

Abstract
In amyotrophic lateral sclerosis (ALS), an adult onset disease in which there is progressive degeneration of motoneurones, it has been suggested that an intrinsic hyperexcitability of motoneurones (i.e. an increase in their firing rates), contributes to excitotoxicity and to disease onset. Here we show that there is no such intrinsic hyperexcitability in spinal motoneurones. Our studies were carried out in an adult mouse model of ALS with a mutated form of superoxide dismutase 1 around the time of the first muscle fibre denervations. We showed that the recruitment current, the voltage threshold for spiking and the frequency–intensity gain in the primary range are all unchanged in most spinal motoneurones, despite an increased input conductance. On its own, increased input conductance would decrease excitability, but the homeostasis for excitability is maintained due to an upregulation of a depolarizing current that is activated just below the spiking threshold. However, this homeostasis failed in a substantial fraction of motoneurones, which became hypoexcitable and unable to produce sustained firing in response to ramps of current. We found similar results both in lumbar motoneurones recorded in anaesthetized mice, and in sacrocaudal motoneurones recorded in vitro, indicating that the lack of hyperexcitability is not caused by anaesthetics. Our results suggest that, if excitotoxicity is indeed a mechanism leading to degeneration in ALS, it is not caused by the intrinsic electrical properties of motoneurones but by extrinsic factors such as excessive synaptic excitation.
Introduction
The intrinsic excitability of a neurone is a primary determinant of its normal function and is probably homeostatically regulated. Preservation of normal excitability is particularly important for spinal motoneurones, which generate the output of all motor commands. A dysfunction of homeostatic control of motoneurone excitability might occur in amyotrophic lateral sclerosis (ALS), in which there is a progressive degeneration of these cells.
ALS is an adult onset neurodegenerative disease leading to the death of both corticospinal motor neurones and spinal motoneurones. Despite the fact that mutations on several genes have been identified in familial forms of the disease, including the superoxide dismutase 1 (SOD1) G93A gene mutation (Rosen et al. 1993), the pathophysiology of ALS remains poorly understood. Many mechanisms have been proposed (reviewed in Ilieva et al. 2009), among them oxidative damage, protein aggregation, axonal transport deficits and astrocyte-mediated cell damage. Glutamate-mediated excitotoxicity has been proposed very early on as a potential mechanism for the degeneration of motoneurones in ALS (Rothstein et al. 1990). According to this hypothesis, an elevation of intracellular calcium by repetitive firing or by calcium-permeable glutamate receptors is harmful and would trigger their apoptotic death. Calcium-related apoptotic death is particularly probable in motoneurones, given their restricted calcium buffering capabilities (von Lewinski & Keller, 2005). As part of the excitotoxicity hypothesis, it was suggested that intrinsic hyperexcitability of motoneurones could, by itself, contribute to excitotoxic stress (Kuo et al. 2005; von Lewinski & Keller, 2009; Ilieva et al. 2009). A chronically hyperexcitable motoneurone would fire more spikes in response to a given synaptic input and consequently more calcium ions would flow into the cytoplasm, eventually leading to its death.
Using mouse models incorporating the mutant human SOD1 gene (mSOD1), several studies have set out to test the intrinsic excitability of the motoneurones during ALS. Very young motoneurones from mSOD1 embryos recorded in whole spinal cords in vitro (Martin et al. 2013) or in culture (Pieri et al. 2003; Kuo et al. 2005) are clearly hyperexcitable due to increased sodium currents. Neonatal motoneurones display a strong upregulation of both voltage-sensitive sodium and calcium currents (Quinlan et al. 2011). Yet unlike motoneurones from embryos, the input conductance of neonatal motoneurones is substantially increased in mSOD1 mice (Bories et al. 2007; Quinlan et al. 2011). This conductance increase compensated for the increased voltage-dependent currents and thereby kept overall excitability of neonatal mSOD1 motoneurones at the same level as controls (Quinlan et al. 2011).
Nevertheless, if intrinsic hyperexcitability is a significant contributor to cell death in ALS, it should become manifest at later developmental stages and it should reach its maximum at the age where neuromuscular junctions start to degenerate. It is thus particularly important to establish whether spinal motoneurones are hyperexcitable just before the degeneration of neuromuscular junctions. A previous study by Meehan et al. (2010) has assessed adult motoneurone excitability in a different SOD1 model (G127X). The authors have shown that, in this model, there were no changes in input conductance or rheobase, but the transition from primary range to secondary range occurred at a lower firing frequency, suggesting an increase in calcium persistent current. However, the time of denervation is unknown in this model and it is therefore difficult to assess if these changes precede or follow the denervation. Jiang et al. (2009) have shown that there were no changes in the monosynaptic reflex loop in the sacrocaudal cord of adult G93A mice, but this study does not allow separation of intrinsic and extrinsic excitability. In the present study, we have used SOD1 G93A mice, in which the times of denervations are well known (Pun et al. 2006; Hegedus et al. 2007, 2008), and we have investigated the intrinsic electrical properties of adult mouse motoneurones both in vivo and in vitro (Jiang & Heckman, 2006; Manuel et al. 2009). We studied the age range of 30–80 days, spanning the time when the first waves of denervation occur without appreciable motoneurone loss in the spinal cord (Gurney et al. 1994; Chiu et al. 1995). Our results show that, in both preparations, excitability was preserved in the majority of cases despite an increase in cell input conductance. In a substantial number of motoneurones, however, hypoexcitability developed as they failed to produce sustained firing in response to quasi-stationary inputs. Altogether, motoneurones do not develop hyperexcitability at stages of the disease just preceding the denervation of neuromuscular junctions. Instead, homeostatic processes maintain excitability in most cells and, in a substantial subpopulation, homeostasis fails and is replaced by hypoexcitability. These results show that, if excitotoxicity is indeed a fundamental cause of degeneration, it is not caused by an intrinsic hyperexcitability of the motoneurones but must instead be due to alterations in their synaptic inputs.
Methods
In vivo experiments
In vivo experiments were performed in accordance with European directives (86/609/CEE and 2010-63-UE) and French legislation. They were approved by the Paris Descartes University ethics committee.
Because of the invasive nature of the in vivo experiments, we chose to backcross the SOD1G93A transgene on a CD1 strain, which is easier to breed and was thought to have a lower mortality to our surgical procedures. B6.Cg-Tg(SOD1*G93A)1Gur/J males (Jackson Laboratory, Bar Harbor, ME) were crossed with CD1 females (Charles River, L'Arbresle, France) and the transgene copy number was measured by quantitative polymerase chain reaction and was found to be >20. The CD1-Tg(SOD1*G93A) mice had a phenotype very similar to the mSOD1 mice on B6 and B6SJL backgrounds. They developed a progressive paralysis starting with the hindlimbs at about postnatal day (P)90, and reached their endstage point at about P120. Motoneurones from mSOD1 mice were compared to a population of wild-type (WT) motoneurones, recorded from their non-transgenic littermates, as well as the CD1 mice used in Manuel et al. (2009).
The surgical procedures have been described previously (Manuel et al. 2009; Iglesias et al. 2011; Manuel & Heckman, 2012a). Briefly, atropine (0.20 mg kg−1; Aguettant, Lyon, France) and methylprednisolone (0.05 mg; Solu-Medrol; Pfizer, Paris, France) were given subcutaneously at the onset of experiment, to prevent salivation and oedema, respectively. Fifteen minutes later, the anaesthesia was induced with an intraperitoneal injection of sodium pentobarbitone (70 mg kg−1; pentobarbital; Ceva Santé Animale, Libourne, France). A tracheotomy was performed, and the mouse was artificially ventilated with pure oxygen (SAR-830/AP ventilator; CWE, Ardmore, PA). The end tidal 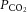 was maintained at about 4% (MicroCapstar; CWE). The heart rate was monitored (CT-1000; CWE), and the central temperature was kept at 37°C using an infrared heating lamp and electric blanket. A catheter was introduced in the external jugular vein, allowing us to supplement the anaesthesia whenever necessary (usually every 10–20 min) by intravenous injections (6 mg kg−1). The adequacy of anaesthesia was assessed on lack of noxious reflexes and on the stability of the heart rate (usually 400–500 bpm) and end-tidal
was maintained at about 4% (MicroCapstar; CWE). The heart rate was monitored (CT-1000; CWE), and the central temperature was kept at 37°C using an infrared heating lamp and electric blanket. A catheter was introduced in the external jugular vein, allowing us to supplement the anaesthesia whenever necessary (usually every 10–20 min) by intravenous injections (6 mg kg−1). The adequacy of anaesthesia was assessed on lack of noxious reflexes and on the stability of the heart rate (usually 400–500 bpm) and end-tidal 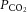 . A slow intravenous infusion (50 μl h−1) of a 4% glucose solution containing NaHCO3 (1%) and gelatine (14%; Plasmion; Fresenius Kabi, Sèvres, France) helped maintain the physiological parameters. In some experiments, the animal was paralysed after the surgery with pancuronium bromide (Pavulon; Schering-Plough, Courbevoie, France). In this case, additional doses of anaesthetics were provided at the same frequency as before the paralysis, and adequacy of the anaesthesia was assessed on the stability of the heart rate and of the
. A slow intravenous infusion (50 μl h−1) of a 4% glucose solution containing NaHCO3 (1%) and gelatine (14%; Plasmion; Fresenius Kabi, Sèvres, France) helped maintain the physiological parameters. In some experiments, the animal was paralysed after the surgery with pancuronium bromide (Pavulon; Schering-Plough, Courbevoie, France). In this case, additional doses of anaesthetics were provided at the same frequency as before the paralysis, and adequacy of the anaesthesia was assessed on the stability of the heart rate and of the 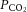 . In all experiments, the sciatic nerve was dissected and mounted on a monopolar electrode for stimulation. The vertebral column was immobilized with two pairs of horizontal bars (Cunningham Spinal Adaptor; Stoelting, Dublin, Ireland) applied on the Th12 and L2 vertebral bodies, and the L3–L4 spinal segments were exposed by a laminectomy at the Th13–L1 level. The tissues in the hindlimb and the spinal cord were covered with pools of mineral oil. At the end of the experiments, animals were killed with a lethal intravenous injection of pentobarbitone.
