

Abstract
In this paper we give the current state of high-throughput virtual screening. We describe a case study of using a task-parallel MPI (Message Passing Interface) version of Autodock4 [1], [2] to run a virtual high-throughput screen of one-million compounds on the Jaguar Cray XK6 Supercomputer at Oak Ridge National Laboratory. We include a description of scripts developed to increase the efficiency of the predocking file preparation and postdocking analysis. A detailed tutorial, scripts, and source code for this MPI version of Autodock4 are available online at http://www.bio.utk.edu/baudrylab/autodockmpi.htm.
Keywords: Virtual high-throughput screening, Automated molecular docking, Drug discovery, High-performance computing
1. Introduction
Many pharmaceuticals act by selectively binding to a specific protein and thus inhibiting a specific process relevant to a disease or illness. Because of this, the early stage of drug discovery consists of identifying potential compounds that bind to a protein of interest with a high affinity and specificity. Experimentally testing a very large number of these compounds is both costly and time consuming. Virtual high-throughput screening is an equivalent computational process that can reduce the time and cost of discovering new drugs [3], [4].
A virtual screen is a task-parallel application in which each ligand from a large library of drug-like compounds is “docked” into a protein of interest. The docking method produces the structure of the ligand docked in the presumed active-site of the protein and a calculated affinity estimating how well the ligand binds to the protein.
The scoring functions in typical docking applications use severe approximations in order to efficiently produce results. Therefore, the calculated binding affinities rarely reproduce accurate experimental affinities. However, docking applications can be very successful at reproducing the correct “docked” conformation as well as scoring active compounds better than inactive compounds[5]. Therefore, virtual screenings are powerful tools to create “enriched” libraries (see Figure 1 for depiction).
Figure 1. Virtual Screening as a tool to create “enriched” libraries of potential novel pharmaceuticals.

Given a large library of diverse drug-like compounds, virtual screening applications can be used to relatively quickly screen the entire library and assign scores to each compound. Compounds with a high affinity (i.e. active) for the protein of interest are more likely to score higher than non-active compounds. Once all of the compounds are scored and ranked, the library can be partitioned by scores thus creating an “enriched” library that is a subset of the original and presumably contains a much higher percent of active compounds. The smaller number of compounds scoring well in the virtual screen can then be tested experimentally with a higher success rate.
Since the true power of docking tools lies in the production of enriched libraries, the capability of distributing a large number of docking tasks and collecting and analyzing the data is extremely important. In this study, a task-parallel MPI implementation of Autodock4 is used that was developed to be used on high-performance computing (HPC) systems. A similar implementation uses the MapReduce paradigm of Hadoop in order to distribute the docking tasks on Hadoop clusters or cloud computing systems [6]. Other methods have been developed to run virtual screens on small clusters and grid platforms. However, some HPC resources require an MPI implementation in order to utilize tens of thousands of high-end processors at once, which is the aim for Autodock4.lga.MPI. These applications require a fair amount of computational expertise to set-up and run. This makes proper documentation and sharing of code and potential pitfalls important in order for these tools to aid researchers and improve the efficiency of drug development.
The main goals of this paper is to illuminate the reader on the current technological state of high-throughput virtual screening and to share the experience of running a very large virtual screen of just over one million compounds and direct the readers' attention to the publicized code and tutorial to bring this powerful tool to a wider community of researchers. In Section 2 we give a review of the current published literature regarding high-throughput virtual screening. In section 3 we describe improvements we have made in the high-throughput virtual screening process (for the predocking and postdocking steps) when using Autodock4.lga.MPI. In section 4 we describe the case study of running a million compound virtual screening in under 24 hours and highlight some areas for future improvements. In Section 5 we give a description of some future directions for virtual screenings.
2. Virtual Screening: Existing Tools and Techniques
The increasing computational power of supercomputers allows for potentially very large chemical databases to be screened against a variety of protein targets. The software technology that is needed to leverage such computing power is the subject of much effort. A recent overview of some virtual screening tools aimed at making the task easier (typically through a graphical user interface - GUI) on mostly smaller computing architectures or individual machines, can be found in [7].
