

Abstract
We investigated a neural reflex that controls the strength of inflammatory responses to immune challenge – the inflammatory reflex. In anaesthetized rats challenged with intravenous lipopolysaccharide (LPS, 60 μg kg−1), we found strong increases in plasma levels of the key inflammatory mediator tumour necrosis factor α (TNFα) 90 min later. Those levels were unaffected by previous bilateral cervical vagotomy, but were enhanced approximately 5-fold if the greater splanchnic sympathetic nerves had been cut. Sham surgery had no effect, and plasma corticosterone levels were unaffected by nerve sections, so could not explain this result. Electrophysiological recordings demonstrated that efferent neural activity in the splanchnic nerve and its splenic branch was strongly increased by LPS treatment. Splenic nerve activity was dependent on inputs from the splanchnic nerves: vagotomy had no effect on the activity in either nerve. Together, these data demonstrate that immune challenge with this dose of LPS activates a neural reflex that is powerful enough to cause an 80% suppression of the acute systemic inflammatory response. The efferent arm of this reflex is in the splanchnic sympathetic nerves, not the vagi as previously proposed. As with other physiological responses to immune challenge, the afferent pathway is presumptively humoral: the present data show that vagal afferents play no measurable part. Because inflammation sits at the gateway to immune responses, this reflex could play an important role in immune function as well as inflammatory diseases.
Introduction
The mammalian innate immune system responds to injuries or infections first by developing inflammation. Mast cells and macrophages are the sentinel cells responsible for the rapid reaction and release of pro-inflammatory cytokines that are necessary for the body to fight and overcome infections (Nathan, 2002). Amongst the various cytokines, tumour necrosis factor-α (TNFα) is a necessary and sufficient mediator of local and systemic inflammation that is produced by activated macrophages (Tracey et al. 1986). Inflammation, however, can come at a price. If excessive and generalized it can cause severe tissue injury and, in extreme cases, death. Therefore, it is important that inflammatory responses to immune challenge are appropriately regulated.
Not surprisingly, the nervous system is involved, and may be regarded as providing a rapid response in advance of cellular regulation by regulatory T cells and by alternatively activated macrophages. The CNS can influence immune function to control inflammation via two main ways: first, humorally, by activating the hypothalamic–pituitary–adrenal (HPA) axis to release glucocorticoids, which suppress the further synthesis of inflammatory cytokines (Besedovsky et al. 1986); second, neurally, via the autonomic nervous system (reviewed by Nance & Sanders, 2007).
A mechanism termed the inflammatory reflex has been proposed as the basis for autonomic regulation of immune function (Tracey, 2002; Andersson & Tracey, 2012). The efferent arm of this reflex – the neural-to-immune link – is thought to be the ‘cholinergic anti-inflammatory pathway’ (Rosas-Ballina & Tracey, 2009). According to this paradigm, an immune challenge is relayed by afferent nerves and/or by humoral signals to the brain, whereupon the brain drives efferent nerve fibres in the vagus that act ultimately to inhibit the release of TNFα by macrophages in the spleen (Andersson & Tracey, 2012). The pathway is complex. Vagal efferent fibres are proposed to activate the (sympathetic) splenic nerves (Rosas-Ballina et al. 2008), which in turn release noradrenaline to activate a population of acetylcholine-synthesizing T cells in the spleen: these then release acetylcholine, which acts via nicotinic receptors on splenic macrophages to inhibit TNFα production and release (Rosas-Ballina et al. 2008, 2011). Despite general acceptance of this theory, the model has been challenged for two main reasons: (1) in the principal test model (rats or mice made endotoxaemic with lipopolysaccharide (LPS) derived from Escherichia coli), it is not clear that cutting the vagus nerves, which should remove the inhibitory neural action, exacerbates inflammation (Caldwell et al. 1999; Bernik et al. 2002; Fuentes et al. 2005; Mihaylova et al. 2012); (2) there is evidently no synaptic connection from the vagus to the splenic sympathetic nerves (Bratton et al. 2012; Martelli et al. 2014).
Therefore, the aims of this study were to demonstrate the existence of a neural reflex able to control the inflammatory response induced by an immune challenge, to test the strength of such a reflex and to find the neural pathways responsible for its actions.
Methods
Ethical approval
All animal experiments were performed in accordance with guidelines of the National Health and Medical Research Council of Australia and were approved by the Animal Experimentation Ethics Committee of the Florey Institute of Neuroscience and Mental Health.
Thirty-eight adult male Sprague Dawley rats (290–350 g) were housed at 22°C on a 12 h light/dark cycle and used for these experiments. At the end of each experiment animals were killed with an overdose injection of pentobarbital sodium (>100 mg kg−1 i.v., Troy Laboratories, Glendenning, NSW, Australia).
Electrophysiological experiments
Anaesthesia was induced with pentobarbital sodium (60 mg kg−1, i.p.), the animal's trunk was shaved and the trachea was cannulated. Anaesthesia was then maintained for the duration of surgery by 2% isoflurane in pure oxygen, delivered by artifical ventilation (rodent ventilator, Ugo Basile, Comerio, Italy), before being replaced by urethane for the experiment proper (see below). Artificial ventilation was set at 65–70 inflations min–1, adjusting the tidal volume to maintain an end-tidal CO2 concentration between 3.5 and 4.5% throughout the experiment. The right femoral artery and vein were cannulated for monitoring blood pressure and intravenous administration of drugs, respectively. A water-perfused silastic jacket was positioned around the animal to maintain its body temperature around 37°C. Core temperature was measured by a thermocouple inserted 5 cm into the rectum. When preparatory surgery was complete, anaesthesia with isoflurane was gradually withdrawn and replaced by urethane (1.0–1.2 g kg−1, i.v.), but artificial ventilation with oxygen was maintained for the rest of the experiment. Animals were subdivided into two experimental groups (n = 4 per group) in which efferent activity was recorded from either the splenic sympathetic nerve (Splenic group) or the greater splanchnic sympathetic nerve (Splanchnic group).