. In all experiments, the sciatic nerve was dissected and mounted on a monopolar electrode for stimulation. The vertebral column was immobilized with two pairs of horizontal bars (Cunningham Spinal Adaptor; Stoelting, Dublin, Ireland) applied on the Th12 and L2 vertebral bodies, and the L3–L4 spinal segments were exposed by a laminectomy at the Th13–L1 level. The tissues in the hindlimb and the spinal cord were covered with pools of mineral oil. At the end of the experiments, animals were killed with a lethal intravenous injection of pentobarbitone.
The motoneurones were impaled with micropipettes (tip diameter, 1.0–1.5 μm) filled with 3 m KCl (resistance, 5–10 MΩ). Recordings were made using an Axoclamp 2B amplifier (Molecular Devices, Sunnyvale, CA) connected to a Power1401 interface and using the Spike2 software (CED, Cambridge, England). After impalement, identification of motoneurones rested on the observation of antidromic action potentials in response to the electrical stimulation of their axon in the sciatic nerve. All motoneurones retained for analysis had a resting membrane potential below −50 mV and an overshooting action potential >65 mV. All recordings were performed using the discontinuous current clamp mode (7–9 kHz) and sampled at 20 kHz.
In vitro experiments
Intracellular recordings were made from motoneurones in the sacrocaudal spinal cord of adult B6SJL-Tg(SOD1*G93A)1Gur/J mice and their non-transgenic littermates (age 40–50 days old). All experimental procedures were approved by Northwestern University animal research committee.
Procedures have been described previously (Jiang & Heckman, 2006; Manuel et al. 2012). Briefly, animals were anaesthetized with urethane (1.8 g kg−1) to dissect out the spinal cord caudal to the T12 vertebra. The sacrocaudal spinal cord was then transferred to a dissection dish containing oxygenated modified artificial cerebrospinal fluid (mACSF) at room temperature (20–21°C). Following a 1 h resting period in mACSF, the cord was transferred to a recording chamber, where it was submerged in normal ACSF flowing at 3–6 ml min−1 at room temperature. The cord was secured by passing insect pins through lateral vasculature and connective tissue and into a silicone elastomer (Sylgard) base. The normal ACSF had the following composition (in mm): 122 NaCl, 24 NaHCO3, 3 KCl, 2.5 CaCl2, 1 MgSO4 and 12 glucose in distilled water, bubbled with 95% O2–5% CO2 and pH 7.4. A modified ACSF (mACSF) was used during dissection and recovery to prevent excitotoxic injury. The mACSF composition was (in mm): 118 NaCl, 24 NaHCO3, 3 KCl, 1.5 CaCl2, 1.3 MgSO4, 25 glucose, 1.4 NaH2PO4, 5 MgCl2 and 1 kynurenic acid.
Intracellular recordings were obtained using sharp electrodes filled with either 3 m KCl or a mixture of 1 m potassium acetate and 1 m KCl and bevelled to a resistance of 25–30 MΩ using a rotary beveller (BV-10; Sutter Instruments, Novato, CA). A stepper-motor (Model 2660 Micropositioner; David Kopf Instruments, Tujunga, CA) was used to advance the electrodes into the ventral horn, and intracellular recordings from motoneurones were made with an Axoclamp 2B intracellular amplifier (Molecular Devices, Sunnyvale, CA) running in bridge mode or in discontinuous current clamp mode (switching rate 5–8 kHz, output bandwidth 10 kHz) and sampled at 20 kHz with a Spike2/1401Plus system (CED). The ventral roots were wrapped around Ag/AgCl stimulating electrodes and sealed with grease, which allowed for antidromic identification of motoneurones. Motoneurones with a resting potential below −55 mV and antidromic spike overshoot >0 mV were retained for analysis. Neurones were recorded at least 2 h after the injection of urethane, and after the spinal cord was immerged for more than 1 h in ACSF, giving ample time to wash out any residual anaesthetics from the spinal cord, as shown by the fact that persistent inward currents can be readily recorded in the same conditions in rat sacrocaudal spinal cord (Bennett et al. 2001).
Electrophysiology
Upon penetration, a series of small amplitude, square current pulses were used to plot the I–V relationship. Motoneurones of mSOD1 mice, as those of WT mice (Manuel et al. 2009), exhibited a voltage sag, leading to a plateau, in response to small (depolarizing or hyperpolarizing) current pulses and a rebound (either hyperpolarizing or depolarizing) after the cessation of the pulse (Fig. 1A). These features suggest the presence of an Ih current (McLarnon, 1995). As a consequence, the input conductance was measured from the peak responses (arrowhead in Fig. 1Aa), where the contribution of the Ih current was negligible. The ‘sag ratio’ of the voltage response was then computed as the ratio of the peak resistance over the plateau resistance (Manuel et al. 2009).
Figure 1.
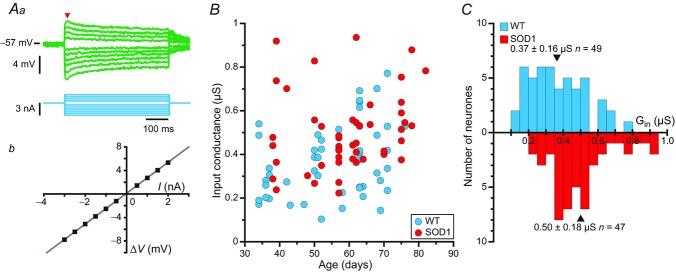
Aa, average responses of a mSOD1 motoneurone (top traces; each trace is an average of five sweeps) to a series of current pulses (bottom traces) lasting 500 ms and ranging from −3 to +2 nA. Notice the sag on the voltage response of the motoneurone: the voltage rapidly reached a peak, before stabilizing to a lower plateau value. Ab, plot of the deflection of the voltage (ΔV, measured at the peak of the response, arrowhead in Aa) versus the intensity of the current pulse. B, plot of the input conductances of WT and mSOD1 motoneurones versus the age of the mice. C, distribution of the input conductances (Gin) of WT (top) and mSOD1 (bottom) motoneurones. In each histogram, arrowheads mark the position of the mean. mSOD1, mutant superoxide dismutase 1; WT, wild-type.
The discharge properties were investigated using triangular ramps of current (rate 0.1–10 nA s−1). F–I curves were obtained by plotting the instantaneous firing frequency versus the intensity of the injected current at the time of the spike. In few cases, long-lasting (500 ms) depolarizing current pulses, repeated at the frequency of 1 Hz, were also used to study the stationary discharge properties.
The voltage threshold for firing was determined on the first spike of a current ramp, as the point at which the first derivative of the voltage reached 10 mV ms−1 (Sekerli et al. 2004). The onset current was determined as the intensity of the current ramp at this time point.
Data analysis
We have compared the electrical properties of 51 mSOD1 (from 19 animals) and 49 WT motoneurones (from 23 animals) recorded in the lumbar cord in vivo and 34 mSOD1 (from 18 animals) and 41 WT (from 23 animals) in the sacrocaudal cord in vitro. Most of the electrical properties described in the Results section did not have a normal distribution (Kolmogorov–Smirnov test), and we therefore used the non-parametric Wilcoxon–Mann–Whitney test to compare averages. P < 0.05 was considered as significant.
Because the disease progresses over the time period used in our in vivo study (WT: 34–76 days old, 55 ± 13 days old, n = 49 vs. mSOD1: 38–82 days old, 59 ± 13 days old, n = 51; distributions not significantly different, Kolmogorov–Smirnov P = 0.24), we tested the effect of age vs. mutation using a non-parametric, non-linear ANCOVA (Young & Bowman, 1995). This was performed in R v.2.11.1 (R Development Core Team, 2010) using the sm package v.2.2–4.1 (Bowman & Azzalini, 2010). Other statistical tests were performed either in Kaleidagraph v4.1 (Synergy Software, Reading, PA) or in R.
The motoneurones recorded in vitro were taken from a more restricted time range that was comparable in both genotypes (WT: 41–51 days old vs. mSOD1: 40–50 days old).
Results
Lumbar motoneurones display an increase of their input conductance
We first compared the input conductance (Fig. 1A) of mSOD1 and WT lumbar motoneurones recorded intracellularly in anaesthetized mice from 34 to 82 days old. The input conductances of mSOD1 and WT motoneurones are plotted against mouse age in Fig. 1B. ANCOVA analysis revealed that the mSOD1 mutation elicited an upward shift in the peak input conductance (P = 0.003) as shown in Fig. 1C. The mutation had the same impact on conductance regardless of the age of the animals (no significant interaction between age and mutation factors; P = 0.64, ANCOVA test for parallelism). Accordingly, the whole distribution of input conductances was shifted towards larger conductances (Fig. 1C). Most notably, we did not find any mSOD1 lumbar motoneurones with input conductances smaller than 0.2 μS whereas WT motoneurones with input conductances smaller than this value were commonly observed. At the other end of the conductance spectrum, a few mSOD1 motoneurones had input conductances larger than 0.8 μS but these large values were never observed in WT motoneurones. On average, the input conductance increased from 0.37 ± 0.16 μS (0.10–0.77 μS, n = 49) in WT mice to 0.50 ± 0.18 μS (0.22–0.93 μS, n = 47, one-tailed Wilcoxon–Mann–Whitney P < 0.001) in mSOD1 mice (i.e. a 35% average increase).
