

Abstract
The mechanistic imperatives for catalysis of deprotonation of α–carbonyl carbon by triosephosphate isomerase (TIM) are discussed. There is a strong imperative to reduce the large thermodynamic barrier for deprotonation of carbon to form an enediolate reaction intermediate; and, a strong imperative for specificity in the expression of the intrinsic phosphodianion binding energy at the transition state for the enzyme-catalyzed reaction. Binding energies of 2 and 6 kcal/mol, respectively, have been determined for formation of phosphite dianion complexes to TIM and to the transition state for TIM-catalyzed deprotonation of the truncated substrate glycolaldehyde [T. L. Amyes, J. P. Richard, Biochemistry 2007, 46, 5841]. We propose that the phosphite dianion binding energy, which is specifically expressed at the transition state complex, is utilized to stabilize a rare catalytically active loop-closed form of TIM. The results of experiments to probe the role of the side chains of Ile172 and Leu232 in activating the loop-closed form of TIM for catalysis of substrate deprotonation are discussed. Evidence is presented that the hydrophobic side chain of Ile172 assists in activating TIM for catalysis of substrate deprotonation through an enhancement of the basicity of the carboxylate side-chain of Glu167. Our experiments link the two imperatives for TIM-catalyzed deprotonation of carbon by providing evidence that the phosphodianion binding energy is utilized to drive an enzyme conformational change, which results in a reduction in the thermodynamic barrier to deprotonation of the carbon acid substrate at TIM compared with the barrier for deprotonation in water. The effects of a P168A mutation on the kinetic parameters for the reactions of whole and truncated substrates are discussed.
Introduction
Experimental studies on the mechanism of enzyme action have long been guided by the results of chemical studies to determine the mechanistic imperatives for enzymatic catalysis. Linus Pauling proposed that all catalysis, but most especially enzymatic catalysis, is due to the development of stabilizing interactions between the catalyst and the transition state.[1, 2] Wolfenden has calculated the transition state binding energies required to account for the rate acceleration for many important enzymatic reactions.[3] These binding energies are so large that they cannot be fully expressed at the Michaelis complex to product, because this would result in the rate of enzyme turnover being strongly limited by the rate of product dissociation.[4] This provides an imperative for enzymes to bind their transition states with a much higher affinity than substrate and product.[5]
Figure 1 shows the free energy profile for thermodynamically unfavorable deprotonation of carbon to form a carbanion reaction intermediate (Scheme 1), where the kinetic barrier ΔGf‡ is due largely to the thermodynamic barrier ΔG° for the proton transfer reaction.[6-9] This provides a strong imperative to reduce the kinetic barrier, through stabilization of the carbanion intermediate that results in a decrease in ΔG° (Figure 1).[8] Other strategies to reduce the kinetic barrier are useful, but not sufficient, to obtain the enormous rate accelerations observed for enzymatic catalysis of deprotonation of carbon. For example, the catalytic side chains at an enzyme catalyst of deprotonation of carbon should be optimally positioned to minimize the entropic barrier associated with the freezing of side chain motion,[10, 11] but such an “optimal alignment” of side chains will not normally affect the thermodynamic reaction barrier ΔG°. The width of the energy barrier for proton transfer reactions (Figure 1) is not much greater than the quantum mechanical wavelength for a C-H bond,[12] so that some hydrogen transfer events at an enzyme-bound substrate should proceed by tunneling through the barrier.[13, 14] However, such tunneling cannot reduce the barrier for deprotonation of a carbon acid below the thermodynamic reaction barrier.
Figure 1.
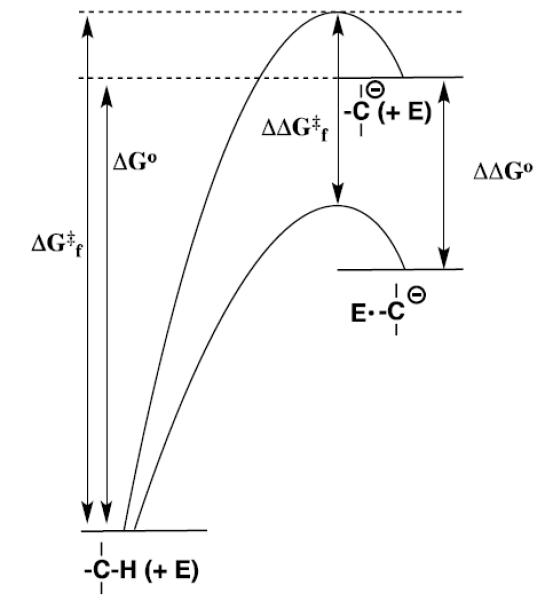
Reaction coordinate profiles for nonenzymatic and enzyme-catalyzed deprotonation of a weak carbon acid to form a carbanion, which show the effect of a reduction in the thermodynamic barrier to intermediate formation (ΔΔGo) on the activation barrier for the enzymatic reaction (ΔΔG‡f).