Splenic group
The spleen was mobilized and its neurovascular attachments were individually ligated and cut close to the spleen, which was then removed. The splenic postganglionic sympathetic nerve was identified, dissected out and placed over a pair of silver hook electrodes for electrophysiological recording of its centripetal activity while the region was bathed in mineral oil. The nerve activity was amplified (10 000-fold), and filtered (100–1000 Hz). Activity was monitored continuously using an oscilloscope and recorded to computer using a CED Power 1401 interface and Spike2 software (Cambridge Electronic Design, Cambridge, UK), using a sampling rate of 5 kHz. Spikes were thresholded and counted in 10 s time bins. After 10 min of stable baseline recording, LPS (from E. coli 0111:B4; Sigma-Aldrich, St Louis, MO, USA; 60 μg kg−1 in 0.5 ml saline) was injected i.v. After 90 min, both cervical vagus nerves were cut and after a further 10 min the left greater splanchnic nerve was cut (Fig. 1).
Figure 1.
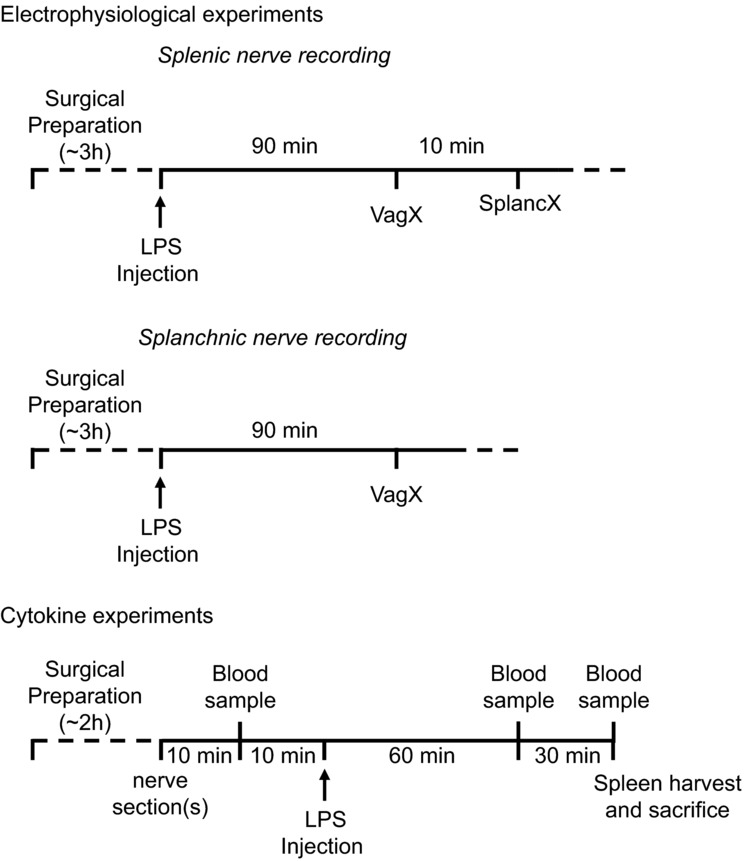
Electrophysiological experiments: surgical preparation and setting up stable nerve recordings took approximately 3 h. Animals were then injected with LPS (60 μg kg−1 i.v.) and followed for 90 min. At that point the cervical vagi were cut bilaterally (VagX). In splenic nerve recording experiments, the ipsilateral (left) splanchnic nerve was cut after a further 10 min (SplancX). Cytokine experiments: after approximately 2 h of surgical preparation, the cervical vagi (VagX), the splanchnic nerves (SplancX), both sets of nerves (VagX + SplancX) or neither (Sham) were severed in those respective groups. After a further 10 min, 1 ml of blood was collected (baseline sample). After a further 10 min, LPS was injected (60 μg kg−1 i.v.). Two further 1 ml blood samples were collected at 60 and 90 min after LPS injection. At 90 min the spleen was also extracted and the rat was killed.
Splanchnic group
The left greater splanchnic sympathetic nerve was identified: using a dorso-lateral approach (Dogan et al. 2003) the adrenal gland was freed from the perirenal fat, and the greater splanchnic nerve was followed along the greater psoas muscle in the centripetal direction – from the adrenal to the diaphragm. It was cut and its proximal part placed over a pair of silver wire hook electrodes while the region was bathed in mineral oil. Further recording details were as described above for splenic nerve recording. After 10 min of stable baseline recording, animals were injected i.v. with LPS (60 μg kg−1). After 90 min, both cervical vagus nerves were cut (Fig. 1).
Cytokine experiments
Rats were anaesthetized and prepared for surgery and artificially ventilated as described above. Animals were subdivided into four experimental groups (n = 5 per group): (1) ‘VagX’ (cervical vagi cut bilaterally); (2) ‘SplancX’ (greater splanchnic sympathetic nerves cut bilaterally); (3) ‘VagX + SplancX’ (bilateral section of vagi and splanchnic nerves); (4) ‘Sham’ (both sets of nerves exposed but not cut). LPS was injected i.v. (60 μg kg−1 dissolved in 0.5 ml saline). Ten further animals (n = 5 per group) were assigned to SplancX or Sham groups and injected with saline (0.5 ml i.v.) to control for any effect of these surgical procedures on our measures of inflammation.
Blood (1 ml) was collected from the right femoral artery at 10 min before (baseline levels), and 60 min and 90 min after LPS (or saline) administration. Immediately after the last blood sample was taken, the spleen was excised and the animal killed with an overdose of i.v. pentobarbitone. Blood samples were immediately centrifuged (15 min, 2000 g), and the plasma together with the spleen stored at −80°C for subsequent analysis (see Fig. 1 for a schema of the experimental timeline).
TNFα and corticosterone measurement
Plasma samples and spleens were assayed for TNFα using enzyme-linked immunosorbent assay kits (R&D Systems Inc., Minneapolis, MN, USA). For spleen preparation, frozen tissue samples were weighed and placed in homogenization buffer (PBS, containing a protease-inhibitor cocktail (Sigma-Aldrich), 0.5% Triton X-100; pH 7.2; 4°C) at a ratio of 100 mg tissue ml–1. Samples were homogenized and subjected to one freeze–thaw cycle and then sonicated for 5 min. The final homogenate was centrifuged at 12,000 g for 10 min. Tissue supernatants were separated and used for TNFα determination. Plasma samples taken at 90 min after LPS administration were also assayed for corticosterone (ELISA; Abnova, Jhongli, Taiwan).
Statistical analysis
Statistical analyses on electrophysiological experiments were performed using repeated-measures one-way analysis of variance (ANOVA). In both cases, post hoc comparisons were made by the Bonferroni test. Statistical analyses on data from cytokine experiments were performed using a repeated measures two-way ANOVA. Factors considered were time and experimental group. P values < 0.05 were considered significant.