The mutation also induced an increase of the plateau conductance compared to controls (P = 0.006, ANCOVA), once again regardless of the age of the animal (no significant interaction between age and mutation factors, P = 0.26, ANCOVA test for parallelism). The plateau input conductance was 0.61 ± 0.28 μS (0.12–1.25 μS, n = 49) on average in control animals, and it increased to 0.70 ± 0.31 μS (0.25–1.61 μS, n = 47) on average in mSOD1 mice (one-tailed Wilcoxon–Mann–Whitney P = 0.001). However, the difference in sag ratio between the two populations of motoneurones was not significant (1.32 ± 0.26, n = 49 vs. 1.38 ± 0.23, n = 47; P = 0.12, ANCOVA), suggesting that the mutation causes the Ih current to increase in parallel with the input conductance.
The increase in average input conductance cannot be explained by the death of the small motoneurones as the whole spectrum of conductances is shifted toward larger values, with the emergence of high conductances never observed in WT motoneurones. Furthermore, at the age at which our recordings were performed, no significant cell loss was observed in the spinal cord (Gurney et al. 1994; Chiu et al. 1995).
Despite the increased input conductance, excitability is unchanged in most mutant superoxide dismutase 1 motoneurones
The excitability of a neurone is determined by the amount of excitation (i.e. current) required to bring its membrane potential to firing threshold, and once firing has started, by the slope of its input–output relationship. To investigate whether the threshold current and the F–I gain were modified, we studied the firing properties of mSOD1 and WT motoneurones in response to slow triangular ramps of currents. Figure 2 shows the responses of two typical motoneurones recorded in the same 39 day old mSOD1 mouse. As in WT animals, the motoneurones of mSOD1 mice exhibited a subprimary range where small subthreshold oscillations at high frequency preceded the emission of a full-blown spike (arrowheads in Fig. 2Ab and Bb; Manuel et al. 2009). This range was followed by the classical primary range where subthreshold oscillations were absent (Fig. 2Ac and Bc). During the descending ramp, oscillations reappeared at the end of the discharge. However, the response to the slow triangular ramps was asymmetric. In most cases (17 of 20 or 85% in mSOD1 mice; 22 of 31 or 71% in WT mice), the de-recruitment current was higher than the recruitment current and the F–I relationship displayed a clockwise hysteresis (Fig. 2Ad). In the other cases (three of 20 or 15% in mSOD1 mice; nine of 31 or 29% in WT mice), the recruitment current was lower and the F–I relationship displayed a counter clockwise hysteresis (Fig. 2Bd; no difference in the proportions of clockwise and counter clockwise hystereses between WT and mSOD1 motoneurones, P = 0.32, Fisher's exact test). All these features (subprimary range preceding the primary range, clockwise or counter clockwise hysteresis) were similar to those that we previously described in lumbar motoneurones of WT mice (Manuel et al. 2009; Iglesias et al. 2011).
Figure 2.
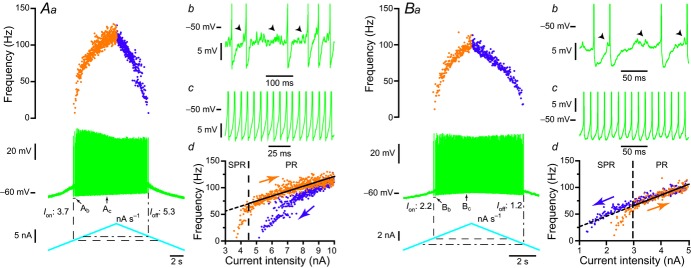
A, clockwise response of an mSOD1 motoneurone to a slow ramp (1 nA s−1) of current. Aa, slow current ramp (bottom trace), voltage response (middle trace) and instantaneous firing frequency (top plot). The onset of the discharge is shown by the vertical dashed line (Ion, 3.7 nA) while the current of de-recruitment (Ioff, 5.3 nA) is indicated by the vertical dash-dotted line. Ab, magnification of the voltage trace at the recruitment (region indicated in Aa). Spikes have been truncated to highlight the high-frequency oscillations (arrowheads) that appeared in the interspike intervals. Note the firing variability that characterizes the SPR. Ac, magnification of the region indicated in Aa. Increasing the injected current resulted in a PR without oscillation between spikes and with less variability. Ad, plot of the instantaneous firing frequency versus the intensity of the injected current for the ascending and descending branches of the ramp shown in Aa. Note that this F–I relationship displayed a clockwise hysteresis. Vertical dashed line indicates the transition between the SPR and the PR on the ascending branch. The gain of the F–I curve can be estimated by the slope of the linear regression (continuous line) in the PR. B, counter-clockwise behaviour of another mSOD1 motoneurone in response to a slow ramp (1 nA s−1) of current. Ba, same organization as in Aa. Note that, in this motoneurone, the de-recruitment current (Ioff, 1.2 nA) is lower than the recruitment current (Ion, 2.2 nA). Bb, magnification of the voltage trace at recruitment (region indicated in Ba). High-frequency voltage oscillations (arrowheads) were present between spikes as for the motoneurone illustrated in A. Bc, magnification of region indicated in Ba. Bd, same organization as in Ad. Note that the F–I relationship of this motoneurone displayed counter-clockwise hysteresis. PR, primary range; mSOD1, mutant superoxide dismutase 1; SPR, subprimary range.
Remarkably, the recruitment current was not different in the motoneurones of WT and mSOD1 mice. The recruitment current was highly correlated to the input conductance: the larger the input conductance, the higher the recruitment current (WT: Pearson product–moment correlation coefficient r = 0.64, P < 0.0001 vs. mSOD1: r = 0.85, P < 0.0001; WT and mSOD1 regressions are not significantly different, P = 0.68, ANCOVA; Fig. 3A), just as in normal motoneurones in other species (Fleshman et al. 1981; Gustafsson & Pinter, 1984; Zengel et al. 1985). The recruitment current spanned the same range in mSOD1 (2.1–11.4 nA) as in WT mice (1.2–12.6 nA) and the average values were not significantly different (WT: 5.0 ± 3.5 nA, n = 37 vs. mSOD1: 6.0 ± 3.0 nA, n = 29; P = 0.11, two-tailed Wilcoxon–Mann–Whitney; Fig. 3B). Moreover, the gain in the primary range was similar in the two populations of motoneurones (Fig. 4A). The gain was 18.0 ± 9.7 Hz nA−1 (7–40 Hz nA−1; n = 23) in WT mice and 16.2 ± 6.7 Hz nA−1 (9–32 Hz nA−1, n = 12; P = 0.85, two-tailed Wilcoxon–Mann–Whitney) in mSOD1 mice. Altogether, excitability in mSOD1 motoneurones is unchanged despite the input conductance increase.
Figure 3.
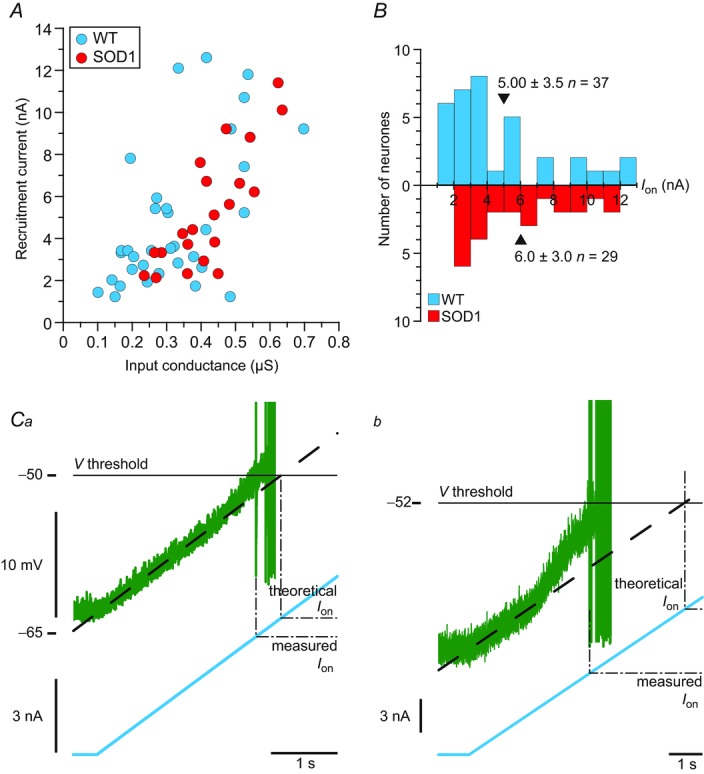
A, plot of the recruitment current of WT and mSOD1 motoneurones versus input conductance. B, distributions of recruitment current of WT and mSOD1 motoneurones. The mean values are indicated by arrowheads. C, magnification of the voltage (top trace) and current (bottom trace) near the recruitment of a WT (Ca) and mSOD1 (Cb) motoneurone (the mSOD1 motoneurone is the one illustrated in Fig. 2A). Ion is measured as the current intensity at which the motoneurone fires the first spike (‘measured Ion’, dashed line), while the ‘theoretical Ion’ is the current intensity that would be needed to reach the same voltage threshold if the membrane was purely passive (dot-dashed line). mSOD1, mutant superoxide dismutase 1; WT, wild-type.
Figure 4.
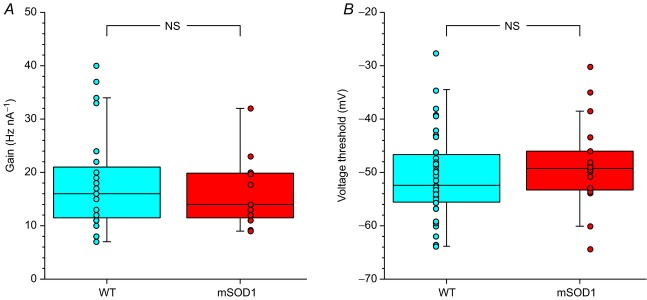
A, box-and-whisker diagram and data points of the distribution of the gain (measured in the primary range) of WT and mSOD1 motoneurones. B, box-and-whisker diagram and data points of the distribution of the voltage threshold of WT and mSOD1 motoneurones. In both diagrams, the central box represents the values from the lower to upper quartile (25–75th percentile). The middle line represents the median. The vertical line extends to the minimum and maximum values that fall within 1.5 times the interquartile distance. mSOD1, mutant superoxide dismutase 1; NS, not significant; WT, wild-type.