Scheme 1.
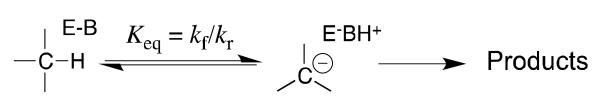
The stabilization of a carbanion intermediate relative to substrate at an enzyme active site will result in a substantial stabilization of the transition state for the uphill proton transfer, because the “late” reaction transition state should closely resemble the carbanion intermediate.[15] Ketosteroid isomerase (KSI, Scheme 2A)[16, 17] and triosephosphate isomerase (TIM, Scheme 2B)[18, 19] respond to the imperative for intermediate stabilization by providing amino acid side chains that selectively stabilize the respective enolate intermediates through the formation of hydrogen bonds or ion pairs to the enolate oxygen.[5]
Scheme 2.
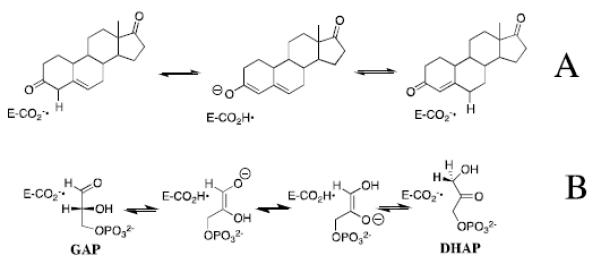
William P. Jencks proposed that enzyme selectivity in transition state binding extends beyond those atoms directly involved in the chemical reaction, and may include nonreactive substrate fragments.[4, 5] This mini-review briefly describes experiments which provide strong evidence that the transition state for TIM-catalyzed deprotonation of carbon is selectively stabilized by interactions with the stable phosphodianion of the whole substrates glyceraldehyde 3-phosphate (GAP) and dihydroxyacetone phosphate (DHAP); and, by interactions with phosphite dianion in catalysis of the reaction of the truncated substrate glycolaldehyde.[20, 21] We then consider the mechanism by which the interactions between TIM and phosphite dianion (HPO32−), are “switched-on” as the reactants proceed from the Michaelis complex to the reaction transition state,[5, 22, 23] and conclude with a presentation of the results of recent mutagenesis studies on TIM, which provide insight into the mechanisms for enzyme activation by dianions.
Enzyme Activation by Phosphite Dianion
Triosephosphate isomerase (TIM) catalyzes the stereospecific and reversible conversion of dihydroxyacetone phosphate (DHAP) to (R)-glyceraldehyde 3-phosphate (GAP), by a proton transfer mechanism through enzyme-bound cis-enediolate reaction intermediates (Scheme 2B).[22, 24-29] Roughly 80% of the rate acceleration for enzymatic catalysis of the aldose-ketose isomerization of (R)-glyceraldehyde 3-phosphate (GAP) by triosephosphate isomerase (TIM) is due to the 12 kcal/mol stabilization of the transition state by interactions with the terminal phosphodianion group of GAP.[30, 31] This raises the question of whether the interactions between TIM and the substrate dianion are utilized simply to anchor the bound substrate to the enzyme (Km effect), or if they activate TIM for catalysis of deprotonation of enzyme-bound substrate (kcat effect). The key to separating anchoring from activating interactions of a substrate dianion is to eliminate the anchoring connection at the substrate.[21, 32, 33] Enzyme activation may then be detected by comparing catalysis of reactions of a truncated substrate piece in the absence and presence of a putative dianion activator. In the case of TIM, we have examined catalysis of the reaction of the truncated substrate glycolaldehyde (GA), and the activation of this enzymatic reaction by HPO32−, because the earlier results of Ray and coworkers suggested that these pieces are good steric match for the whole substrates GAP and DHAP.[34]
We first showed that rabbit muscle TIM catalyzes the slow exchange of the α-carbonyl hydrogen of GA for deuterium from solvent D2O,[21] and later that the enzyme from chicken muscle catalyzes the full range of formal isomerization and deuterium exchange reactions of [1-13C]-GA to form [2-13C]-GA, [2-13C, 2-2H]-GA and [1-13C, 2-2H]-GA (Scheme 4),[35] that were initially characterized for the enzyme-catalyzed reactions of the substrates GAP and DHAP.[28, 29] The TIM-catalyzed reactions of GA and [1-13C]-GA are strongly activated by exogenous phosphite dianion.[21, 35] Figure 2 shows representative data for activation of the slow Trypanosoma brucei brucei (TbbTIM) catalyzed reaction of [1-13C]-GA by phosphite dianion.[20] We have characterized similar activation of TIMs from chicken muscle,[35] rabbit muscle,[21] and yeast.[36]
Scheme 4.