2.1 Parallelization approaches of docking of large chemical databases
Two large docking initiatives on the EGEE Grid infrastructure have been reported [8], the first targeting malaria and the second to target Influenza [9]. The study referenced in [9] screened about 300,000 ZINC [10] compounds against eight variants of neuraminidase from homology models on more than 2000 CPUs, generating about 600 Gigabytes of data. This study used two different Grid tools: an enhanced version of WISDOM [9] on 2000 worker nodes for a 6 week period, and a lightweight framework called DIANE. Due to Grid scheduling overhead and problems with the Grid resource Broker, WISDOM had a distribution frequency of only 38%. In addition, about 30% of the jobs failed during Grid scheduling and had to be resubmitted. DIANE had a similar failure rate but could automate the resubmission of tasks and had a much higher distribution efficiency of 80%. However, DIANE is not very scalable due to communication needs between the DIANE master and DIANE workers (which are limited to a few hundred).
Closer to our MPI development reported in the present paper is the multi-level parallelization of Autodock4.2 (mpAD4) [11] which uses MPI to parallelize the distribution of individual docking jobs and OpenMP to multi-thread the Lamarkian Genetic Algorithm (conformational search mechanism) in Autodock. This implementation was only tested on up to 16,384 CPU cores.
Approaches to develop better ways to connect to and utilize multiple computational resources such as High-Performance Computing (HPC) and High-Throughput Computing (HTC) systems have been repored [12], [13]. This work is applicable to the WISDOM [8] project which uses HTC resources for virtual screens and HPC resources to run Molecular Dynamics simulations to more accurately rescore the top hits resulting from the initial screen.
Computing tasks are often classified as either high-throughput computing (HTC) or high-performance computing (HPC). An emerging classification is many-task computing (MTC) and it differs from traditional HTC tasks typically done on a grid in that their metric of interest is the time to completion of the job and can take advantage of more traditionally HPC systems. Virtual screening is recognized as an important MTC application [14], and there is continued interest on the best paradigms for MTC applications to run optimally on HPC architectures [15]. In [16], a virtual screening tool was developed using DOCK [17], that incorporated HTC features available on the Blue Gene/L supercomputer. The development included single processor optimization with compiler flag optimization and optimized libraries and optimization of the parallel implementation by increasing load balance by presorting the jobs by complexity and decreasing I/O by storing temporary files in memory. Instead of a using a parent-child (master-slave) scheme to distribute the individual docking jobs, a HTC mode available on Blue Gene/L was used in which a work dispatcher runs on the front end of the supercomputer. While the traditional MPI parent-child implementation had a major drop in performance between 8,192 (which had near linear performance) and 16,384 processors, the HTC version maintained near linear performance on 16,384 processors. Another large-scale implementation of DOCK was done on Falcon [18] which is an execution framework that allows for loosely coupled programs to run on petascale systems and can scale up to 160,000 cores. In a case study, after making some changes to I/O patterns, DOCK scaled to 116,000 cores while being executed through Falcon.
2.2 Current GPU Development
Graphical processing units (GPUs) which have been developed for efficient rendering of high-quality graphics can be used in order to accelerate some scientific codes and are currently being incorporated into many large computing systems because of their increased available floating point operations to power ratio. In a recent review of GPU development in the computational molecular sciences [19], it is noted that GPU development in docking has not been as popular as other areas such as Molecular Dynamics because the important metric in docking is the time to completion of an entire screening, not the individual docking task and large GPU clusters are still rare. However, there is a trend for some of the largest supercomputer centers in the world to incorporate GPUs in their systems and utilizing them to speed-up individual docking tasks can reduce the time taken for large scale screenings if proper load balancing is ensured. In [20], the authors use GPUs to accelerate the precalculation of potential grids. With the docking engine FlexScreen, this calculation dominates 80% of the runtime for an individual docking. In a typical screen where a large library of compounds is docked into the same structure, this information can be reused. However, when using multiple receptor files, it becomes increasingly more important to accelerate this portion of the calculation. They were able to accelerate the grid calculations by a factor of 60. Also, as mentioned above in [21], the authors here achieved a speed-up of 213× when accelerating the calculation of the non-bonded electrostatic interactions.