Results
Electrophysiological experiments – LPS increases efferent activity in the splenic and splanchnic sympathetic nerves in rats
The spleen is a major organ mediating inflammatory responses to LPS (Huston et al. 2006) and splenic nerves have been implicated in the neural control of that inflammation (Rosas-Ballina & Tracey, 2009; Vida et al. 2011). In a first set of experiments, we therefore recorded the activity of the splenic sympathetic nerve in anaesthetized rats to confirm that this pathway was indeed activated by inflammatory challenge. In agreement with previous work (MacNeil et al. 1997), i.v. administration of LPS (60 μg kg−1) was followed by a strong rise in efferent splenic nerve activity (Fig. 2A, B). This was accompanied by classic signs of a febrile response: an increase in heart rate and body core temperature (Fig. 2A).
Figure 2.
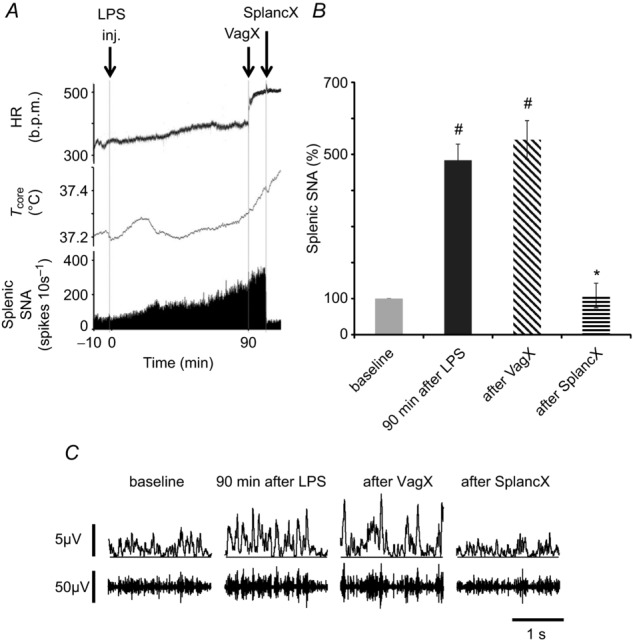
A, example of a chart record showing heart rate (HR, in beats per minute (b.p.m.)), body core temperature (Tcore, °C) and efferent splenic sympathetic nerve activity (SNA) of a urethane-anaesthetized rat injected i.v. with LPS (60 μg kg−1, at ‘inj’). Splenic nerve activity was quantified as spikes per 10 s that exceeded a selected threshold value. Ninety minutes after LPS injection, both cervical vagi were cut (at VagX). Subsequently, the left (ipsilateral) greater splanchnic sympathetic nerve was cut (at SplancX). B, mean splenic SNA in four rats, expressed as a percentage of baseline levels. Statistics were performed on log-transformed absolute values, using a repeated-measures one-way ANOVA followed by a Bonferroni post hoc test. #P < 0.01 compared to baseline levels. *P < 0.01 compared to levels 90 min after LPS. C, examples of raw splenic SNA recordings (lower traces) and rectified, smoothed recordings (10 ms time constant, upper traces) taken at the times indicated. Splenic SNA was significantly increased 90 min after LPS injection, was undiminished by bilateral vagotomy but was significantly reduced by cutting the left greater splanchnic nerve.
To test the hypothesis that it is the vagus nerves that drive the splenic nerves under conditions of immune challenge (Rosas-Ballina et al. 2008), we bilaterally cut the cervical vagus nerves 90 min after LPS administration. Heart rate rose abruptly after vagotomy (Fig. 2A), demonstrating that cardioinhibitory vagal fibres had been cut, but there was no reduction in efferent splenic nerve activity. By contrast, splenic nerve activity dropped dramatically when the left (ipsilateral) greater splanchnic nerve was cut (Fig. 2A–C). The minor contribution from the contralateral splanchnic nerve, which was not cut for technical reasons, can account for the remaining activity (Bratton et al. 2012).
Next, we recorded the efferent activity of the left greater splanchnic sympathetic nerve, and found that this too was strongly activated by LPS (60 μg kg−1, Fig. 3A, B). Splanchnic nerve activity was also unaffected by bilateral vagotomy (Fig. 3A–C).
Figure 3.
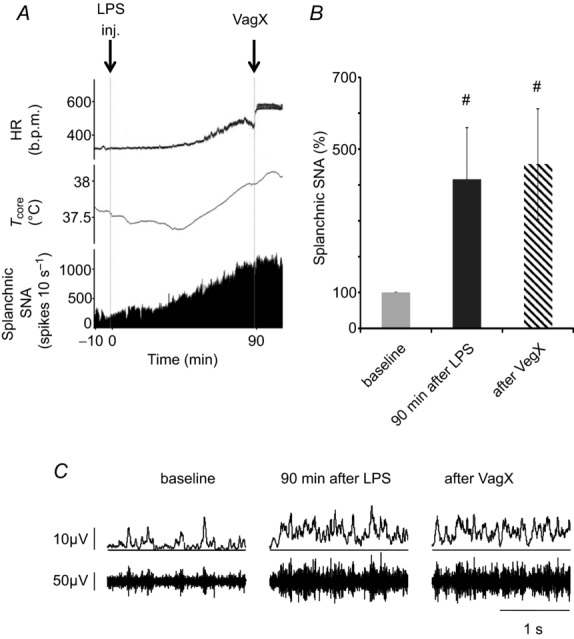
A, example of a chart record showing heart rate (HR, in beats per minute (b.p.m.)), body core temperature (Tcore, °C) and efferent activity in the splanchnic sympathetic nerve (SNA) of a urethane-anaesthetized rat injected i.v. with LPS (60 μg kg−1, at ‘inj’). Splanchnic nerve activity was quantified as spikes per 10 s that exceeded a selected threshold value. Ninety minutes after LPS injection, both cervical vagi were cut (at VagX). B, mean splanchnic SNA, expressed as a percentage of baseline levels. Statistics were performed on log-transformed absolute values, using a repeated measures one-way ANOVA followed by a Bonferroni post hoc test. #P < 0.05 compared to baseline levels. C, examples of raw splanchnic SNA recordings (lower traces) and rectified, smoothed recordings (10 ms time constant, upper traces) taken at the times indicated. Splanchnic SNA was increased significantly 90 min after LPS treatment but was undiminished by bilateral vagotomy.