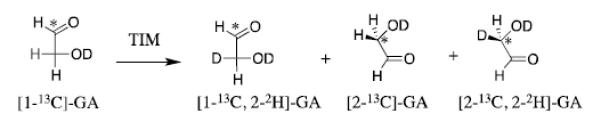
Figure 2.
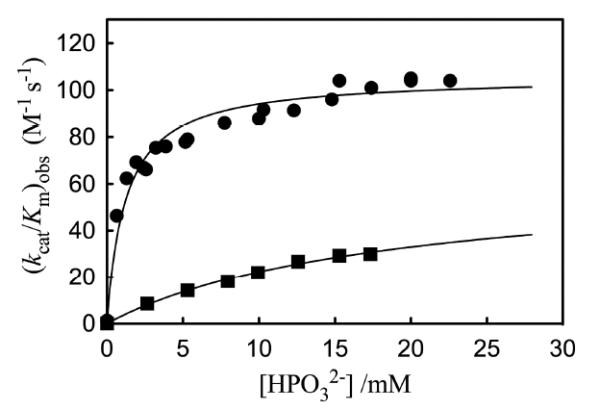
The dependence of the observed second-order rate constant (kcat/Km)obs for the proton transfer reactions of the free carbonyl form of [1-13C]-GA catalyzed by wildtype and L232A mutant Tbb TIM on the concentration of phosphite dianion in D2O, pD 7.0 and 25 °C. Key: (■) Wildtype Tbb TIM; (③) L232A mutant Tbb TIM. Reprinted with permission from Ref. 45. Copyright 2011 American Chemical Society.
An analysis of the kinetic data from Figure 2 provides the following kinetic parameters (Scheme 5): (kcat/Km)E = 0.07 M−1 s−1, the second-order rate constant for the unactivated TIM catalyzed reactions of [1-13C]-GA; (kcat/Km)E•HPi = 64 M−1 s−1, the limiting second-order rate constant at saturating phosphite dianion for the reaction catalyzed by the binary enzyme-phosphite dianion complex; Kd = 19 mM, the dissociation constant for breakdown of the enzyme-phosphite dianion complex; and, [(kcat/Km)E•HPi/Kd] = 3400 M−2 s−1 the third-order rate constant for enzyme activation by low concentrations of phosphite dianion.
Scheme 5.
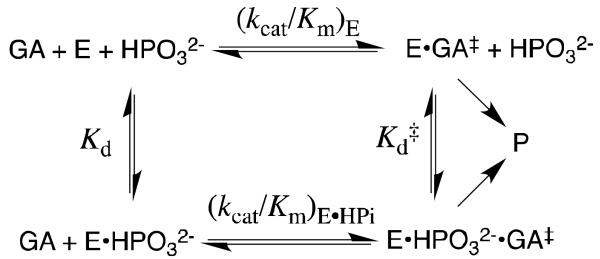
Scheme 5 shows that the strong activation of TIM by phosphite dianion is a direct result of the dianion specificity for transition state binding. The expression (kcat/Km)E•HPi/(kcat/Km)E = Kd/Kd‡ = 900 relates the magnitude of enzyme activation to dianion specificity for transition state binding (Scheme 5). The total stabilization of the transition state by interactions with phosphite dianion, -RTlnKd‡ = 6.4 kcal/mol, is 50% of the 12 kcal/mol dianion binding energy observed for the whole substrate GAP; and phosphite dianion binds to the transition state complex E•GA‡, with a 4.1 kcal/mol larger binding energy than for binding to free enzyme. The larger 12 kcal/mol dianion binding energy for the reaction of the whole substrate GAP, than the 6.4 kcal/mol phosphite dianion binding energy, represents the smaller entropic cost associated with the binding and unimolecular reaction of GAP, compared to the bimolecular reaction of the substrate pieces GA + HPO32−.[37]
Model for Enzyme Activation
The selectivity of phosphite dianion in binding to the transition state complex E•GA‡ for TIM catalyzed isomerization of GA might be due to direct stabilizing interactions between GA‡ and the dianion. However, the electrostatic interaction between phosphite dianion and the enediolate-like transition state for deprotonation of carbon are destabilizing and only weakly stabilizing intermolecular Van der Waals interactions are expected for these small polar molecules. Just a single intermolecular hydrogen bond between the C-2 hydroxyl of GA and phosphite dianion is possible; but, inspection of X-ray crystal structures shows that the substrate DHAP and other ligands adopt an extended conformation when bound to TIM.[38-43] A stabilizing intermolecular hydrogen bond will therefore not form at a transition state ternary complex E•GA•HPO32− that adopts a similar extended conformation.