3. Virtual Screening Using Task-Parallel MPI Autodock4
We report here on recent development of Autodock4.lga.MPI [2] and on its application to screen quickly a very large database of compounds.Autodock4.lga.MPI is a task-parallel version of Autodock4 that allows for independent and simultaneous docking of a large number of ligands into a single specified protein on up to thousands of processors at one time. The original serial version has many different input files: 1) a parameter file for each ligand, 2) 3-D coordinate files for both the ligand and protein, and 3) pre-calculated affinity maps of the protein (one for each atom type in the ligand library). The I/O is reduced in the parallel version by creating only two parameter files for the entire screening, instead of one file for each of the ligands in the screening. One parameter file contains all of the parameters specific to the docking job; these are the same for all of the ligands. The other file contains a list of the ligand specific parameters and is used for distributing the tasks. The I/O is also reduced by having a single binary file for the precalculated affinity maps using HDF5 [22].
3.1 Improvements in Efficiency
Autodock4.lga.MPI only distributes the actual docking tasks. While this is the computationally expensive aspect of a virtual screening, when a virtual screening is scaled-up to handle millions of compounds on thousands of processors, file preparation and result analysis become the bottlenecks in decreasing the overall screening time (the time it takes to deliver the results).
3.1.1 PreDocking
The process in Figure 1 of [2] shows that the standard procedure is done for predocking plus two additional steps. The standard procedure includes 1) preparing the ligand and receptor PDBQT (similar to a Protein Data Bank (PDB) file with Autodock charge (Q) and atom type (T) information) files, 2) creating the affinity maps (grids), and 3) creating the Docking Parameter Files (DPF). The PDBQT files are the coordinate files that Autodock4 takes as input for the ligands and receptor. They include charge and atom type information needed by Autodock4 and are generated using AutoDockTools (ADT) [23]. The affinity maps are created using Autogrid (program packaged with Autodock) and one file is needed for each atom type in the ligand library. The DPF files are parameter files used by Autodock4. One file is used for each individual docking of a ligand into a receptor. These files are also created by ADT. ADT includes scripts in order to automate this process as well.
The additional predocking steps required by Autodock4.lga.MPI include running 1) make_binary.exe and 2) make_dpf_input.exe. (1) creates the single binary file used by Autodock4.lga.MPI from the ASCII affinity maps created in the standard procedure. (2) creates the two input files needed by Autodock4.lga.MPI from the DPF files previously required by Autodock4. Both of these programs were developed along with Autodock4.lga.MPI.
C shell scripts were developed along with Autodock4.lga.MPI to automate the predocking process including the standard procedure. One of the major pitfalls of this method is that it was developed to run sequentially either locally or from a login node on a large machine, such as a supercomputer. If the predocking process is done on a local machine then a large amount of data will have to be transferred to the supercomputer between the predocking and docking procedures. Since several users could be using the same login node on a supercomputer, predocking from a login node can create severe contention. Therefore, we have developed python scripts that automate the predocking process including a submission script so that the predocking can be done from a compute node. This allows the input data to be generated at the location it needs to be for the docking process without creating extra contention on the login nodes.
Another pitfall of the previous process is that it only ran sequentially. Since the same process must be done for a large amount of ligands, the predocking process is quite straightforward to parallelize. The modified process can use multiple processors to partition the ligand library and process the input in parallel. The benefit of this method is two-fold because not only is the data being prepared in parallel, but the precursor files are created in a subdirectory for each processor. This reduces the load on the file system when millions of files need to be written to and read from. The final files needed by Autodock4.lga.MPI are moved to the parent directory once they no longer need to be read from. This is where they will need to be for the actual docking procedure and they will not need to be moved again. A partial ligand specific parameter file is made during this predocking process per processor. Every ligand processed by a particular processor is included in the partial parameter file associated with that processor. These partial files are already ordered correctly (ligands with the most torsional degrees of freedom listed first). We include a script to combine these files which runs very quickly since the individual files are already correctly ordered.