Cytokine experiments – the splanchnic nerves operate to damp down the levels of TNFα in endotoxemic rats
To examine how the activity transmitted by autonomic nerves controls the level of inflammation, we tested the effects of cutting the greater splanchnic sympathetic nerves and the vagi on inflammatory responses to immune challenge in urethane-anaesthetized rats. Four groups of rats were injected with LPS (60 μg kg−1 i.v.). From undetectable levels at baseline, plasma TNFα was significantly elevated at both 60 and 90 min after LPS injection in each group (Fig. 4A). Previous section of the greater splanchnic sympathetic nerves (SplancX) enhanced plasma TNFα levels ∼4-fold at 60 min and ∼5-fold at 90 min, compared with animals given sham surgery (Sham) (Fig. 4A). Vagotomy had no significant effect, either alone (VagX) or in combination with splanchnic nerve section (VagX + SplancX) (Fig. 4A). Rises in body temperature were similar across LPS-treated groups (Fig. 5), confirming the previous finding that severing splanchnic nerves or vagi does not alter the increase in core body temperature induced by comparable doses of LPS (Caldwell et al. 1999; Dogan et al. 2003; Romanovsky et al. 2005). Heart rate showed an upward trend after LPS treatment, but only reached statistical significance in the two groups whose splanchnic nerves had been cut (SplancX and VagX + SplancX; Fig. 5). Plasma corticosterone levels at +90 min were comparable across groups (Fig. 4B). Tissue concentrations of TNFα in spleens of SplancX and SplancX + VagX animals followed the same trend as their plasma levels, being nearly double those of Sham and VagX animals (Fig. 4C). Plasma TNFα concentrations remained undetectable in a further five SplancX and five Sham animals that received saline instead of LPS (data not shown).
Figure 4.
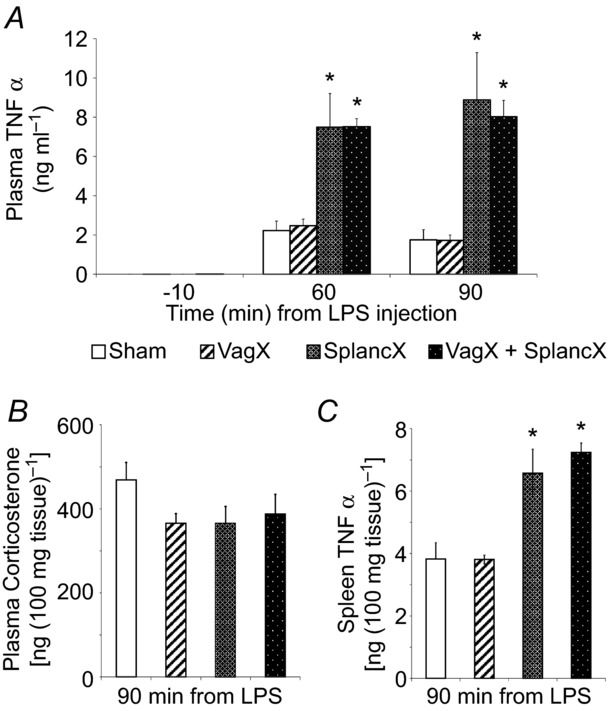
A, plasma TNFα levels in four groups of animals (VagX, SplancX, VagX + SplancX and Sham; each n = 5) in arterial blood samples taken 10 min before, and 60 and 90 min after LPS injection (60 μg kg−1 i.v.). Data were analysed using a repeated-measures two-way ANOVA followed by a Bonferroni post hoc test. *P < 0.001 compared to Sham and VagX levels at the same time point. B, plasma corticosterone levels in the same groups of rats measured in blood samples taken 90 min after LPS administration. No differences were detected between groups (P = 0.194, one-way ANOVA). C, TNFα levels measured in spleens of the same four groups of rats (removed 90 min after LPS injection). *P < 0.01 compared to Sham and VagX treatment (one-way ANOVA followed by Bonferroni post hoc comparison).
Figure 5.
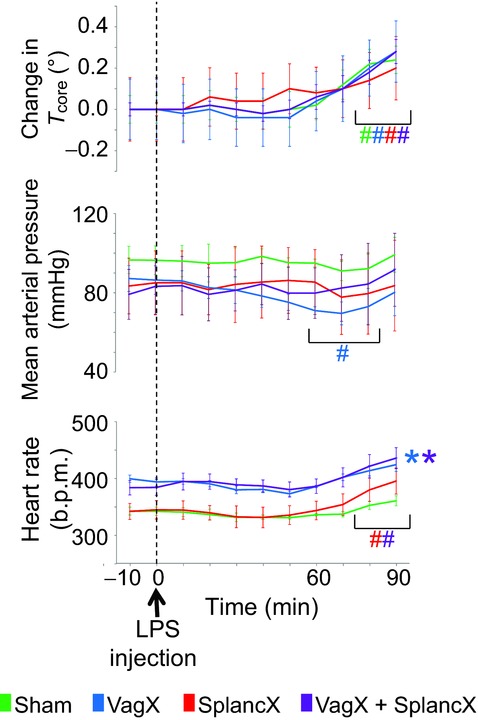
Changes of core body temperature, mean arterial pressure and heart rate in response to LPS treatment (60 μg kg−1 i.v.) in four groups of rats, as indicated. Results are expressed as mean ± SEM. Data were analysed using a repeated-measures two-way ANOVA followed by Bonferroni post hoc test. #P < 0.05 compared to baseline levels. *P < 0.05 compared to sham group.
Discussion
Our findings confirm that i.v. LPS triggers a strong systemic inflammatory response. Following an established paradigm (e.g. Borovikova et al. 2000), we measured systemic inflammation by circulating levels of TNFα, which is recognized as ‘a necessary and sufficient’ early mediator of inflammation (Tracey et al. 1986; Tracey, 2002). Our findings support the hypothesis (Tracey, 2002) that inflammation is under the control of an inhibitory neural reflex. But that reflex, we find, is more powerful than previously thought, and is mediated by sympathetic nerves rather than the vagi. LPS treatment caused a sustained rise in efferent activity in the greater splanchnic nerve and its splenic branch, activity that came through conventional sympathetic nerve pathways, not the vagi. That activity caused a roughly 80% suppression of the plasma TNFα response to LPS, as shown by the 5-fold increase in animals whose splanchnic nerves were cut. The reflex suppression cannot be attributed to the HPA axis, because plasma corticosterone levels were unchanged by splanchnic nerve section. Moreover, the clear lack of an effect of vagotomy shows that, at least under these experimental conditions, the vagus nerves play no discernible part – afferent or efferent – in the reflex control of inflammation.