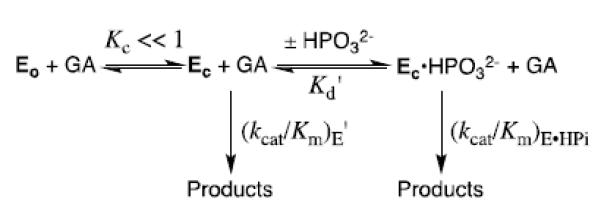
X-ray crystallographic analyses show that the binding of substrate DHAP [39, 40] and transition state analogs phosphoglycolate (PGA)[38, 41] and phosphoglycolohydroxamate (PGH)[42, 43] to TIM is accompanied by a large change in enzyme conformation, from a loop-open form that allows for fast ligand binding, to a loop-closed form that sequesters the ligand from interaction with bulk solvent. We have incorporated this enzyme conformational change into Scheme 6,[5, 22, 35, 44, 45] where TIM is shown to exist as a dominant inactive open form EO and the reaction is catalyzed by a rare active closed form EC, which shows selectivity in binding to both the transition state GA‡ and to phosphite dianion. Enzyme activation by the binding of HPO32− is proposed to occur because the active closed form EC is stabilized by interactions with the dianion activator.
Scheme 6.
The most prominent motion that occurs upon ligand binding to TIM is closure of an 11-residue phosphodianion gripper loop 6 that runs from Pro166 to Ala176 for TIM from chicken muscle, or from Pro168 to Ala178 for TIM from Trypanosoma brucei brucei.[46] There is also significant motion of loop 7 [Try208 to Ser211 for chicken muscle TIM], which involves mainly rotation about peptide bonds, as described below. The overall conformational change is driven energetically by the formation of hydrogen bonds between the ligand phosphodianion and backbone amides of Gly171 (loop 6) and Ser211 (loop 7). In addition, a network of interloop hydrogen bonds that involve amino acid side chains and backbone amides from loop 6 and loop 7 stabilizes the closed form of TIM. Wierenga has examined this complex ligand-driven conformational change in great detail, and his discussions of its role in catalysis have enriched the biochemical literature.[41, 46, 47] McDermott and coworkers have carried out elegant studies to examine the dynamics of closure of loop 6 over the enzyme active site,[48-50] while Loria and coworkers have probed the role in catalysis of hydrogen bonding interactions between loop 6 and loop 7.[51, 52]
The changes that occur upon ligand binding to TIM involve so many different interactions, that it is difficult to recognize those interactions critical to the process of enzyme activation by phosphite dianion. We were struck by the following observation of Wierenga; “In the closed form of loop 6, the side chain of Ile172 folds over the substrate, shielding it and the catalytic glutamate from bulk solvent. Glu167 and the substrate are also contacted by Leu232 (see Figure 3). In this way, the Glu167 side chain in the closed conformation is buried in a pocket formed by the side chains of Ile172 and Leu232”.[41] We also noticed that the carboxylate side chain of the active site base Glu165/167 is well solvated at unliganded TIM (Figure 4A), but that loop closure results in the extrusion of several water molecules to bulk solvent (Figure 4B). These observations prompted the proposal that the activation of TIM by the dianion-driven conformational change from EO to EC is partly or entirely the result of desolvation of the carboxylate side chain of Glu165/167, and the placement of this side chain in a clamp that consists of the hydrophobic side chains of Ile172 and Leu232 (Figure 3).[23]
Figure 3.
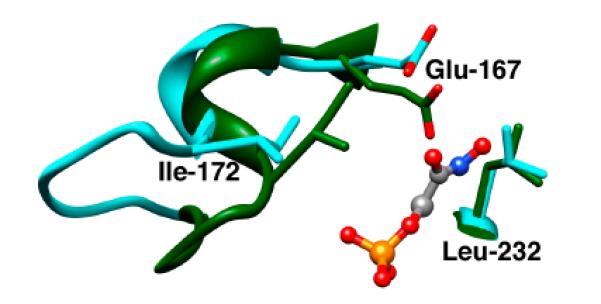
Models, from X-ray crystal structures, of the unliganded open (cyan, PDB entry 5TIM) and the PGH-liganded closed (green, PDB entry 1TRD) forms of TIM from Trypanosoma brucei brucei in the region of the enzyme active site. Reprinted with permission from Ref. 45. Copyright 2011 American Chemical Society.
Figure 4.