The lack of change in recruitment current was not due to a parallel hyperpolarization of the voltage threshold for spiking (WT: −64 to −28 mV, −51 ± 8 mV, n = 35 vs. mSOD1: −64 to −30 mV, −49 ± 8 mV n = 18; P = 0.3, two-tailed Wilcoxon–Mann–Whitney; Fig. 4B), nor to a depolarization of the resting membrane potential (WT: −81 to −50 mV, −66 ± 9 mV, n = 49 vs. mSOD1: −83 to −50 mV, −63 ± 9 mV, n = 51; P = 0.15, two-tailed Wilcoxon–Mann–Whitney). The resting membrane potential did not change despite the increase in Ih current. This suggests that the leak conductance increased in parallel with Ih as both conductances have an opposite effect on the resting membrane potential.
Instead, the fact that recruitment current was unaltered despite the increased input conductance might be due to an upregulation of subthreshold depolarizing currents. Supporting this assumption, we often observed an acceleration of the voltage trajectory before the first spike on a current ramp (compare Fig. 3Ca and Cb). As a consequence, the recruitment current (measured current) was smaller than the theoretical current that would be expected if only leak currents were present before the initiation of the first spike (i.e. linear voltage trajectory). The difference between the theoretical current (estimated by extrapolating the linear voltage trajectory close to rest until it reached the voltage threshold) and the measured current was larger in mSOD1 motoneurones (1.9 ± 1.6 nA, n = 14) than in WT motoneurones (0.7 ± 1.0; n = 33; P = 0.002, one-tailed Wilcoxon–Mann–Whitney). This suggests that some subthreshold depolarizing currents are upregulated in mSOD1 motoneurone providing the basis for homeostatic regulation of motoneurone excitability. This acceleration in membrane potential in motoneurones is probably due to the action of a persistent sodium current (Harvey et al. 2006; Kuo et al. 2006). An increase in persistent sodium current has previously been demonstrated in mSOD1 motoneurones in the neonate (Quinlan et al. 2011) and thus the present result suggests that this increase persists in the adult state.
A fraction of motoneurones are hypoexcitable
In contrast, not all mSOD1 motoneurones were able to produce a repetitive discharge during the ramp of current. Some motoneurones only fired a few spikes during the ascending ramps (Fig. 5Aa) while others were not able to fire at all (Fig. 5Ba) despite the fact that all motoneurones were nevertheless able to emit a single spike in response to a brief pulse of current (see Fig. 5Ab and Bb).
Figure 5.
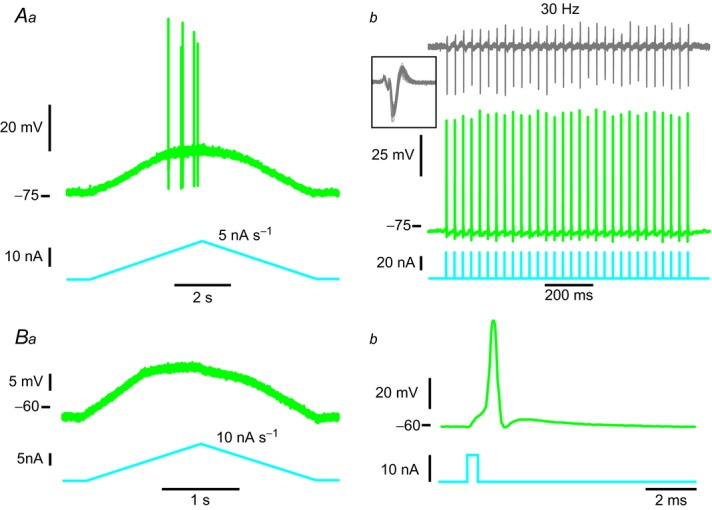
A, example of an mSOD1 motoneurone that fired only a few spikes in response to slow current ramps. Aa, current ramp (bottom trace) and voltage response (top trace). Ab, this motoneurone was nevertheless able to fire single spikes (middle trace) in response to a train of brief current pulses (bottom trace) and its neuromuscular junction was still functional, as shown by the fact that electromyography activity (top trace) was consistently observed following each spike (inset: superposition of all the electromyography sweeps showing very little variability). B, example of an mSOD1 motoneurone that was unable to fire any spike in response to slow current ramps. Ba, current ramp (bottom trace) and voltage response (top trace). Bb, this motoneurone was nevertheless able to fire a single spike (top trace) in response to a brief pulse of current (bottom trace). mSOD1, mutant superoxide dismutase 1.
Remarkably, these motoneurones lost their ability to produce a sustained discharge before their neuromuscular junctions stopped working. Figure 5Ab shows that the neuromuscular junctions of this motoneurone can be reliably activated at a rate of 30 Hz when triggering spikes with short pulses of current. This suggests that the loss of function in the motoneurone cell body (inability to produce sustained firing) preceded the peripheral functional impairment.
Among 38 tested motoneurones in mSOD1 mice, only 22 (58%) were able to produce sustained firing during the slow ramps of currents (bottom solid bars and pie chart, Fig. 6A). Twelve (32%) failed to discharge a single action potential during the slow ramps despite the fact that they were still able to fire one spike in response to a short current pulse (bottom crosshatched bars and pie chart, Fig. 6A). Four more motoneurones (10%) produced only a few spikes during the ramp (bottom hatched bars and pie chart, Fig. 6A). Interestingly, the input conductances of those motoneurones (that failed to respond to a slow ramp or that exhibited a poor response) were spread in the full range of input conductances of mSOD1 motoneurones. In sharp contrast, 36 motoneurones of the 41 tested (88%) in WT mice produced sustained firing during a slow ramp of current (top solid bars, Fig. 6A) whereas only three (7%) did not discharge at all during the ramp (though they could fire one spike in response to a current pulse, top crosshatched bars, Fig. 6A) and only two (5%) discharged only a few spikes during the ramp (top hatched bars; Fig. 6A). Moreover, those motoneurones that failed to produce sustained firing during the ramp were restricted to those that had the highest input conductance (above 0.42 μS). Overall, the proportion of motoneurones in mSOD1 mice that failed to produce a good repetitive firing in response to a slow ramp of current was much higher than in WT mice (16 of 38 for mSOD1 mice vs. five motoneurones of 41 for WT mice; P = 0.004, Fisher's exact test) and their input conductances are spread across the full spectrum of input conductances, unlike in WT mice.
Figure 6.
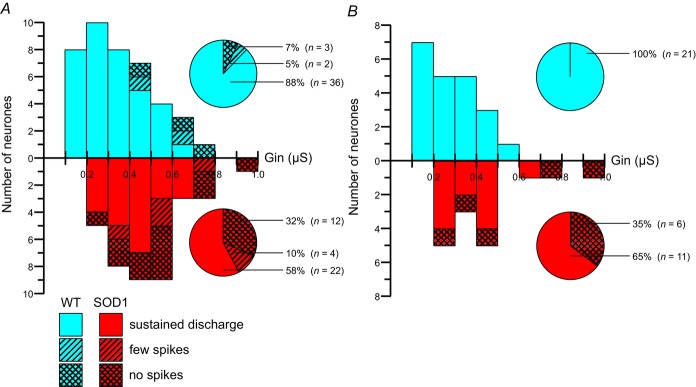
A, distribution of input conductance (Gin) for different categories of WT (top) and mSOD1 (bottom) motoneurones aged between 34 and 82 days old. Solid bars correspond to motoneurones that were able to produce sustained firing, hatched bars correspond to motoneurones that fired only a few spikes, and crosshatched bars correspond to motoneurones unable to fire any spike during ramps of current. Pie charts indicate the proportions of each category among our population of WT (top pie chart) and mSOD1 (bottom pie chart) motoneurones. B, same organization as in A for a time window restricted to P34–P60. mSOD1, mutant superoxide dismutase 1; WT, wild-type.
The contrast between motoneurones from WT and mSOD1 mice is even more striking in the time window P34–P60 (Fig. 6B), that is to say the period just preceding and during the very first denervations of neuromuscular junction (Pun et al. 2006; Hegedus et al. 2007, 2008). At this time period, all 21 WT lumbar motoneurones displayed full repetitive firing in response to a slow ramp (top solid bars and pie chart, Fig. 6B). However, only 11 of 17 mSOD1 lumbar motoneurones (65%) were able to produce sustained firing (bottom solid bars and pie chart, Fig. 6B). The six others (35%) were unable to discharge a single spike during the ramp despite being able to fire one spike in response to a short pulse (bottom crosshatched bars and pie chart, Fig. 6B; P = 0.004, Fisher's exact test). Again their input conductances spanned the full range of input conductances of mSOD1 motoneurones. These results clearly indicate that, in mSOD1 mice, a large fraction of motoneurones were unable to fire repetitively and therefore were less excitable.
Changes in spike generating mechanisms in hypoexcitable motoneurones
In neonatal cells, a primary compensation for the increase in input conductance is an increase in persistent inward sodium currents. As demonstrated by Kuo et al. (2006), Miles et al. (2005) and others (Lee & Heckman, 2001; Harvey et al. 2006; Bouhadfane et al. 2013), a deficit in persistent sodium current prevents a motoneurone from producing sustained firing in response to long current pulses or slow ramps of current. If the failure of spike generation in hypoexcitable cells is indeed due to failure of the persistent sodium current to keep up with increased input conductance, then the hypoexcitable cells should exhibit two behaviours: there should be a lack of a upswing in membrane depolarization just before spike onset (a behaviour generated by persistent sodium current) and the cells should still spike to sharp transient inputs (as these rapidly rising inputs do not need the assistance of the upswing in membrane potential).