The previous make_dpf_input.exe process used to create the ligand specific parameter file made extensive use of AWK scripts (an interpreted language common on most Unix-like operating systems). While AWK scripts can be quite powerful, they complicate this file preparation procedure. AWK is not very reusable as it is difficult to trace what the script is doing, and it also necessitates temporary files to be made during the process when the amount of data needed in this step is small enough to hold in memory until the file can be written in its final form. In our current predocking procedure, the necessary information is parsed out of the DPF files and stored in a python dictionary. This makes it very easy to sort the data and write it only once in the final form after all of the DPF files have been parsed.
Previously, the C shell predocking scripts assumed that there were already PDB (Protein Data Bank) coordinate files for every ligand in the library. This would mean that, even if the files are being prepared on the supercomputer, a large number of files will have to be collected and transferred before the predocking stage even begins. In the present updated procedure, the input is a concatenated MOL2 file which contains the entire ligand library in one file. This is the only file that needs to be transferred to the supercomputer in order to start the predocking process. Also, a MOL2 file is commonly used to store the information on a library of ligands and would more likely be the initial input when starting a virtual screening. A script is used in order to partition the library into subdirectories for the parallel predocking process as described above. To illustrate the improvements here, the million compound library that is screened in this study has a concatenated MOL2 file size of 2.3 GB and a compressed file size (gzip) of 406 MB. The directory containing the PDBQT input files of the library is 4.0 GB and the compressed and archived file size (tar) is 531 MB. While the file size of the compressed data is close, the limiting factor here is the time it takes to compress and archive the files. It takes on the order of minutes to compress the concatenated MOL2 file and hours to compress and archive the PDBQT files. In addition to that, uncompressing the archived PDBQT files on a login node of a busy supercomputer could take longer than a day.
Another key difference in the new procedure is completely separating the receptor preparation from the ligand library preparation. The previous predocking method prepared the receptor files and ligand files from the same script. However, the receptor preparation is much simpler as there is only one receptor per screening and so it is much more straightforward to prepare these files manually then to make changes in a script to adapt it to a different project. Also, ligand libraries may be prepared separately and used in different screenings with different receptors.
The complete workflow of the new predocking procedure is given in Figure 2. A concatenated file containing the 3D coordinates for all of the ligands in the screening is split into individual files for each worker processor. Each processor parses out the individual molecules in their workset and uses ADT scripts in order to create the PDBQT input files and the DPF files needed to create the list_par.dpf input file.
Figure 2.
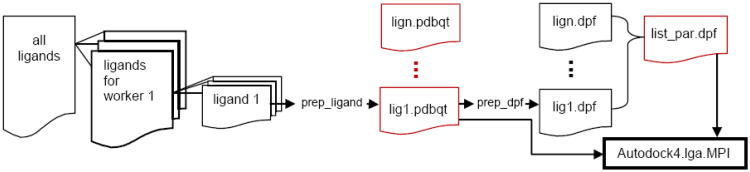
Workflow for PreDocking procedure. The files outlined in red are input files for Autodock4.lga.MPI. The entire workflow is managed by custom scripts available in the online tutorial.
In addition to increasing the efficiency of the predocking process, the new procedure also aims at simplifying it. The previous C shell scripts needed many changes in order to adapt them to a new project, which required lots of code deciphering. In contrast, the new scripts require minimal changes, such as changing path names. There is a thorough tutorial that documents all necessary changes for a first time user.
3.1.2 PostDocking
Similar to the previous predocking procedure, C shell scripts were developed along with Autodock4.lga.MPI in order to analyze the results and create a ranked list of all the ligands in the library. Again, these scripts make extensive use of AWK and are very hard to decipher and reuse, even though modifications will always need to be made to adapt them to a new project. These scripts require many temporary files while manipulating the data to extract the interesting results. The scripts are also developed to either run locally or from a login node, and this requires either a large amount of data to be transferred that there is no need to save or extra contention on a login node.
The new postdocking procedure uses python scripts that can run locally or be submitted to a compute node via a submission script. Initially, we developed the script to parse all of the result files and keep the needed information in a python dictionary in order to quickly sort and write the ranked list once after all the results have been parsed. However, due to wall-clock time limits, we developed a second way that splits the parsing and sorting. One temporary file is kept with the needed information to maintain it if the job is killed. Once all of the results are collected, the entire temporary file is read into memory, sorted, and written in the correct order very quickly.