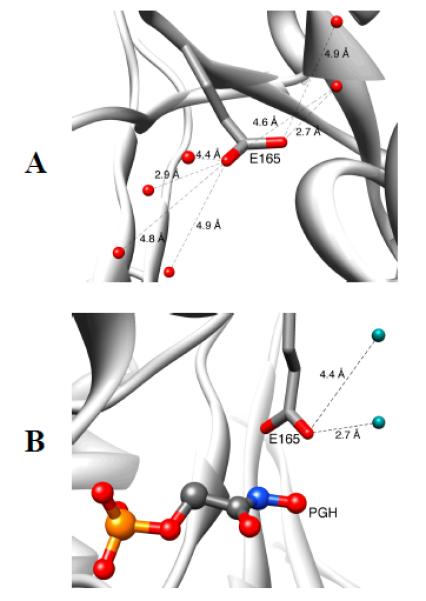
Models, from X-ray crystal structures, which show the open form of TIM from Trypanosoma brucei brucei (TbbTIM) where the carboxylate side chain of Glu167 is well solvated (Panel A, PDB entry 5TIM); and, the desolvated closed form of TIM liganded to the intermediate analog phosphoglycolohydroxamate (Panel B, PDB entry 2VXN).
I172A and L232A Mutants of TIM
We tested our proposal by replacing the hydrophobic side chains of Ile172 and Leu232 by the small −CH3 side chain of alanine.[44, 45] The I172A and L232A mutations of TbbTIM result in 105- and 5.5-fold reductions, respectively, in kcat/Km for enzyme-catalyzed isomerization of GAP, which shows that Ile172 plays a more important role than Leu232 in ensuring optimal catalysis of the reaction of the whole substrate. The I172A and L232A mutations, likewise, show different effects on the values of the kinetic parameters for the TIM-catalyzed reactions of the substrate pieces [1-13C]-GA and phosphite dianion. The I172A mutation results in > 20 and 280 fold decreases, respectively, in the second-order rate constants (kcat/Km)E and (kcat/Km)E•HPi for the unactivated and the phosphite dianion activated TIM-catalyzed reactions of [1-13C]-GA, but little change in Kd for breakdown of the binary enzyme-phosphite complex (Scheme 5). These results show that the side chain of Ile172 enhances, the reactivity of the free enzyme and the binary complex E•HPO32 toward catalysis of reactions of GA. By comparison, the I172A mutation results in essentially no change in Kd for binding of phosphite dianion to TIM, and only a small increase in Kd‡ for dianion binding to the transition state complex [E•HPO32−]‡, so that the side chain of Ile172 plays little or no role in stabilizing complexes between TIM and HPO32−.[44]
Figure 2 compares the effect of increasing [HPO32−] on (kcat/Km)obs for deprotonation of [1-13C]-GA catalyzed by wildtype and L232A mutant TbbTIM.[45] Our data show that the L232A mutation results in the following changes in the kinetic parameters for wildtype TbbTIM (the reaction of GA and HPO32−): a 17-fold increase in (kcat/Km)E for the TIM-catalyzed reactions of [1-13C]-GA in D2O; a 25-fold increase in (kcat/Km)E•HPi/Kd for reaction of the TIM-catalyzed reaction substrate pieces GA + HPO32−; and a 16-fold decrease in Kd for dissociation of HPO32− from TIM (Scheme 5). The mutation leads to only a small 35% increase in (kcat/Km)E•HPi, and an overall 11-fold decrease in the (kcat/Km)E•HPi/(kcat/Km)E that defines enzyme activation by HPO32− (Scheme 5).
TIM has by two criteria achieved perfection in catalysis of isomerization of GAP,[26] and mutations of wildtype TIM were therefore not expected to result in an increase in the efficiency for enzymatic catalysis of the reactions of truncated substrates. This increase in catalytic activity can be rationalized by eq 1-3, derived for Scheme 6 by assuming that phosphite dianion functions solely to hold TIM in an active closed conformation, so that (kcat/Km)E = (kcat/Km)E•HPi. These relationships predict that mutations which cause an increase in KC for conversion of inactive open enzyme EO to the active closed form EC will result in the observed increase in (kcat/Km)E, decrease in Kd and increase in [(kcat/Km)E•HPi]/Kd]. The effects of the L232A mutation are therefore consistent with a ca 1.7 kcal/mol stabilization of a catalytically active loop-closed enzyme (EC) relative to an inactive open form (EO) at L232A mutant compared to wildtype TbbTIM, which corresponds to a ca. 17-fold increase in KC (Scheme 6).
| (1) |
| (2) |
| (3) |
We have proposed that the role of Leu232 in catalysis by TIM is to ensure a large barrier to the enzyme conformational change,[5, 44, 45] so as to maximize enzyme activation observed upon binding of the substrate phosphodianion. This has the effect of ensuring a large enzyme activation by phosphite dianion and provides a rationalization for the decrease from the 900-fold activation of the wildtype TIM-catalyzed reaction of GA by phosphite dianion to the smaller 80-fold activation of the L232A TIM mutant.