Consistent with these predictions, all the motoneurones that failed to produce a sustained discharge were able to emit a single spike in response to a brief pulse of current (see above). Furthermore, the voltage upswing observed in motoneurones that fire repetitively is absent in the motoneurones that fire only a few spikes in response to a slow current ramp. Figure 7 shows the upswings (empty arrowheads) in two motoneurones that are able to produce a sustained firing (mSOD1 and WT motoneurone, top two traces, respectively). In sharp contrast the voltage trajectory of the mSOD1 motoneurone unable to produce a sustained firing (bottom trace) plateaus below the expected passive trajectory (dashed line) just before firing the first spike (filled arrowhead). These motoneurones needed more current to reach their firing threshold than what would be expected if the motoneurones were purely passive (the difference between the theoretical current and the measured current was negative: −0.5 ± 0.4 nA, n = 4). These results support a role for a relative reduction of the persistent sodium current as a major mechanism of the development of hypoexcitability.
Figure 7.
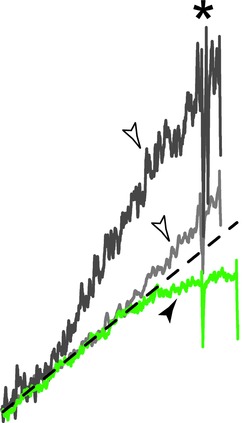
Traces were smoothed using a 10 ms window, aligned to the first spike (asterisk) and scaled so that the passive voltage trajectory (dashed line) was the same in all traces. The top trace corresponds to a mutant superoxide dismutase 1 motoneurone that was able to produce repetitive firing (same motoneurone as in Figs 2A and 3Cb), the middle trace is from a wild-type motoneurone (same motoneurone as in Fig. 3Ca), and the bottom trace is from the motoneurone in Fig. 5Aa.
The lack of hyperexcitability is not due to anaesthetics: in vitro study of sacrocaudal motoneurones of mutant superoxide dismutase 1 mice
Our results show that lumbar motoneurones either successfully maintain their excitability or become hypoexcitable as suggested by their inability to produce sustained firing in response to ramps of current. Altogether these results indicate that lumbar motoneurones are not hyperexcitable in mSOD1 mice. However, we performed these experiments on deeply anaesthetized mice, and it has been shown that the anaesthetics that we used can suppress voltage-dependent currents that have a profound effect on the excitability of motoneurones (Guertin & Hounsgaard, 1999). One might therefore wonder whether mSOD1 motoneurones could turn out to be hyperexcitable in non-anaesthetized conditions. For technical reasons, it is extremely difficult to perform intracellular recordings of adult lumbar motoneurones in conditions where no anaesthetics are used (lumbar cord in vitro or decerebrate mouse in vivo). Instead, we recently developed a preparation in which it is possible to record adult motoneurones in vitro in the sacrocaudal region of the spinal cord (Jiang & Heckman, 2006; Manuel et al. 2012). Even though we cannot record lumbar motoneurones in these conditions, tail muscles are atrophied and sacrocaudal motoneurones degenerate with a time course similar to the lumbar motoneurones in the SOD1 mouse model of ALS (Alves et al. 2011).
The sacrocaudal motoneurones have been recorded in a restricted time window (P40–P50), i.e. just before denervation. They displayed smaller input conductances than lumbar motoneurones, but, as above, the motoneurone input conductance was 46% larger in mSOD1 mice than in WT mice (P < 0.001, one-tailed Wilcoxon–Mann–Whitney; Fig. 8A and Table 1). The plateau input conductance was also significantly larger in sacrocaudal motoneurones of mSOD1 mice (P < 0.001, one-tailed Wilcoxon–Mann–Whitney; Table 1).
Figure 8.

Aa, voltage response (top traces) to a series of subthreshold current pulses (bottom traces). Each trace is the average of five sweeps. Ab, plot of the voltage deviation (ΔV) measured at the peak of the response (arrowhead in Aa) versus the intensity of the current pulse, used to estimate the input conductance of the cell. Ac, histogram of the input conductances (Gin) of WT (top histogram) and mSOD1 (bottom histogram) sacrocaudal motoneurones. The mean values are indicated by arrowheads. B, example of a sacrocaudal WT motoneurone that was able to sustain a repetitive discharge in response to a stationary stimulus. Ba, response (top trace) to a slow current ramp. The dashed lines indicate the onset and de-recruitment currents. Bb, F–I relationship of that motoneurone. Note that it displayed a clockwise hysteresis. C, example of a sacrocaudal mSOD1 motoneurone that was unable to produce sustained firing in response to stationary stimuli. Ca, this motoneurone was unable to fire a single spike (top trace) in response to a slow ramp of current (bottom trace). Cb, this motoneurone was nevertheless able to fire at most a doublet of spikes (top trace) at the onset of a square pulse (bottom trace). D, input conductances (Gin) of sacrocaudal motoneurones that fired (solid bars) or that failed to fire (crosshatched bars) during ramps in WT and mSOD1 mice. Pie charts indicate the proportions of each category among our population of WT (top pie chart) and mSOD1 (bottom pie chart) motoneurones. mSOD1, mutant superoxide dismutase 1; WT, wild-type.
Table 1.
Comparison of electrophysiological properties between mSOD1 and WT sacrocaudal motoneurones
| WT | mSOD1 | Significance | |
|---|---|---|---|
| Resting membrane potential | −61 ± 4 mV | −62 ± 5 mV | P = 0.61 (NS) |
| [−69 to −55] | [−75 to −55] | ||
| n = 41 | n = 33 | ||
| Peak input conductance | 0.13 ± 0.07 μS | 0.19 ± 0.08 μS | P < 0.001 (***) |
| [0.05 – 0.35] | [0.09 – 0.38] | ||
| n = 41 | n = 33 | ||
| Plateau input conductance | 0.16 ± 0.10 μS | 0.22 ± 0.10 μS | P < 0.001 (***) |
| [0.05 – 0.45] | [0.09 – 0.55] | ||
| n = 41 | n = 33 | ||
| Sag ratio | 1.17 ± 0.14 | 1.17 ± 0.14 | P = 0.73 (NS) |
| [1.00 – 1.68] | [1.00 – 1.56] | ||
| n = 41 | n = 32 | ||
| Recruitment current (Ion) | 3.5 ± 2.4 nA | 3.3 ± 1.7 nA | P = 0.88 (NS) |
| [0.4 – 9.1] | [0.7 – 6.4] | ||
| n = 24 | n = 18 | ||
| Gain in primary range | 8 ± 5 Hz nA−1 | 10 ± 4 Hz nA−1 | P = 0.15 (NS) |
| [2 – 20] | [5 – 20] | ||
| n = 17 | n = 14 | ||
| Voltage threshold | −44 ± 6 mV | −41 ± 8 mV | P = 0.40 (NS) |
| [−53 to −33] | [−52 to −21] | ||
| n = 14 | n = 18 |
In each cell is given the average value ± s.d., the range of values, and the number of observations. Abbreviations: mSOD1, mutant superoxide dismutase 1; NS, not significant; WT, wild-type.
As with lumbar motoneurones, excitability was assessed using slow ramps of current. Sacrocaudal motoneurones usually responded to ramps of current with the same clockwise (Fig. 8Ba and Bb) or counter-clockwise hysteresis (not shown) as lumbar motoneurones. The recruitment current remained unchanged in the sacrocaudal motoneurones of mSOD1 mice compared to WT mice (Table 1). In addition, the F–I gain was not statistically different between mSOD1 mice and WT mice (Table 1). This suggests that excitability is maintained in these motoneurones. As in lumbar motoneurones, the lack of effect on recruitment current was not due to a decrease of the voltage threshold for spiking, nor to an increase in the resting membrane potential (see Table 1).
Similarly to lumbar motoneurones, a fraction of sacrocaudal motoneurones failed to respond to slow ramps of current (Fig. 8Ca), despite the fact that they could fire one spike at the onset of a current pulse. These motoneurones were never able to produce a strong repetitive firing, even in response to a strong current pulse (Fig. 8Cb; eight of 27 in mSOD1 mice, four of 32 in WT mice, Fig. 8D). Furthermore the input conductances of the motoneurones that were unable to produce sustained firing were distributed among the full input conductance range in mSOD1 mice whereas they were restricted to the highest input conductances in WT mice (Fig. 8D).
Discussion
We showed that, in either the lumbar or the sacrocaudal region of the spinal cord, motoneurones of adult SOD1G93A mice are not hyperexcitable. Instead, most of the motoneurones successfully maintained their excitability despite an increase of their input conductance. Their voltage threshold for spiking, their recruitment current and their F–I gain in the primary range were unchanged. Yet this excitability homeostasis failed in a substantial fraction of mSOD1 motoneurones, which lost their ability to fire repetitively in response to a quasi-stationary input, and therefore became less excitable. The lack of hyperexcitability was not caused by the use of anaesthetics, as we found the same results during in vitro experiments, in which no anaesthetics were used. Moreover, the lack of hyperexcitability cannot be explained by the death of the more excitable cells as, at the age at which our recordings were performed, no significant cell loss is observed in the spinal cord (Gurney et al. 1994; Chiu et al. 1995). For the same reason, the increase in input conductance cannot be explained by the death of small motoneurones. In spinal motoneurones of neonatal mSOD1 mice, the increased input conductance was shown to be caused in part by morphological alterations (Elbasiouny et al. 2010). Similar explanations might hold true in adults.