The current scripts utilize the python glob module which finds all pathnames matching the given pattern. The glob() function creates a list of file names that can be used to iterate through all of the files. This makes restarting the job very easy (if it was killed due to wall-clock time restrictions, etc) because the file list can be truncated to start at the index past the last file parsed in the previous job.
The postdocking procedure returns the file names for any result files with incomplete results. Another script ensures that all the result files have been created. Any missing or incomplete docking tasks can then be restarted to ensure a complete screening.
The improvements described above increased the efficiency of postdocking by an order of magnitude, so the postdocking procedure has not been parallelized. However, as with the predocking, it would be very easy to parallelize this step as well. The result files could be partitioned, then parsed and sorted on individual processors. If enough processors are used to guarantee that the postdocking would finish within a systems wall-clock time limit, then a temporary file would not be necessary. Each processor could hold the results in memory and write the partial ranked list once. Then partial sorted lists could be combined very quickly on one processor.
In addition to increasing the efficiency of the postdocking process, the new procedure aims at simplifying it and making it more thorough. Just as with the predocking, a detailed tutorial outlines all the changes that need to be made in order to adapt the scripts for a new project. The new scripts also have more features and options.
Autodock4 results cluster the individual trials for one docking between one ligand and the receptor. Trials with similar ligand conformations are placed in the same cluster and the coordinates of the conformation with the lowest energy are reported for each cluster. A ranked list can be created in two different ways: 1) using the lowest energy cluster or 2) using the largest cluster. If the docking parameters are set-up correctly, the largest cluster should be the converged result. Whereas, the previous scripts only ranked the results using the largest cluster, the new scripts create sorted lists both ways: however, the largest cluster is typically used.
The Autodock4.lga.MPI scripts collected only the structural information for the top specified percentage of the ranked library. However, the new procedure allows for the structure of a range of scored ligands to be collected. If a known active ligand is included in the input library, then the ranked ligands with scores similar to the known active ligand may be collected.
The previous scripts did not include any ligands in the ranked list that had a positive binding free energy. This is because Autodock4 cannot calculate the binding constant for any ligand with a positive free energy and the inhibition constant is also reported in the list and a secondary sort key in the ranking. The new method gets around this by assigning a null value of 0 for the inhibition constant for any ligand with a positive binding free energy and not attempting to parse the information from the file. This allows all of the ligands to be included in the ranked list.
Finally, the new method includes scripts and detailed instructions to create a concatenated SDF (Structure Data File) of the enriched database. SDF files are structural files that can include associated data. This method uses the <UNIQUE_ID> associated data tag to include the rank of each ligand from the screening. This enables the delivery of the results in an easy and efficient manner.
The complete workflow for the new postdocking procedure is given in Figure 3. All the result files from Autodock4.lga.MPI are parsed in order to create a ranked list of compounds. The result files corresponding to the highest ranking ligands are parsed in order to generate PDB files containing the 3D coordinates of the final docked ligand poses. Options are given in order to generate a concatenated file of the results.
Figure 3.

Workflow for PostDocking procedure. Files outlined in red are the output files from Autodock4.lga.MPI. Custom scripts available in the tutorial manage the workflow.
3.2 Improvements in Accuracy
When examining log files, we discovered some random errors of reading the binary affinity maps. These appear to be memory errors as they are not reproducible and occur at different rates on different architectures. The errors occurred while running 105 ligand screenings on the Newton cluster at University of Tennessee as well as while running a 1 million ligand screening on the Jaguar Cray XT5 at Oak Ridge National Laboratory. However, we did not receive the errors while running a 1 million ligand screening on the new XK6 architecture.
During these errors, a message is written in the result file claiming that the wrong size grid spacing is used. This is due to the binary file not being read correctly. However, using incorrect (null) input, the calculations are still performed and results are reported that appear normal during the postdocking process. If these files are not searched for explicitly, the erroneous results will end up in the analysis and may skew the final rankings. Therefore, we have developed a script to search for the errors. Detailed instructions are included in the tutorial.