Origin and Magnitude of the Enhanced Basicity of the Catalytic Side Chain for Glu165/167
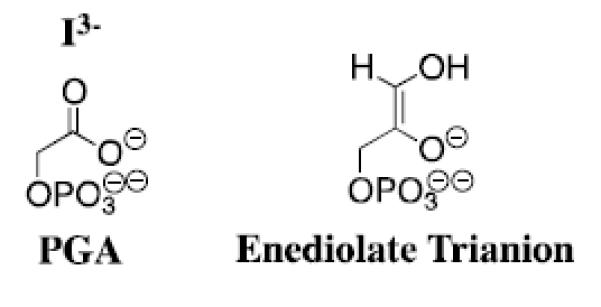
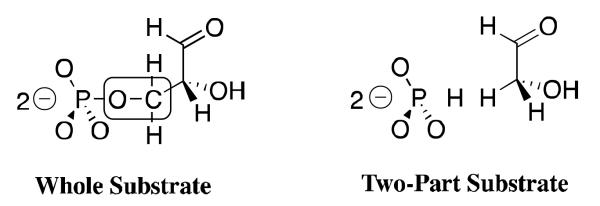
Wolfenden and coworkers showed that phosphoglycolate trianion (PGA, Scheme 7) is a high affinity competitive inhibitor of TIM, and proposed that the high affinity of this inhibitor reflects the high specificity of TIM for binding the transition state for deprotonation of DHAP, which presumably resembles the enediolate reaction intermediate (Scheme 7).[53, 54] The 31P and 13C NMR chemical shifts of PGA at the enzyme-inhibitor complex are essentially the same as for free PGA trianion.[55, 56] However the binding of PGA trianion is coupled to the net uptake of a proton by the inhibitor complex (Scheme 8A).[57] It can be concluded that the binding of PGA trianion to TIM results in an increase in the basicity of a catalytic side chain. The X-ray crystal structure of the TIM•PGA3− complex (Figure 5) shows the complex to be stabilized by a hydrogen bond between the carboxylic acid side chain of Glu165/167 and the carboxylate anion of PGA3−.[38, 41] These results are expected for an enediolate intermediate analog, because the side chain of Glu165/167 exists in the basic form for free enzyme, in order to catalyze deprotonation of the carbon acid substrate, and, in the protonated form at the enediolate intermediate complex (Scheme 8B).[58]
Scheme 7.
Scheme 8.
Figure 5.
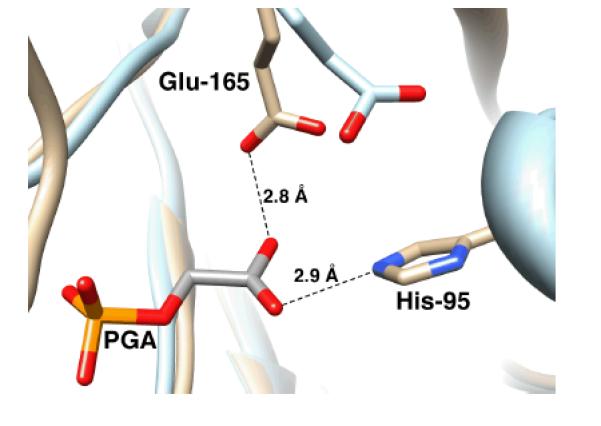
Models, from X-ray crystal structures, of the active site of unliganded yeast TIM (light blue, PDB entry 1YPI) and yeast TIM complexed with PGA (tan, PDB entry 2YPI). Ligand binding is accompanied by a 2 Å shift in the position of the carboxylate side chain of Glu-165 towards the bound ligand. Reprinted with permission from Ref. 59. Copyright 2013 American Chemical Society.
If the binding of PGA to TIM is accompanied by the uptake of a proton by the carboxylate side chain of Glu165/167, then binding must induce a large increase in the side chain basicity. This raises the question of the origin and magnitude of the enhancement in side-chain basicity.[59] Figure 6 shows the pH-profile for values of log Ki for competitive inhibition of wildtype TbbTIM by PGA trianion. The values of the inhibition constant Ki are first-order in [H+], consistent with the formal binding of PGA trianion plus a proton at TIM (Scheme 8A). The extended linear pH profile in Figure 6 shows that the carboxylate side chain at unliganded wildtype TbbTIM is deprotonated at pH ≥ 4.9, but is protonated at the inhibitor complex (EH•I3−) at pH ≤ 9.3 (Scheme 9). The profile is therefore consistent with a ca. 6-unit increase in the pKa of the catalytic side chain of Glu165/167 from pKa ≈ 4 at low pH (at the free enzyme),[57] to pH ≥ 9.3 at the inhibitor complex (EH•I3−) (Scheme 9).[59] A similar increase in the basicity of this side chain on proceeding from free TIM to the enediolate reaction intermediate will contribute to the observed rate enhancement for TIM-catalyzed deprotonation of GAP, if the enhanced side chain basicity is expressed at the transition state for the TIM-catalyzed deprotonation of GAP.[58, 60]
Figure 6.