The excitotoxicity hypothesis in amyotrophic lateral sclerosis
The excitotoxicity hypothesis rests on several indirect findings, such as the fact that riluzole, the only drug shown to have a beneficial, albeit small, effect on the progression of the disease (Lacomblez et al. 1996), is a glutamate release inhibitor and a blocker of voltage-activated sodium channels (Bellingham, 2011), which could decrease glutamate toxicity. The excitotoxicity hypothesis has received a lot of support from the fact that patients with ALS have elevated levels of glutamate in their plasma and cerebrospinal fluid (Rothstein et al. 1990) as well as from transcranial magnetic stimulation studies in patients with sporadic and familial ALS (Vucic et al. 2008).
So far, the excitotoxicity hypothesis has only been investigated in mSOD1 mice. It remains the best model currently available to study the progressive and selective motoneurone death, the hallmark of ALS, as other models (e.g. based on TDP-43 transgenes) do not adequately replicate some aspects of neuromuscular degeneration (Van Den Bosch, 2011).
Motoneurone hyperexcitability: an early but transient event in mutant superoxide dismutase 1 mice
Spinal motoneurones recorded in whole spinal cords in vitro or in culture from mSOD1 embryos have been shown to be hyperexcitable: they are recruited at lower currents and display higher F–I gains (Pieri et al. 2003; Kuo et al. 2005; Martin et al. 2013). This is partly due to an upregulation of the sodium persistent inward current (Kuo et al. 2005). In addition, the recovery from fast inactivation of the sodium current proved to be faster in mSOD1 motoneurones contributing to their hyperexcitability (Zona et al. 2006).
At a later stage, in neonatal animals, both sodium and calcium persistent currents were shown to be upregulated (Quinlan et al. 2011). However, this upregulation did not result in hyperexcitability as the recruitment current and the F–I gain were the same in mSOD1 and WT mice (Quinlan et al. 2011). Similarly, in another ALS model, the SOD1G85R mouse, spinal motoneurones did not exhibit changes in rheobase or stationary F–I gain (Pambo-Pambo et al. 2009). This might be due to the fact that, at this time point, the input conductance of spinal motoneurones is significantly larger in mSOD1 mice (in both the G93A and G85R mutants) than in WT mice (Bories et al. 2007; Quinlan et al. 2011). It thus appears that the upregulation of inward currents just compensates for the increase of the input conductance to preserve the net excitability.
This still holds true in adults as we have shown that excitability is unchanged in most spinal motoneurones despite the continuing conductance increase. The fact that the voltage upswing before the initiation of the first spike is larger in mSOD1 motoneurones compared to WT motoneurones strongly suggests an upregulation of inward currents. However, a substantial fraction of motoneurones was not able to fire in response to a quasi-stationary input (slow ramp of current) indicating that the homeostatic regulation of excitability has failed in these motoneurones. The fact that the voltage trajectory levels off just before emission of the first spike suggests that the upregulation of the persistent sodium current is unable to keep up with the increase in leak current in those motoneurones. Indeed, a deficit in persistent sodium current prevents repetitive firing of motoneurones but still allows spiking in response to transient inputs (Lee & Heckman, 2001; Kuo et al. 2006). However we cannot exclude a contribution from an upregulation of potassium currents. At this point in time, it is not possible to directly measure the amplitude of currents in adult mouse motoneurones. Voltage clamp recordings of spinal motoneurones are extremely difficult to perform in vivo and pharmacological manipulation of ionic currents cannot be achieved in these conditions. Moreover, we have been unsuccessful in performing reliable voltage clamp recordings of sacrocaudal motoneurones in our in vitro preparation.
Hypoexcitability occurred before the motor units have started to degenerate as we found that their neuromuscular junctions can still work normally. However, the reduced activity of hypoexcitable motoneurones probably does not cause the degeneration of neuromuscular junctions, as a block of the axons by TTX has no impact on the degeneration time course of neuromuscular junctions in SOD1G93A mice (Carrasco et al. 2012). One interesting possibility is that the hypoexcitability may help preserve motoneurone survival by restricting the calcium inflow to the cell body. On the other hand hypoexcitability would contribute to weak force production. Whatever the case, the progression of hypoexcitability within the motor pools might be considered as a marker of the disease progression.
In another model (SOD1G127X mice), the input conductance was not increased in adults, but the excitability was also unchanged as the rheobase and the gain was not modified (Meehan et al. 2010). In these mice, a secondary range of firing (i.e. an increase of the F–I gain following the primary range, supposedly due to activation of a calcium persistent current) was observed and appeared at lower frequencies in mSOD1 animals (on average 133 Hz) than in WT (167 Hz) suggesting an increase of the calcium persistent inward current. However, such high-frequency firing is probably unphysiological. In mouse motor units the tetanic force is reached near the transition between the subprimary range and the primary range, which occurs at much lower frequencies (32–75 Hz; Manuel & Heckman, 2011). Our results differ substantially from those of Meehan et al. (2010), as we observed a proportion of motoneurones that become hypoexcitable. However, the comparison between our respective results is difficult given the fact that they used a different model, in which the time of denervation is unknown. It is therefore possible that they have studied motoneurone excitability at a different stage of the disease.
Our present results, obtained in adult SOD1G93A mice, demonstrate therefore that the hyperexcitability observed at very early developmental stages does not persist into adulthood. The transient nature of the intrinsic hyperexcitability in mSOD1 mice is not restricted to spinal motoneurones. It also occurs in hypoglossal motoneurones. However, the time course of the hyperexcitability may somehow be different than in spinal motoneurones. Indeed, hypoglossal motoneurones are hyperexcitable in neonates (van Zundert et al. 2008). However at end stage, they are no longer hyperexcitable; the recruitment current, the F–I gain and the intensity of persistent inward currents are the same in mSOD1 mice and in controls (Fuchs et al. 2013).
Excitotoxicity might be induced by alterations of excitatory or inhibitory inputs to motoneurones
As spinal motoneurones are intrinsically hyperexcitable only transiently, weeks before they start to degenerate, it is unlikely that hyperexcitability contributes by itself to the degeneration process. However, excitotoxicity could still be induced by an alteration in the synaptic inputs received by the motoneurone, leading to an excessive excitation. A reduction of the inhibitory inputs or an increase of the excitatory inputs would lead to higher firing rates, thereby increasing the calcium concentration in the cytoplasm. Moreover, an increased glutamatergic excitation could by itself induce an excessive calcium influx through the AMPA/Kainate receptors because the GluR2 subunit is lacking in motoneurones (Rao & Weiss, 2004). Evidence for alterations of both the inhibitory and excitatory synaptic inputs impinging on motoneurones, as well as the properties of their postsynaptic receptors have been reported (Carunchio et al. 2008; Jiang et al. 2009; Chang & Martin, 2011; Sunico et al. 2011).
Motoneurones are also under the external influence of neuromodulatory descending pathways that can affect their excitability and firing. In particular, spinal motoneurones are highly sensitive to serotoninergic inputs that tend to increase excitability through modulation of (among others) calcium-activated potassium currents, hyperpolarization activated inward currents and persistent inward currents (Perrier et al. 2013). In our experimental conditions, these effects are minimal due to the depth of the anaesthesia (in vivo experiments) or absence of serotonin in the recording solution (in vitro experiments). Yet, motoneurones in ALS might not become hyperexcitable under the action of serotoninergic inputs as there is evidence that serotoninergic pathways degenerate in ALS (Sandyk, 2006; Dentel et al. 2013). However, degeneration of serotoninergic axons could lead to a modification of postsynaptic receptors causing them to become constitutively active (Murray et al. 2010), and eventually leading to spasticity (Bennett et al. 2001). Evidence for this phenomenon has been found in ALS, but only at very advanced stages of the disease (Dentel et al. 2013), which makes it unlikely a primary cause of the degeneration. Indeed, at the time points studied in our in vitro experiments, we did not find any motoneurones displaying the firing profiles characteristic of spasticity due to loss of serotoninergic inputs (type 4 F–I relationship in Bennett et al. 2001).
The superoxide dismutase 1 mutation affects all spinal motoneurones, regardless of their physiological type
There is a well-known relationship between the input conductance of a motoneurone and the physiological type of its motor unit: small motoneurones of slow contracting motor units (S) display the smallest input conductances whereas large motoneurones of fast contracting and fatigable motor units (FF) have the largest input conductances, motoneurones of fast contracting fatigue-resistant motor units (FR) being of intermediate size (Burke, 1981; Gustafsson & Pinter, 1984; Zengel et al. 1985; see also Manuel & Heckman, 2012b for preliminary data on mouse motor units). A salient result of our present work is that the whole spectrum of input conductances is shifted towards larger values. Moreover, the input conductances of motoneurones that turned into a less excitable state are distributed over the full range of input conductances. Hypoexcitability is therefore not restricted to a motoneurone subtype. Altogether our results strongly suggest that all motoneurones are affected regardless of the physiological type of their motor unit.
However, in SOD1G93A mice, FF motor units degenerate first, followed by FR motor units; whereas S motor units do not degenerate (Pun et al. 2006; Hegedus et al. 2007, 2008). As the ratio of excitatory versus inhibitory synapses is larger in FF motoneurones than in S motoneurones (Conradi et al. 1979; Kellerth et al. 1983; Brannstrom, 1993), we might speculate that FF motoneurones would be much more affected than S motoneurones by a shift in the balance between excitation and inhibition (Sunico et al. 2011), which might explain their greater vulnerability.
Key points
Intrinsic hyperexcitability of spinal motoneurones is thought to contribute to excitotoxicity during amyotrophic lateral sclerosis (ALS), but it has never been demonstrated that adult motoneurones become hyperexcitable before disconnection from their muscle fibres.
We found an increased input conductance in motoneurones recorded in a mouse model of ALS. Yet, most cells retained normal excitability as measured by current onset for firing and input–output gain. This indicates successful regulation of excitability, compensating for the increase in conductance.
In contrast, some cells became hypoexcitable, losing their ability to fire repetitively to quasi-stationary inputs before denervation. Hypoexcitability might therefore be an early marker of disease progression.