The concept that the nervous system influences the immune system via nerves is not new. Felten et al. (1985) provided the initial descriptions of the sympathetic innervation of the thymus and spleen in mice. Since then, a number of studies have implicated an inhibitory action of the sympathetic nervous system on inflammation, based on anatomical (Nance & Sanders, 2007) as well as in vivo physiological and pharmacological evidence (Katafuchi et al. 1993b; Szelenyi et al. 2000b). It was in this context that Tracey (2002) introduced the idea of the inflammatory reflex, and used this as a basis to promote the theory that the parasympathetic and sympathetic nervous systems act together to maintain immunological homeostasis. According to this model, immune challenge reflexly activates the ‘cholinergic anti-inflammatory pathway’ via the vagus nerves, which then drive the splenic nerves to inhibit excessive release of TNFα (for a comprehensive review see Andersson & Tracey, 2012). The spleen and the splenic nerves are essential for this action (Huston et al. 2006; Rosas-Ballina et al. 2008).
Our evidence shows that this theory needs to be revised. We found here that the vagus nerves do not drive the splenic efferent nerve activity that follows immune challenge with LPS; that activity is driven by the conventional sympathetic pathway though the splanchnic nerves. This finding fits with our previous report that there is no synaptic connection from the vagus to the splenic sympathetic nerves (Bratton et al. 2012). Critically, we found that cutting the cervical vagi caused no increase in the plasma TNFα response to LPS, whether or not the splanchnic nerves were cut. This means that any anti-inflammatory pathways in the vagi, however and wherever they might act, were functionally silent. We therefore conclude that the ‘cholinergic anti-inflammatory pathway’ and the vagus nerves do not constitute the efferent arm of the inflammatory reflex. That efferent arm is purely sympathetic. This is not to deny that vagal pathways can exert anti-inflammatory actions in acute inflammation, but these are best regarded as phenomena to be engaged experimentally by exogenous electrical (Borovikova et al. 2000) or pharmacological stimuli (Bernik et al. 2002; Martelli et al. 2014). Any physiological function they might serve is unknown.
We showed directly that immune challenge with LPS caused a sustained increase in the efferent activity of the greater splanchnic nerve, confirming previous findings on urethane-anaesthetized rabbits (Iriki & Saigusa, 1998). When combined with the observation that cutting that nerve strongly disinhibits TNFα production, the most likely explanation is that the immune challenge activates CNS pathways that reflexly drive the sympathetic anti-inflammatory pathway in the splanchnic nerves. Less likely possibilities include the following. (1) The splanchnic anti-inflammatory nerve fibres are not the same ones that we recorded and found to be excited by LPS. If so, the activity of those hypothetical fibres would be tonic, rather than driven by LPS. Under baseline conditions they would be undetected, but their anti-inflammatory action would be revealed after LPS challenge. (2) The splanchnic nerves contain both afferent and efferent pathways of the inflammatory reflex. This seems unlikely because the increased splanchnic activity following LPS challenge was recorded from the central cut end of the nerve, when splanchnic afferent pathways on that side had already been disconnected.
The main efferent target organ for this sympathetic anti-inflammatory action is most likely the spleen, principally macrophages located in the white pulp (Rosas-Ballina et al. 2008, 2011; Vida et al. 2011). The spleen is the major source of plasma TNFα following systemic LPS challenge (Huston et al. 2006). We found, as have others (MacNeil et al. 1997), that LPS challenge causes a strong and sustained increase in efferent activity of the splenic nerve. We additionally demonstrated that this activity was driven from the splanchnic nerve, but not the vagi. It is already known that direct electrical stimulation of the splenic nerve can suppress the cytotoxicity of splenic natural killer cells (Katafuchi et al. 1993b) and reduce the TNFα response to LPS challenge (Kees et al. 2003; Vida et al. 2011). Those suppressive actions are attributable to stimulation of beta adrenergic receptors on splenocytes (Katafuchi et al. 1993b; Szelenyi et al. 2000a,b2000b; Kees et al. 2003; Vida et al. 2011). All the components are therefore in place to support an efferent action of the inflammatory reflex on the spleen. But it is important to note that our data do not exclude the involvement of other organs supplied by the greater splanchnic sympathetic nerves, such as the adrenal gland, the liver and the immune tissue associated with the gut (see Fig. 6). These possibilities remain to be explored.
Figure 6.
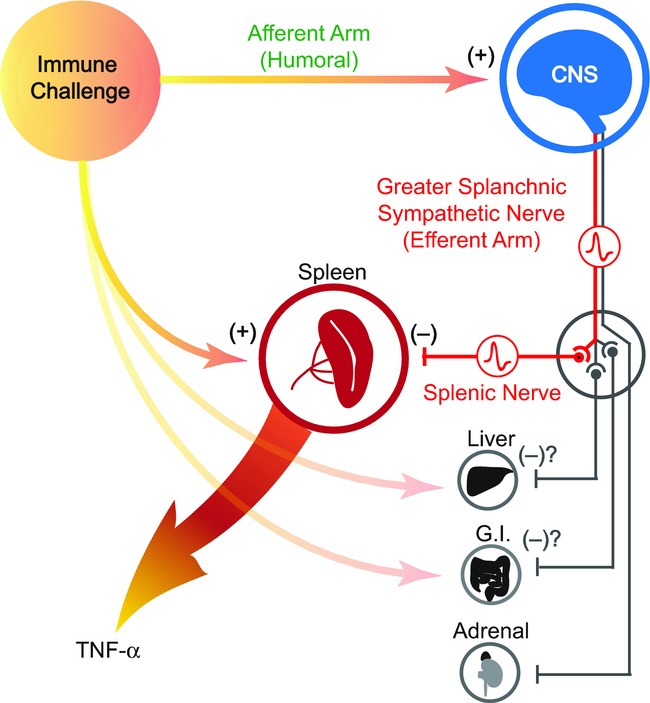
An immune challenge such as lipopolysaccharide (LPS) is detected by cells of the innate immune system, which respond by releasing pro-inflammatory cytokines such as TNFα. In response to systemic LPS, the spleen is known to be the major source of TNFα, which in turn plays a pivotal role in driving the full inflammatory response. At the same time, the immune challenge is detected by the CNS. As in the case of fever, this afferent signal is mediated by humoral factors such as inflammatory cytokines and/or prostaglandins. The CNS then responds by activating a specific subset of sympathetic nerves, whose function is to suppress excessive production of TNFα by the spleen. Other possible targets include the liver, cells in the gastrointestinal (G.I.) tract and the adrenal glands. Those anti-inflammatory nerve fibres run in the greater splanchnic nerve, which constitutes the efferent arm of the inflammatory reflex. Nerve pathways in the vagus do not contribute.