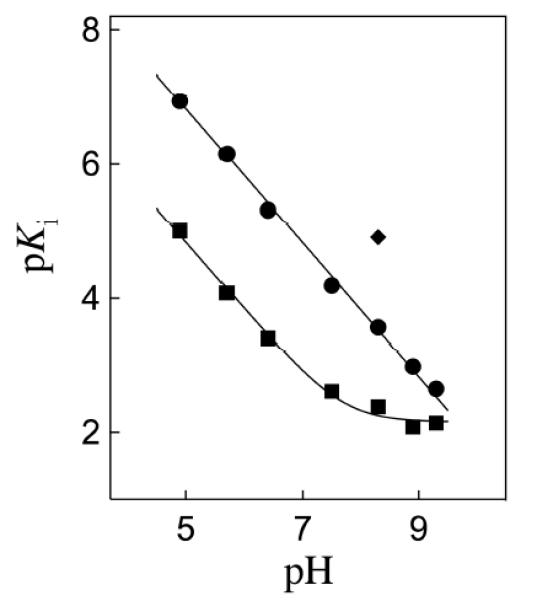
The pH dependence of the dissociation constant Ki (M) for breakdown of the complex between I3− and wildtype TbbTIM (③), I172A mutant TbbTIM (■), and L232A mutant TbbTIM (◆). Reprinted with permission from Ref. 59. Copyright 2013 American Chemical Society
Scheme 9.

Figure 6 shows data for inhibition of I172A and L232A mutant forms of TbbTIM by PGA3−. The fit of the data for I172A mutant TIM to the appropriate equation derived for Scheme 9 gives pKEHI = 7.7 for ionization of the EH•I3− complex. The effect of the I172A mutation of the pH profile for wildtype TIM is consistent with the conclusion that: “the ~2 pK unit higher pKa of the carboxylate group of Glu-167 at the EH•I3− complex for wildtype compared with I172A mutant TbbTIM results from the "clamping" action of the side chain of Ile172 that leads to destabilization of E•I3− by unfavorable electrostatic interactions between the neighboring carboxylate anions of Glu-167 and bound I3−, and stabilization of EH•I3− by the formation of a hydrogen bond between the carboxylate group of I3− and the protonated side chain of Glu-167. Thus the bulky hydrophobic side chain of Ile-172 restricts the movement of the basic carboxylate side chain of Glu-167 relative to I3− at E•I3−, resulting in an increase in the driving force for protonation to give EH•I3−. The I172A mutation then lifts this restriction, allowing separation of the carboxylate anions of the enzyme and bound I3− and relief of the destabilizing electrostatic interactions (Figure 3).”[59]
By contrast, the L232A mutation results in a 20-fold increase in the affinity of TbbTIM for PGA trianion at pH 8.3 (Figure 6). This result is consistent with the proposal that the L232A mutation results in a substantial increase in the concentration of a catalytically active closed form of TIM (EC) relative to the open form (EO, Scheme 6); and, that the closed enzyme shows specificity for binding the reaction transition state and the transition state analog PGA. The net result of the enhanced and reduced affinity, respectively, of the L232A and I172A mutant enzymes for PGA is a ≈ 1000-fold tighter binding of PGA trianion to the L232A compared with the I172A mutant enzyme.[59]
P168A Mutant
Movement of loop 6 from the open to closed conformation is accompanied by 90° and 180° rotation, respectively, of the peptide bonds at Gly211-Gly212 and at Gly212-Ser213 of loop 7 (Figure 7A). This motion leads to a clash between the carbonyl oxygen of Gly211 and the side chain of Pro168, that is relieved by movement of the Glu167-Pro168 depeptide unit that places the side chain of Glu167 in the catalytically active “swung-in” position.[46, 61] Wierenga and coworkers have prepared the P168A mutant of TbbTIM and determined X-ray crystal structures for the unliganded and liganded forms of the mutant enzyme.[61] The mutation causes only small changes in the structure of the unliganded TIM. The steric clash between the carbonyl oxygen of Gly211 and the side chain of Pro168 is absent for the P168A, and the only significant effect of the P168A mutation on the structure of the closed E•PGA complex is to cause the carboxylate side chain to remain in the swung-out conformation for the unliganded enzyme (Figure 7B).[61]
Figure 7.