We thereby demonstrate that, if excitotoxicity is indeed a mechanism leading to degeneration in ALS, it is not caused by changes in the intrinsic electrical properties of the motoneurones but most probably by extrinsic factors such as excessive synaptic excitation.
Acknowledgments
The authors wish to thank Drs Jack Miller and Dave Bennett for technical assistance with the in vitro recordings, Ms Audrey Goulian for the genotyping of mice, Dr Sylvain Hanneton for his help with the statistical analysis and Dr Bradon Stell for his careful proofreading of the manuscript.
Glossary
- ALS
amyotrophic lateral sclerosis
- SOD1
superoxide dismutase 1
- WT
wild-type
- ACSF
artificial cerebrospinal fluid
- ANCOVA
analysis of covariance
- TTX
tetrodotoxin
- AMPA
2-amino-3-(3-hydroxy-5-methyl-isoxazol-4-yl)propanoic acid
- S
slow contracting
- FR
fast contracting, fatigue resistant
- FF
fast contracting, fatigable
Additional information
Competing interests
The authors declare no competing financial interests.
Author contributions
N.D., M.M., C.I. and D.Z. performed the in vivo experiments. M.M. and S.M.E. performed the in vitro experiments. N.D., M.M. and D.Z. analysed the data. N.D, M.M., C.J.H. and D.Z. wrote the article. All authors approved the final version of the manuscript.
Funding
Financial supports provided by AFM (MNM2 2009 Grant 14229), ARS-SLA (Grant LS_051481), ANR ‘Hyper-MND’ (ANR-2010-BLAN-1429-01), NIH-NINDS (R01NS077863), Thierry Latran Foundation (OHEX Project), and Target-ALS are gratefully acknowledged. N.D. was supported by a Contrat Doctoral Université Paris Descartes; M.M. was supported by the Foundation pour la Recherche Médicale and the Milton Safenowitz Post Doctoral Fellowship for ALS Research (ALS Association); C.I. was supported by the Association Française contre les Myopathies; and S.M.E. was supported by the Tim E. Noel fellowship from the ALS Society of Canada.
Author's present address
Sherif M. Elbasiouny: Department of Neuroscience, Cell Biology, and Physiology, Boonshoft School of Medicine, Wright State University, Dayton, OH 45435, USA.
References
- Alves CJ, de Santana LP, dos Santos AJ, de Oliveira GP, Duobles T, Scorisa JM, Martins RS, Maximino JR, Chadi G. Early motor and electrophysiological changes in transgenic mouse model of amyotrophic lateral sclerosis and gender differences on clinical outcome. Brain Res. 2011;1394:90–104. doi: 10.1016/j.brainres.2011.02.060. [DOI] [PubMed] [Google Scholar]
- Bellingham MC. A review of the neural mechanisms of action and clinical efficiency of riluzole in treating amyotrophic lateral sclerosis: what have we learned in the last decade. CNS Neurosci Ther. 2011;17:4–31. doi: 10.1111/j.1755-5949.2009.00116.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett DJ, Li Y, Siu M. Plateau potentials in sacrocaudal motoneurons of chronic spinal rats, recorded in vitro. J Neurophysiol. 2001;86:1955–1971. doi: 10.1152/jn.2001.86.4.1955. [DOI] [PubMed] [Google Scholar]
- Bories C, Amendola J, Lamotte d’Incamps B, Durand J. Early electrophysiological abnormalities in lumbar motoneurons in a transgenic mouse model of amyotrophic lateral sclerosis. Eur J Neurosci. 2007;25:451–459. doi: 10.1111/j.1460-9568.2007.05306.x. [DOI] [PubMed] [Google Scholar]
- Bouhadfane M, Tazerart S, Moqrich A, Vinay L, Brocard F. Sodium-mediated plateau potentials in lumbar motoneurons of neonatal rats. J Neurosci. 2013;33:15626–15641. doi: 10.1523/JNEUROSCI.1483-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowman A, Azzalini A. 2010. R package ‘sm’: nonparametric smoothing methods, version 2.2–4 edn.
- Brannstrom T. Quantitative synaptology of functionally different types of cat medial gastrocnemius alpha-motoneurons. J Comp Neurol. 1993;330:439–454. doi: 10.1002/cne.903300311. [DOI] [PubMed] [Google Scholar]
- Burke R. Motor units: anatomy, physiology, and functional organization. In: Brooks V, editor. Handbook of Physiology The Nervous System Motor Control. Bethesda, MD: American Physiological Society; 1981. [Google Scholar]
- Carrasco DI, Bichler EK, Rich MM, Wang X, Seburn KL, Pinter MJ. Motor terminal degeneration unaffected by activity changes in SOD1(G93A) mice; a possible role for glycolysis. Neurobiol Dis. 2012;48:132–140. doi: 10.1016/j.nbd.2012.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carunchio I, Mollinari C, Pieri M, Merlo D, Zona C. GAB(A) receptors present higher affinity and modified subunit composition in spinal motor neurons from a genetic model of amyotrophic lateral sclerosis. Eur J Neurosci. 2008;28:1275–1285. doi: 10.1111/j.1460-9568.2008.06436.x. [DOI] [PubMed] [Google Scholar]
- Chang Q, Martin LJ. Glycine receptor channels in spinal motoneurons are abnormal in a transgenic mouse model of amyotrophic lateral sclerosis. J Neurosci. 2011;31:2815–2827. doi: 10.1523/JNEUROSCI.2475-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu AY, Zhai P, Dal Canto MC, Peters TM, Kwon YW, Prattis SM, Gurney ME. Age-dependent penetrance of disease in a transgenic mouse model of familial amyotrophic lateral sclerosis. Mol Cell Neurosci. 1995;6:349–362. doi: 10.1006/mcne.1995.1027. [DOI] [PubMed] [Google Scholar]
- Conradi S, Kellerth JO, Berthold CH, Hammarberg C. Electron microscopic studies of serially sectioned cat spinal alpha-motoneurons. IV. Motoneurons innervating slow-twitch (type S) units of the soleus muscle. J Comp Neurol. 1979;184:769–782. doi: 10.1002/cne.901840409. [DOI] [PubMed] [Google Scholar]
- Dentel C, Palamiuc L, Henriques A, Lannes B, Spreux-Varoquaux O, Gutknecht L, Rene F, Echaniz-Laguna A, Gonzalez de Aguilar JL, Lesch KP, Meininger V, Loeffler JP, Dupuis L. Degeneration of serotonergic neurons in amyotrophic lateral sclerosis: a link to spasticity. Brain. 2013;136:483–493. doi: 10.1093/brain/aws274. [DOI] [PubMed] [Google Scholar]
- Elbasiouny SM, Amendola J, Durand J, Heckman CJ. Evidence from computer simulations for alterations in the membrane biophysical properties and dendritic processing of synaptic inputs in mutant superoxide dismutase-1 motoneurons. J Neurosci. 2010;30:5544–5558. doi: 10.1523/JNEUROSCI.0434-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleshman JW, Munson JB, Sypert GW, Friedman WA. Rheobase, input resistance, and motor-unit type in medial gastrocnemius motoneurons in the cat. J Neurophysiol. 1981;46:1326–1338. doi: 10.1152/jn.1981.46.6.1326. [DOI] [PubMed] [Google Scholar]
- Fuchs A, Kutterer S, Muhling T, Duda J, Schutz B, Liss B, Keller BU, Roeper J. Selective mitochondrial Ca2+ uptake deficit in disease endstage vulnerable motoneurons of the SOD1G93A mouse model of amyotrophic lateral sclerosis. J Physiol. 2013;591:2723–2745. doi: 10.1113/jphysiol.2012.247981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guertin PA, Hounsgaard J. Non-volatile general anaesthetics reduce spinal activity by suppressing plateau potentials. Neuroscience. 1999;88:353–358. doi: 10.1016/s0306-4522(98)00371-6. [DOI] [PubMed] [Google Scholar]
- Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, Caliendo J, Hentati A, Kwon YW, Deng HX. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science. 1994;264:1772–1775. doi: 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]
- Gustafsson B, Pinter MJ. Relations among passive electrical properties of lumbar alpha-motoneurones of the cat. J Physiol. 1984;356:401–431. doi: 10.1113/jphysiol.1984.sp015473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey PJ, Li Y, Li X, Bennett DJ. Persistent sodium currents and repetitive firing in motoneurons of the sacrocaudal spinal cord of adult rats. J Neurophysiol. 2006;96:1141–1157. doi: 10.1152/jn.00335.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegedus J, Putman CT, Gordon T. Time course of preferential motor unit loss in the SOD1 G93A mouse model of amyotrophic lateral sclerosis. Neurobiol Dis. 2007;28:154–164. doi: 10.1016/j.nbd.2007.07.003. [DOI] [PubMed] [Google Scholar]
- Hegedus J, Putman CT, Tyreman N, Gordon T. Preferential motor unit loss in the SOD1 G93A transgenic mouse model of amyotrophic lateral sclerosis. J Physiol. 2008;586:3337–3351. doi: 10.1113/jphysiol.2007.149286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iglesias C, Meunier C, Manuel M, Timofeeva Y, Delestree N, Zytnicki D. Mixed mode oscillations in mouse spinal motoneurons arise from a low excitability state. J Neurosci. 2011;31:5829–5840. doi: 10.1523/JNEUROSCI.6363-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilieva H, Polymenidou M, Cleveland DW. Non-cell autonomous toxicity in neurodegenerative disorders: ALS and beyond. J Cell Biol. 2009;187:761–772. doi: 10.1083/jcb.200908164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang MC, Heckman CJ. In vitro sacral cord preparation and motoneuron recording from adult mice. J Neurosci Methods. 2006;156:31–36. doi: 10.1016/j.jneumeth.2006.02.002. [DOI] [PubMed] [Google Scholar]