The afferent arm of the inflammatory reflex has not yet been specifically defined, but it probably shares the humoral afferent pathway established for two other CNS responses to immune challenge: fever and HPA axis activation. Those responses are mediated by cytokines and prostaglandins acting on the brain, notably the preoptic area (Besedovsky et al. 1986; Turrin & Rivest, 2004; Romanovsky et al. 2005; Saper et al. 2012). In this context, it has been shown that reflex activation of the splenic nerve by LPS is also mediated by central prostaglandin synthesis (MacNeil et al. 1997). Furthermore, the preoptic area has been shown to exert a tonic inhibitory action on the cytotoxicity of splenic natural killer cells, and this is mediated by activity in the splenic nerve (Katafuchi et al. 1993a). Further details remain to be discovered. As noted above, it seems clear that vagal afferent pathways play no part in the inflammatory reflex, at least when it is stimulated with moderate or high doses of LPS (Caldwell et al. 1999; Bernik et al. 2002; Fuentes et al. 2005; Mihaylova et al. 2012; this study). But we cannot exclude the possibility that with very low doses of LPS, vagal afferent pathways might contribute to the inflammatory reflex, as they can to fever (Romanovsky et al. 2005).
Limitations
Our investigation was restricted to the earliest stages of inflammation (1–2 h). TNFα was chosen as the most appropriate index of early inflammation because of its rapid release ahead of other pro-inflammatory cytokines such as interleukin-1, interleukin-6 and interferon-γ, which we did not measure (Givalois et al. 1994; Kakizaki et al. 1999). We can make no conclusions about the effects of splanchnic nerve section on these later inflammatory markers, or on the longer term progression of the inflammatory response.
Second, we only studied the systemic inflammatory response to i.v. LPS at a single mid-range dose. The positive effect of cutting the splanchnic nerves was strong and clear, but we do not know if this would apply to all doses of LPS. With regard to the lack of an effect of cutting the vagi, others have used higher doses of LPS in anaesthetized animals and tested the effect of vagotomy. The original study by Borovikova et al. (2000) reported that 15 mg kg−1 LPS caused TNFα levels that were 40% higher in vagotomized animals, but those animals had 33% lower corticosterone levels than controls, confounding the result. Subsequent studies with higher doses of LPS than those used here found either no significant effect of vagotomy (Bernik et al. 2002; Mihaylova et al. 2012) or a reduced TNFα response (Fuentes et al. 2005). The choice of LPS dose therefore seems unlikely to have influenced our conclusion that the vagi are not involved.
Note that the neural factors affecting acute systemic inflammation may not be identical to those in models of localized (abdominal) or longer-term inflammation. While the present experiments showed no effect of vagotomy, this has been shown to enhance the inflammatory response of mice 6 h after they were given septic peritonitis (van Westerloo et al. 2005), and to increase chronic inflammation of the colon over a time course of weeks (Ghia et al. 2007; O'Mahony et al. 2009).
Finally, this study was performed on anaesthetized animals. It is known that anaesthesia both depresses autonomic reflexes (Ebert et al. 1995; Umehara et al. 2006) and has an anti-inflammatory action (Kotanidou et al. 1996; Fuentes et al. 2005; Rodriguez-Gonzalez et al. 2013). Both these factors are likely to have influenced our results quantitatively. The inflammatory reflex is evidently robust enough to display itself despite those sources of bias, but it would be worthwhile to extend these experiments in future to unanaesthetized animals.
Significance
Neural influences on immune function can be profound (Meisel et al. 2005). The pivotal position of inflammation in immune responses (Nathan, 2002) means that if this reflex acts too strongly, it could compromise resistance to infection. If, on the other hand, it acts too weakly, it could predispose the body to inflammatory disease. Identification of the efferent neural pathway responsible for this action now opens the way to direct study of its role in these and other conditions.
Key points
It is believed that the CNS controls inflammation via the autonomic nervous system, but the strength of this action and the neural pathways responsible are unclear.
In anaesthetized rats we measured the inflammatory response to lipopolysaccharide (LPS, 60 μg kg−1, i.v.) by plasma tumour necrosis factor α (TNFα) levels 90 min later.
Bilateral section of the splanchnic sympathetic nerves before LPS treatment resulted in a 5-fold increase in the plasma TNFα response, but bilateral vagotomy had no effect.
LPS treatment strongly increased efferent activity in the splanchnic sympathetic nerve and its splenic branch; vagotomy did not affect this.
These results show that, besides directly stimulating inflammation, LPS engages a powerful anti-inflammatory reflex that can inhibit the plasma TNFα response by 80%.
The reflex efferent arm is in the splanchnic sympathetic nerves; the vagi play no part.
Acknowledgments
We thank David Trevaks for technical assistance.
Glossary
- HPA
hypothalamic–pituitary–adrenal
- LPS
lipopolysaccharide
- SplancX
greater splanchnic nerve cut
- TNFα
tumour necrosis factor-α
- VagX
cervical vagus nerve cut
Additional information
Competing interests
The authors have no conflict of interest to declare.
Author contributions
D.M. and R.M.M. designed the study; D.M. and S.T.Y. performed the experiments; D.M., S.T.Y., M.J.M. and R.M.M. interpreted the data and wrote the paper.
Funding
This work was supported by project grant 1051102 from the National Health and Medical Research Council (NHMRC) of Australia and from the Victorian Government Operational Infrastructure Support Program. D.M. was supported by the Fondazione Cassa di Risparmio Bologna. R.M.M. was supported by Principal Research Fellowship 566667 from NHMRC.