4. Case Study
Recently, we completed a screening of one million ligands. The details and biological results of the screening are not discussed here. In this section, the focus is on the lessons learned while performing such a large screening and ideas to further improve the virtual screening of very large ligand libraries.
The one million compound screening ran on the Jaguar Cray XT5 Supercomputer at Oak Ridge National Laboratory using 65 thousand processors. The entire one million compound library was screened just under 24 hours, with half of the library finishing in about 5 hours (see Figure 4). The library consisted of ligands ranging from 0 rotatable bonds to 32 rotatable bonds, with an average of 4.9. About 5% of the library consisted of ligands with at least 10 rotatable bonds (not “drug-like” molecules). Almost the entire library was screened in 10 hours with the large, extremely flexible ligands dominating the screening time.
Figure 4. Actual time to screen one million compounds.

4.1 Lessons Learned
4.1.1 Millions of files is a lot!
Handling millions of compounds is extremely time consuming, even tasks such as transferring files to a new location is very taxing on the file system. When a directory is overloaded, it takes the file system longer to find necessary files. Some of this stress can be removed by creating a directory hierarchy to store files. Also, Autodock4.lga.MPI writes the output files in the same directory as the input files. This creates twice as many files to deal with and necessitates an extra step to separate the files in order to process them more efficiently. Future virtual screening tools that are developed to handle such large jobs should use a file system hierarchy to both reduce the number of input files in one directory and separate the output from the input.
4.1.2 Naming conventions are important
Autodock4.lga.MPI names the result files by the following naming convention:
where n is the worker number (processor) and m is the ligand number (from distribution list). There are two problems with this convention. 1) If a screening is killed (due to wall-clock time, etc.) then the result files from the first round must be renamed before the screening is restarted to avoid files being overwritten. There will be at least x files with the same name where x is the number of worker processors. This could potentially lead to a lot of data loss when using thousands of processors. 2) There is no way to link a result file to a particular ligand without reading the results.
4.2 Additional Improvements
In order to decrease the amount of necessary I/O, Autodock4.lga.MPI uses binary file creation and reading (with HDF5). This requires additional libraries to be available on the system in which the screen will run and increase the complexity of the software. A better solution would be to just eliminate the need to pass such a large amount of I/O in the first place.
Autodock Vina [24] is another docking program similar to Autodock4. Using Vina as the docking engine would eliminate the need to pass the binary files. Vina does not use precalculated affinity maps, but calculates the information efficiently during the docking process. Therefore, only the ligand and receptor files need to be passed to the compute node.
4.3 Tutorial
In order to make high-throughput virtual screening of large chemical libraries more accessible to researchers, we have provided a tutorial detailing the entire process. It includes the predocking (file preparation), docking (calculations), and postdocking (analysis). The tutorial was written relative to running on the Jaguar supercomputer and Lens analysis cluster (smaller cluster with shared file system with Jaguar). Compilation instructions are included in the tutorial.
5. Possible Future Directions
Flexible Receptors: Due to the large number of degrees of freedom in a protein, they are usually considered to be rigid or at least mostly rigid in molecular docking. However, proteins are not static structures and can exist in an ensemble of different conformational states, each of which a different chemical could potentially bind. Reference [25] reviews how molecular dynamics simulations (MD) can be included in the drug discovery process to take into account protein flexibility, such as the Relaxed Complex Scheme (RCS) [26]. In RCS, multiple representative snapshots are obtained from a MD trajectory and used in docking to efficiently capture the diversity of protein's conformational states. Methods to create “super” enriched lists from multiple ranked lists from different conformational states are an interesting and active area of research.
A novel drug must not only bind well to its target protein but also have limited side effects and toxicity. The earlier these adverse effects are found in the drug discovery pipeline, the more cost efficient the entire process becomes. Therefore, a means to virtually test for toxicity and side-effects before the synthesis and laboratory testing of a new chemical would be of great financial benefit. Previous studies have shown that 83% of the experimentally known toxicity and side effects for a drug target could be predicted by an inverse-docking approach of docking the potential drug into a library of proteins [27] and are more novel than the traditional one receptor – many ligands virtual screening [28].
