doi: 10.1016/0306-4522(87)90256-9. [DOI] [PubMed] [Google Scholar]
- Sheffler DJ, Williams R, Bridges TM, Xiang Z, Kane AS, Byun NE, Jadhav S, Mock MM, Zheng F, Lewis LM, Jones CK, Niswender CM, Weaver CD, Lindsley CW, Conn PJ. A novel selective muscarinic acetylcholine receptor subtype 1 antagonist reduces seizures without impairing hippocampus-dependent learning. Mol Pharmacol. 2009;76:356–368. doi: 10.1124/mol.109.056531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen KZ, Johnson SW. Presynaptic dopamine D2 and muscarine M3 receptors inhibit excitatory and inhibitory transmission to rat subthalamic neurones in vitro. J Physiol. 2000;525:331–341. doi: 10.1111/j.1469-7793.2000.00331.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soja PJ, López-Rodríguez F, Morales FR, Chase MH. The postsynaptic inhibitory control of lumbar motoneurons during the atonia of active sleep: effect of strychnine on motoneuron properties. J Neurosci. 1991;11:2804–2811. doi: 10.1523/JNEUROSCI.11-09-02804.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanderHorst VG, Ulfhake B. The organization of the brainstem and spinal cord of the mouse: relationships between monoaminergic, cholinergic, and spinal projection systems. J Chem Neuroanat. 2006;31:2–36. doi: 10.1016/j.jchemneu.2005.08.003. [DOI] [PubMed] [Google Scholar]
- Velazquez-Moctezuma J, Shalauta M, Gillin JC, Shiromani PJ. Cholinergic antagonists and REM sleep generation. Brain Res. 1991;543:175–179. doi: 10.1016/0006-8993(91)91064-8. [DOI] [PubMed] [Google Scholar]
- Vetrivelan R, Chang C, Lu J. Muscle tone regulation during REM sleep: neural circuitry and clinical significance. Arch Ital Biol. 2011;149:348–366. doi: 10.4449/aib.v149i4.1272. [DOI] [PubMed] [Google Scholar]
- Vetrivelan R, Fuller PM, Tong Q, Lu J. Medullary circuitry regulating rapid eye movement sleep and motor atonia. J Neurosci. 2009;29:9361–9369. doi: 10.1523/JNEUROSCI.0737-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber CR, Piacentino V, 3rd, Ginsburg KS, Houser SR, Bers DM. Na+-Ca2+ exchange current and submembrane [Ca2+] during the cardiac action potential. Circ Res. 2002;90:182–189. doi: 10.1161/hh0202.103940. [DOI] [PubMed] [Google Scholar]
- Xu C, Wu M, Morozova E, Alreja M. Muscarine activates the sodium–calcium exchanger via M3 receptors in basal forebrain neurons. Eur J Neurosci. 2006;24:2309–2313. doi: 10.1111/j.1460-9568.2006.05118.x. [DOI] [PubMed] [Google Scholar]