Translational Perspective
There is two-way communication between the immune and nervous systems. Following an immune challenge, it is believed that a neural reflex – the inflammatory reflex – is activated, and this regulates the grade of the inflammatory response in a negative feedback manner. What remains unclear is the strength of such a reflex and the neural pathways responsible for its actions. Here we show that endotoxaemic challenge (lipopolysaccharide, 60 μg kg−1, given i.v.) activates a neural reflex that is powerful enough to suppress the systemic inflammatory response by 80%. The efferent arm of this reflex is in the splanchnic sympathetic nerves, not the vagi as previously proposed. The afferent arm of this reflex is presumptively humoral: it does not involve the vagi. Neural influences on immune function can be profound, but the full importance of this reflex has yet to be explored. The key position of inflammation at the gateway of immune responses means that this reflex could contribute materially on the one hand to susceptibility to infection and on the other to inflammatory disease. The influence of the CNS in resistance to infection, sepsis and inflammatory disease has been insufficiently explored. Identifying the peripheral neural pathway for this action opens the way for rigorous study.
References
- Andersson U, Tracey KJ. Reflex principles of immunological homeostasis. Ann Rev Immunol. 2012;30:313–335. doi: 10.1146/annurev-immunol-020711-075015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernik TR, Friedman SG, Ochani M, DiRaimo R, Ulloa L, Yang H, Sudan S, Czura CJ, Ivanova SM, Tracey KJ. Pharmacological stimulation of the cholinergic antiinflammatory pathway. J Exp Med. 2002;195:781–788. doi: 10.1084/jem.20011714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besedovsky H, del Rey A, Sorkin E, Dinarello CA. Immunoregulatory feedback between interleukin-1 and glucocorticoid hormones. Science. 1986;233:652–654. doi: 10.1126/science.3014662. [DOI] [PubMed] [Google Scholar]
- Borovikova LV, Ivanova S, Zhang M, Yang H, Botchkina GI, Watkins LR, Wang H, Abumrad N, Eaton JW, Tracey KJ. Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature. 2000;405:458–462. doi: 10.1038/35013070. [DOI] [PubMed] [Google Scholar]
- Bratton BO, Martelli D, McKinley MJ, Trevaks D, Anderson CR, McAllen RM. Neural regulation of inflammation: no neural connection from the vagus to splenic sympathetic neurons. Exp Physiol. 2012;97:1180–1185. doi: 10.1113/expphysiol.2011.061531. [DOI] [PubMed] [Google Scholar]
- Caldwell FT, Jr, Graves DB, Wallace BH. Humoral versus neural pathways for fever production in rats after administration of lipopolysaccharide. J Trauma. 1999;47:120–129. doi: 10.1097/00005373-199907000-00025. [DOI] [PubMed] [Google Scholar]
- Dogan MD, Kulchitsky VA, Patel S, Petervari E, Szekely M, Romanovsky AA. Bilateral splanchnicotomy does not affect lipopolysaccharide-induced fever in rats. Brain Res. 2003;993:227–229. doi: 10.1016/j.brainres.2003.09.010. [DOI] [PubMed] [Google Scholar]
- Ebert TJ, Harkin CP, Muzi M. Cardiovascular responses to sevoflurane: a review. Anesth Analg. 1995;81:S11–22. doi: 10.1097/00000539-199512001-00003. [DOI] [PubMed] [Google Scholar]
- Felten DL, Felten SY, Carlson SL, Olschowka JA, Livnat S. Noradrenergic and peptidergic innervation of lymphoid tissue. J Immunol. 1985;135:755s–765s. [PubMed] [Google Scholar]
- Fuentes JM, Hanly EJ, Aurora AR, De Maio A, Talamini MA. Anesthesia-specific protection from endotoxic shock is not mediated through the vagus nerve. Surgery. 2005;138:766–771. doi: 10.1016/j.surg.2005.06.057. [DOI] [PubMed] [Google Scholar]
- Ghia JE, Blennerhassett P, Collins SM. Vagus nerve integrity and experimental colitis. Am J Physiol Gastrointest Liver Physiol. 2007;293:G560–567. doi: 10.1152/ajpgi.00098.2007. [DOI] [PubMed] [Google Scholar]
- Givalois L, Dornand J, Mekaouche M, Solier MD, Bristow AF, Ixart G, Siaud P, Assenmacher I, Barbanel G. Temporal cascade of plasma level surges in ACTH, corticosterone, and cytokines in endotoxin-challenged rats. Am J Physiol. 1994;267:R164–170. doi: 10.1152/ajpregu.1994.267.1.R164. [DOI] [PubMed] [Google Scholar]
- Huston JM, Ochani M, Rosas-Ballina M, Liao H, Ochani K, Pavlov VA, Gallowitsch-Puerta M, Ashok M, Czura CJ, Foxwell B, Tracey KJ, Ulloa L. Splenectomy inactivates the cholinergic antiinflammatory pathway during lethal endotoxemia and polymicrobial sepsis. J Exp Med. 2006;203:1623–1628. doi: 10.1084/jem.20052362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iriki M, Saigusa T. Regional differentiation of sympathetic efferents during fever. Prog Brain Res. 1998;115:477–497. doi: 10.1016/s0079-6123(08)62048-8. [DOI] [PubMed] [Google Scholar]
- Kakizaki Y, Watanobe H, Kohsaka A, Suda T. Temporal profiles of interleukin-1β, interleukin-6, and tumor necrosis factor-α in the plasma and hypothalamic paraventricular nucleus after intravenous or intraperitoneal administration of lipopolysaccharide in the rat: estimation by push-pull perfusion. Endocr J. 1999;46:487–496. doi: 10.1507/endocrj.46.487. [DOI] [PubMed] [Google Scholar]
- Katafuchi T, Ichijo T, Take S, Hori T. Hypothalamic modulation of splenic natural killer cell activity in rats. J Physiol. 1993a;471:209–221. doi: 10.1113/jphysiol.1993.sp019898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katafuchi T, Take S, Hori T. Roles of sympathetic nervous system in the suppression of cytotoxicity of splenic natural killer cells in the rat. J Physiol. 1993b;465:343–357. doi: 10.1113/jphysiol.1993.sp019680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kees MG, Pongratz G, Kees F, Scholmerich J, Straub RH. Via β-adrenoceptors, stimulation of extrasplenic sympathetic nerve fibers inhibits lipopolysaccharide-induced TNF secretion in perfused rat spleen. J Neuroimmunol. 2003;145:77–85. doi: 10.1016/j.jneuroim.2003.09.011. [DOI] [PubMed] [Google Scholar]