6. Conclusions
In this paper we describe an improved predocking and postdocking procedure that works with Autodock4.lga.MPI and give details on a case study of running a million ligand library screening on the Jaguar supercomputer. Autodock4.lga.MPI, a previously published high-throughput screening application, is to the best of our knowledge the most scalable screening application that does not require a non-standard distribution framework (such as Falcon [18]) but relies on MPI which is standard on most high-performance computers. The work here focuses on further improving the total time to complete a screening, from preparing files to obtaining meaningful results for experimental validation. These steps are often not the focus of screening applications and become the bottleneck of very large screenings.
Acknowledgments
We thank Barbara Collignon for useful discussion and Kristina Thiagarajan for support. The National Center for Computational Sciences (NCCS) is acknowledged for a computational time grant (project BIP015). This work was financially supported by NIH grant 1KL2RR031974, Georgetown-Howard Universities Center for Clinical and Translational Science.
Contributor Information
Sally R. Ellingson, Email: sellings@utk.edu, Genome Science and Technology, University of Tennessee, Center for Molecular Biophysics, Oak Ridge National Laboratory.
Sivanesan Dakshanamurthy, Email: sd233@georgetown.edu, Lombardi Comprehensive, Cancer Center, Georgetown University Medical Cntr.
Milton Brown, Email: mb544@georgetown.edu, Lombardi Comprehensive, Cancer Center, Georgetown University Medical Cntr.
Jeremy C. Smith, Email: smithjc@ornl.gov, Biochemistry and Cellular and Molecular Biology, University of Tennessee, Center for Molecular Biophysics, Oak Ridge National Laboratory.
Jerome Baudry, Email: jbaudry@utk.edu, Biochemistry and Cellular and Molecular Biology, University of Tennessee, Center for Molecular Biophysics, Oak Ridge National Laboratory.
References
- 1.Autodock. [Online]. Available: http://autodock.scripps.edu/downloads.
- 2.Collignon B, Schulz R. Task-↓parallel message passing interface implementation of Autodock4 for docking of very large databases of compounds using high-↓performance super↓computers. Journal of computational Chemistry. 2011;32, no. 6:1202–1209. doi: 10.1002/jcc.21696. [DOI] [PubMed] [Google Scholar]
- 3.Shoichet BK. Virtual screening of chemical libraries. Nature. 2004 Dec;432, no. 7019:862–5. doi: 10.1038/nature03197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Werner T, Morris MB, Dastmalchi S, Church WB. Structural modelling and dynamics of proteins for insights into drug interactions. Advanced drug delivery reviews. 2012 Mar;64, no. 4:323–43. doi: 10.1016/j.addr.2011.11.011. [DOI] [PubMed] [Google Scholar]
- 5.Gilson MK, Zhou HX. Calculation of protein-ligand binding affinities. Annual review of biophysics and biomolecular structure. 2007 Jan;36:21–42. doi: 10.1146/annurev.biophys.36.040306.132550. [DOI] [PubMed] [Google Scholar]
- 6.Ellingson SR, Baudry J. High-throughput virtual molecular docking: Hadoop implementation of AutoDock4 on a private cloud; Proceedings of the second international workshop on Emerging computational methods for the life sciences; 2011. [Google Scholar]
- 7.Jacob R, Anderson T, McDougal OM. Accessible High-Throughput Virtual Screening Molecular Docking Software for Students and Educators. PLoS Computational Biology. 2012;8, no. 5:e1002499. doi: 10.1371/journal.pcbi.1002499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jacq N, Breton V, Chen H, Ho L. Virtual screening on large scale grids. Parallel Computing. 2007 [Google Scholar]
- 9.Lee H, Salzemann J, Jacq N, Ho LY, Chen HY, Breton V, Merelli I, Milanesi L, Lin S, Wu YT. Grid-enabled high-throughput in silico screening against influenza A neuraminidase. IEEE Trans Nanobioscience. 2006;5, no. 4:288–95. doi: 10.1109/tnb.2006.887943. [DOI] [PubMed] [Google Scholar]