- Jiang M, Schuster JE, Fu R, Siddique T, Heckman CJ. Progressive changes in synaptic inputs to motoneurons in adult sacral spinal cord of a mouse model of amyotrophic lateral sclerosis. J Neurosci. 2009;29:15031–15038. doi: 10.1523/JNEUROSCI.0574-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellerth JO, Conradi S, Berthold CH. Electron microscopic studies of serially sectioned cat spinal α-motoneurons: V. motoneurons innervating fast-twitch (type FF) units of the gastrocnemius muscle. J Comp Neurol. 1983;214:451–458. doi: 10.1002/cne.901840408. [DOI] [PubMed] [Google Scholar]
- Kuo JJ, Siddique T, Fu R, Heckman CJ. Increased persistent Na+ current and its effect on excitability in motoneurones cultured from mutant SOD1 mice. J Physiol. 2005;563:843–854. doi: 10.1113/jphysiol.2004.074138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo JJ, Lee RH, Zhang L, Heckman CJ. Essential role of the persistent sodium current in spike initiation during slowly rising inputs in mouse spinal neurones. J Physiol. 2006;574:819–834. doi: 10.1113/jphysiol.2006.107094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacomblez L, Bensimon G, Leigh PN, Guillet P, Powe L, Durrleman S, Delumeau JC, Meininger V. A confirmatory dose-ranging study of riluzole in ALS. ALS/Riluzole Study Group-II. Neurology. 1996;47:S242–S250. doi: 10.1212/wnl.47.6_suppl_4.242s. [DOI] [PubMed] [Google Scholar]
- Lee RH, Heckman CJ. Essential role of a fast persistent inward current in action potential initiation and control of rhythmic firing. J Neurophysiol. 2001;85:472–475. doi: 10.1152/jn.2001.85.1.472. [DOI] [PubMed] [Google Scholar]
- Manuel M, Heckman CJ. Adult mouse motor units develop almost all of their force in the subprimary range: a new all-or-none strategy for force recruitment. J Neurosci. 2011;31:15188–15194. doi: 10.1523/JNEUROSCI.2893-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manuel M, Heckman CJ. Simultaneous intracellular recording of a lumbar motoneuron and the force produced by its motor unit in the adult mouse in vivo. J Vis Exp. 2012a:e4312. doi: 10.3791/4312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manuel M, Heckman CJ. Program No 15510 2012 Neuroscience Meeting Planner. New Orleans, LA: Society for Neuroscience, 2012. Online; 2012b. Single motor unit properties in SOD1 mice. [Google Scholar]
- Manuel M, Iglesias C, Donnet M, Leroy F, Heckman CJ, Zytnicki D. Fast kinetics, high-frequency oscillations, and subprimary firing range in adult mouse spinal motoneurons. J Neurosci. 2009;29:11246–11256. doi: 10.1523/JNEUROSCI.3260-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manuel M, Li Y, Elbasiouny SM, Murray K, Griener A, Heckman CJ, Bennett DJ. NMDA induces persistent inward and outward currents that cause rhythmic bursting in adult rodent motoneurons. J Neurophysiol. 2012;108:2991–2998. doi: 10.1152/jn.00518.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin E, Cazenave W, Cattaert D, Branchereau P. Embryonic alteration of motoneuronal morphology induces hyperexcitability in the mouse model of amyotrophic lateral sclerosis. Neurobiol Dis. 2013;54:116–126. doi: 10.1016/j.nbd.2013.02.011. [DOI] [PubMed] [Google Scholar]
- McLarnon JG. Potassium currents in motoneurones. Prog Neurobiol. 1995;47:513–531. doi: 10.1016/0301-0082(95)00032-1. [DOI] [PubMed] [Google Scholar]
- Meehan CF, Moldovan M, Marklund SL, Graffmo KS, Nielsen JB, Hultborn H. Intrinsic properties of lumbar motor neurones in the adult G127insTGGG superoxide dismutase-1 mutant mouse in vivo: evidence for increased persistent inward currents. Acta Physiol (Oxf) 2010;200:361–376. doi: 10.1111/j.1748-1716.2010.02188.x. [DOI] [PubMed] [Google Scholar]
- Miles GB, Dai Y, Brownstone RM. Mechanisms underlying the early phase of spike frequency adaptation in mouse spinal motoneurones. J Physiol. 2005;566:519–532. doi: 10.1113/jphysiol.2005.086033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray KC, Nakae A, Stephens MJ, Rank M, D'Amico J, Harvey PJ, Li X, Harris RL, Ballou EW, Anelli R, Heckman CJ, Mashimo T, Vavrek R, Sanelli L, Gorassini MA, Bennett DJ, Fouad K. Recovery of motoneuron and locomotor function after spinal cord injury depends on constitutive activity in 5-HT2C receptors. Nat Med. 2010;16:694–700. doi: 10.1038/nm.2160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pambo-Pambo A, Durand J, Gueritaud JP. Early excitability changes in lumbar motoneurons of transgenic SOD1G85R and SOD1G(93A-Low) mice. J Neurophysiol. 2009;102:3627–3642. doi: 10.1152/jn.00482.2009. [DOI] [PubMed] [Google Scholar]
- Perrier JF, Rasmussen HB, Christensen RK, Petersen AV. Modulation of the intrinsic properties of motoneurons by serotonin. Curr Pharm Des. 2013;19:4371–4384. doi: 10.2174/13816128113199990341. [DOI] [PubMed] [Google Scholar]
- Pieri M, Albo F, Gaetti C, Spalloni A, Bengtson CP, Longone P, Cavalcanti S, Zona C. Altered excitability of motor neurons in a transgenic mouse model of familial amyotrophic lateral sclerosis. Neurosci Lett. 2003;351:153–156. doi: 10.1016/j.neulet.2003.07.010. [DOI] [PubMed] [Google Scholar]
- Pun S, Santos AF, Saxena S, Xu L, Caroni P. Selective vulnerability and pruning of phasic motoneuron axons in motoneuron disease alleviated by CNTF. Nat Neurosci. 2006;9:408–419. doi: 10.1038/nn1653. [DOI] [PubMed] [Google Scholar]
- Quinlan KA, Schuster JE, Fu R, Siddique T, Heckman CJ. Altered postnatal maturation of electrical properties in spinal motoneurons in a mouse model of amyotrophic lateral sclerosis. J Physiol. 2011;589:2245–2260. doi: 10.1113/jphysiol.2010.200659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Development Core Team. R: A Language and Environment for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing; 2010. [Google Scholar]
- Rao SD, Weiss JH. Excitotoxic and oxidative cross-talk between motor neurons and glia in ALS pathogenesis. Trends Neurosci. 2004;27:17–23. doi: 10.1016/j.tins.2003.11.001. [DOI] [PubMed] [Google Scholar]
- Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O'Regan JP, Deng HX. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- Rothstein JD, Tsai G, Kuncl RW, Clawson L, Cornblath DR, Drachman DB, Pestronk A, Stauch BL, Coyle JT. Abnormal excitatory amino acid metabolism in amyotrophic lateral sclerosis. Ann Neurol. 1990;28:18–25. doi: 10.1002/ana.410280106. [DOI] [PubMed] [Google Scholar]
- Sandyk R. Serotonergic mechanisms in amyotrophic lateral sclerosis. Int J Neurosci. 2006;116:775–826. doi: 10.1080/00207450600754087. [DOI] [PubMed] [Google Scholar]
- Sekerli M, Del Negro CA, Lee RH, Butera RJ. Estimating action potential thresholds from neuronal time-series: new metrics and evaluation of methodologies. IEEE Trans Biomed Eng. 2004;51:1665–1672. doi: 10.1109/TBME.2004.827531. [DOI] [PubMed] [Google Scholar]
- Sunico CR, Dominguez G, Garcia-Verdugo JM, Osta R, Montero F, Moreno-Lopez B. Reduction in the motoneuron inhibitory/excitatory synaptic ratio in an early-symptomatic mouse model of amyotrophic lateral sclerosis. Brain Pathol. 2011;21:1–15. doi: 10.1111/j.1750-3639.2010.00417.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Den Bosch L. Genetic rodent models of amyotrophic lateral sclerosis. J Biomed Biotechnol. 2011;2011:348765. doi: 10.1155/2011/348765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Zundert B, Peuscher MH, Hynynen M, Chen A, Neve RL, Brown RH, Jr, Constantine-Paton M, Bellingham MC. Neonatal neuronal circuitry shows hyperexcitable disturbance in a mouse model of the adult-onset neurodegenerative disease amyotrophic lateral sclerosis. J Neurosci. 2008;28:10864–10874. doi: 10.1523/JNEUROSCI.1340-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Lewinski F, Keller BU. Ca2+, mitochondria and selective motoneuron vulnerability: implications for ALS. Trends Neurosci. 2005;28:494–500. doi: 10.1016/j.tins.2005.07.001. [DOI] [PubMed] [Google Scholar]
- Vucic S, Nicholson GA, Kiernan MC. Cortical hyperexcitability may precede the onset of familial amyotrophic lateral sclerosis. Brain. 2008;131:1540–1550. doi: 10.1093/brain/awn071. [DOI] [PubMed] [Google Scholar]
- Young S, Bowman A. Non-parametric analysis of covariance. Biometrics. 1995;51:920–931. [Google Scholar]
- Zengel JE, Reid SA, Sypert GW, Munson JB. Membrane electrical properties and prediction of motor-unit type of medial gastrocnemius motoneurons in the cat. J Neurophysiol. 1985;53:1323–1344. doi: 10.1152/jn.1985.53.5.1323. [DOI] [PubMed] [Google Scholar]
- Zona C, Pieri M, Carunchio I. Voltage-dependent sodium channels in spinal cord motor neurons display rapid recovery from fast inactivation in a mouse model of amyotrophic lateral sclerosis. J Neurophysiol. 2006;96:3314–3322. doi: 10.1152/jn.00566.2006. [DOI] [PubMed] [Google Scholar]