- Kotanidou A, Choi AM, Winchurch RA, Otterbein L, Fessler HE. Urethan anaesthesia protects rats against lethal endotoxemia and reduces TNF-α release. J Appl Physiol. 1996;81:2305–2311. [PubMed] [Google Scholar]
- MacNeil BJ, Jansen AH, Janz LJ, Greenberg AH, Nance DM. Peripheral endotoxin increases splenic sympathetic nerve activity via central prostaglandin synthesis. Am J Physiol. 1997;273:R609–614. doi: 10.1152/ajpregu.1997.273.2.R609. [DOI] [PubMed] [Google Scholar]
- Martelli D, McKinley MJ, McAllen RM. The cholinergic anti-inflammatory pathway: a critical review. Auton Neurosci. 2014 doi: 10.1016/j.autneu.2013.12.007. in Press. [DOI] [PubMed] [Google Scholar]
- Meisel C, Schwab JM, Prass K, Meisel A, Dirnagl U. Central nervous system injury-induced immune deficiency syndrome. Nat Rev Neurosci. 2005;6:775–786. doi: 10.1038/nrn1765. [DOI] [PubMed] [Google Scholar]
- Mihaylova S, Killian A, Mayer K, Pullamsetti SS, Schermuly R, Rosengarten B. Effects of anti-inflammatory vagus nerve stimulation on the cerebral microcirculation in endotoxinemic rats. J Neuroinflammation. 2012;9:183. doi: 10.1186/1742-2094-9-183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nance DM, Sanders VM. Autonomic innervation and regulation of the immune system (1987–2007) Brain Behav Immun. 2007;21:736–745. doi: 10.1016/j.bbi.2007.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathan C. Points of control in inflammation. Nature. 2002;420:846–852. doi: 10.1038/nature01320. [DOI] [PubMed] [Google Scholar]
- O'Mahony C, van der Kleij H, Bienenstock J, Shanahan F, O'Mahony L. Loss of vagal anti-inflammatory effect: in vivo visualization and adoptive transfer. Am J Physiol Regul Integr Comp Physiol. 2009;297:R1118–1126. doi: 10.1152/ajpregu.90904.2008. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Gonzalez R, Baluja A, Veiras Del Rio S, Rodriguez A, Rodriguez J, Taboada M, Brea D, Alvarez J. Effects of sevoflurane postconditioning on cell death, inflammation and TLR expression in human endothelial cells exposed to LPS. J Transl Med. 2013;11:87. doi: 10.1186/1479-5876-11-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romanovsky AA, Almeida MC, Aronoff DM, Ivanov AI, Konsman JP, Steiner AA, Turek VF. Fever and hypothermia in systemic inflammation: recent discoveries and revisions. Front Biosci. 2005;10:2193–2216. doi: 10.2741/1690. [DOI] [PubMed] [Google Scholar]
- Rosas-Ballina M, Ochani M, Parrish WR, Ochani K, Harris YT, Huston JM, Chavan S, Tracey KJ. Splenic nerve is required for cholinergic antiinflammatory pathway control of TNF in endotoxemia. Proc Natl Acad Sci U S A. 2008;105:11008–11013. doi: 10.1073/pnas.0803237105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosas-Ballina M, Olofsson PS, Ochani M, Valdes-Ferrer SI, Levine YA, Reardon C, Tusche MW, Pavlov VA, Andersson U, Chavan S, Mak TW, Tracey KJ. Acetylcholine-synthesizing T cells relay neural signals in a vagus nerve circuit. Science. 2011;334:98–101. doi: 10.1126/science.1209985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosas-Ballina M, Tracey KJ. The neurology of the immune system: neural reflexes regulate immunity. Neuron. 2009;64:28–32. doi: 10.1016/j.neuron.2009.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saper CB, Romanovsky AA, Scammell TE. Neural circuitry engaged by prostaglandins during the sickness syndrome. Nat Neurosci. 2012;15:1088–1095. doi: 10.1038/nn.3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szelenyi J, Kiss JP, Puskas E, Szelenyi M, Vizi ES. Contribution of differently localized α2-and β-adrenoceptors in the modulation of TNF-α and IL-10 production in endotoxemic mice. Ann N Y Acad Sci. 2000a;917:145–153. doi: 10.1111/j.1749-6632.2000.tb05378.x. [DOI] [PubMed] [Google Scholar]
- Szelenyi J, Kiss JP, Vizi ES. Differential involvement of sympathetic nervous system and immune system in the modulation of TNF-α production by α2-and β-adrenoceptors in mice. J Neuroimmunol. 2000b;103:34–40. doi: 10.1016/s0165-5728(99)00234-9. [DOI] [PubMed] [Google Scholar]
- Tracey KJ. The inflammatory reflex. Nature. 2002;420:853–859. doi: 10.1038/nature01321. [DOI] [PubMed] [Google Scholar]
- Tracey KJ, Beutler B, Lowry SF, Merryweather J, Wolpe S, Milsark IW, Hariri RJ, Fahey TJ, 3rd, Zentella A, Albert JD. Shock and tissue injury induced by recombinant human cachectin. Science. 1986;234:470–474. doi: 10.1126/science.3764421. [DOI] [PubMed] [Google Scholar]
- Turrin NP, Rivest S. Unraveling the molecular details involved in the intimate link between the immune and neuroendocrine systems. Exp Biol Med. 2004;229:996–1006. doi: 10.1177/153537020422901003. [DOI] [PubMed] [Google Scholar]
- Umehara S, Tanaka M, Nishikawa T. Effects of sevoflurane anaesthesia on carotid-cardiac baroreflex responses in humans. Anesth Analg. 2006;102:38–44. doi: 10.1213/01.ane.0000183651.10514.9a. [DOI] [PubMed] [Google Scholar]
- van Westerloo DJ, Giebelen IA, Florquin S, Daalhuisen J, Bruno MJ, de Vos AF, Tracey KJ, van der Poll T. The cholinergic anti-inflammatory pathway regulates the host response during septic peritonitis. J Infect Dis. 2005;191:2138–2148. doi: 10.1086/430323. [DOI] [PubMed] [Google Scholar]
- Vida G, Pena G, Deitch EA, Ulloa L. α7-cholinergic receptor mediates vagal induction of splenic norepinephrine. J Immunol. 2011;186:4340–4346. doi: 10.4049/jimmunol.1003722. [DOI] [PMC free article] [PubMed] [Google Scholar]