- 10.Irwin JJ, Sterling T, Mysinger MM, Bolstad ES, Coleman RG. ZINC: A Free Tool to Discover Chemistry for Biology. Journal of chemical information and modeling. 2012 Jul; doi: 10.1021/ci3001277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Norgan A, Coffman P, Kocher J, Katzmann D, Sosa C. Multilevel Parallelization of AutoDock 4.2. Journal of Cheminformatics. 2011;3, no. 12 doi: 10.1186/1758-2946-3-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Riedel M, Memon A, Memon MS, Mallmann D, Streit A, Wolf F, Lippert T. Improving e-Science with Interoperability of the e-Infrastructures EGEE and DEISA; Proceedings of the …; 2008. [Google Scholar]
- 13.Riedel M, Memon M. e-Science Infrastructure Integration Invariants to Enable HTC and HPC Interoperability Applications. 2011 IEEE International Parallel & Distributed Processing Symposium. 2011:922–931. [Google Scholar]
- 14.Raicu I. Many-task computing for grids and supercomputers. Many-Task Computing on Grids …. 2008:1–11. [Google Scholar]
- 15.Katz DS, Armstrong TG, Zhang Z, Wilde M, Wozniak JM. Many-Task Computing and Blue Waters. Technical Report CI-TR-13-0911 Computation Institute, University of Chicago & Argonne National Laboratory. 2012:1–47. [Google Scholar]
- 16.Peters A, Lundberg M, Lang P, Sosa C. High throughput computing validation for drug discovery using the DOCK program on a massively parallel system. 1st Annual MSCBB. 2007 [Google Scholar]
- 17.The Official UCSF DOCK Web-site: DOCK6. [Online]. Available: http://dock.compbio.ucsf.edu/DOCK_6/index.htm.
- 18.Raicu I, Zhang Z, Wilde M, Foster I. Toward loosely coupled programming on petascale systems; SC '08 Proceedings of the 2008 ACM/IEEE conference on Supercomputing; 2008. [Google Scholar]
- 19.Harvey MJ, De Fabritiis G. A survey of computational molecular science using graphics processing units. WIREs Computational Molecular Science. 2012 [Google Scholar]
- 20.Sánchez-Linares I, Perez-Sanchez H, Guerrero GD, Cecilia JM, Garcia JM. Accelerating multiple target drug screening on GPUs; Proceedings of CMSB; 2011. pp. 95–102. [Google Scholar]
- 21.Guerrero G, Perez-Sanchez HE, Cecilia JM, Garcia JM. Parallelization of Virtual Screening in Drug Discovery on Massively Parallel Architectures; 2012 20th Euromicro International Conference on Parallel, Distributed and Network-based Processing; 2012. [Google Scholar]
- 22.The HDF Group. Hierarchical data format version 5, 2000-2010. [Online]. Available: http://www.hdfgroup.org/HDF5.
- 23.AutoDockTools (ADT) [Online]. Available: http://mgltools.scripps.edu/
- 24.Trott O, Olson A. AutoDock Vina: Improving The Speed And Accuracy Of Docking With a New Scoring Function, Efficient Optimization, and Multithreading. Journal of Computational Chemistry. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Durrant JD, McCammon JA. Molecular dynamics simulations and drug discovery. BMC biology. 2011;9, no. 71 doi: 10.1186/1741-7007-9-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cheng LS, Amaro RE, Xu D, Li WW, Arzberger PW, McCammon JA. Ensemble-based virtual screening reveals potential novel antiviral compounds for avian influenza neuraminidase. Journal of medicinal chemistry. 2008 Jul;51, no. 13:3878–94. doi: 10.1021/jm8001197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen YZ, Ung CY. Prediction of potential toxicity and side effect protein targets of a small molecule by a ligand-protein inverse docking approach. Journal of molecular graphics modelling. 2001;20, no. 3:199–218. doi: 10.1016/s1093-3263(01)00109-7. [DOI] [PubMed] [Google Scholar]
- 28.Hui-fang L, Qing S, Jian Z, Wei F. Evaluation of various inverse docking schemes in multiple targets identification. Journal of molecular graphics & modelling. 2010 Nov;29, no. 3:326–30. doi: 10.1016/j.jmgm.2010.09.004. [DOI] [PubMed] [Google Scholar]