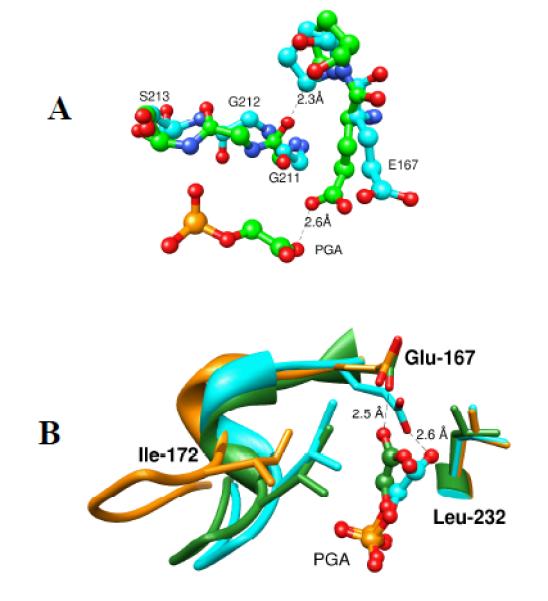
A. Ball and stick models, from X-ray crystal structures, of the active sites of unliganded wildtype TbbTIM (cyan, PDB entry 5TIM) and PGA-liganded TIM from Leishmania mexicana (green, PDB entry 1N55). The figure shows the 90° and 180° rotation, respectively, about the peptide bonds at Gly211-Gly212 and at Gly212-Ser213 of loop 7 as loop 6 moves from the open to closed conformation. The hypothetical distance between the carbonyl oxygen of Gly211 of liganded TIM and the side chain of Pro168 for the unliganded enzyme is 2.3 Å. B. Superposition of models, from the X-ray crystal structures, which show the active sites of unliganded wildtype TbbTIM (gold, PDB entry 5TIM), the PGA-liganded TIM from Leishmania mexicana (cyan, PDB entry 1N55), and PGA-liganded P168A mutant TbbTIM (green, PDB entry 2J27).
The P168A mutation was found to result in a 60-fold decrease in kcat but little change in Km for TbbTIM-catalyzed isomerization of GAP. It was therefore concluded that the ca. 2 Å shift in the position of the carboxylate side of Glu-167 at the P168A mutant results in a significant falloff in enzymatic activity.[61] We have examined the effect of the P168A mutation of TbbTIM on enzyme activation by phosphite dianion.[62] This mutation results in 50-fold and 80-fold decreases, respectively, in (kcat/Km)E and (kcat/Km)E•HPi for deprotonation of the truncated substrate [1-13C]-GA by the free enzyme and the E•HPO32− complex (Scheme 5).[62] By contrast, the mutation has only a small effect on the phosphite dianion dissociation constants Kd and Kd‡ for dianion binding to free enzyme and the transition state complex, respectively (Scheme 5). Consequently, the magnitude of dianion activation of the P168A mutant by the binding of phosphite dianion (600-fold) is nearly the same as observed for wildtype TIM (900-fold).[62]
The observed decreases in (kcat/Km) for TIM-catalyzed isomerization of GAP, and in (kcat/Km)E and (kcat/Km)E•HPi for TIM-catalyzed reactions of [1-13C]-GA reflect a ca. 2 – 3 kcal/mol increase in the activation barrier for deprotonation of bound carbon acid substrates associated with the shift in the side-chain base of Glu167 away from its optimally aligned “swung-in” position at wildtype TIM.[40] The P168A mutation has no effect on the important protein-phosphodianion interactions, so that no change in the intrinsic phosphite dianion binding energy is observed.
A simple interpretation for these results is that catalysis of proton transfer and dianion activation of this proton transfer reaction occurs at separate sites on the protein catalyst; and, that the effect of the P168A mutation is localized to the former catalytic site. The catalytic and activating sites at TIM must interact to the extent that dianion binding at the activator site triggers a conformational change that extends to the catalytic site. We have proposed that GA and HPO32− bind essentially independently at TIM and that: (a) the binding of HPO32− at the dianion site plays the passive role of stabilizing the preexisting active enzyme EC, but does not affect the structure or intrinsic catalytic activity of EC. (b) The binding of GA to EC has little or no effect on the affinity of HPO32− at the second site. The result of the independent binding of these ligands is to lock TIM into its catalytically active closed form.[62]
Concluding Remarks
The primary mechanistic imperative for TIM is that the protein catalyst reduce the large thermodynamic barrier observed for deprotonation of the carbon acid in water. We have provided evidence that this is accomplished through a ligand-driven enzyme conformational change that results in desolvation of the carboxylate side chain and a large enhancement of side chain basicity. A second imperative is that a portion of the stabilizing binding interactions of the nonreacting phosphodianion of substrate be specifically expressed at the transition state for the TIM catalyzed reaction. We propose that this imperative is met through the utilization of dianion binding interaction to stabilize an active closed form of the catalyst, present at a low concentration, which shows specificity in binding of the transition state. The common thread in satisfying these imperatives is the utilization of the intrinsic phosphodianion binding energy to drive an unfavorable enzyme conformational change, because the conformational change creates an enzyme active site where the thermodynamic barrier for deprotonation of bound substrate is reduced compared to the barrier in water (Figure 1).
These mechanistic themes are central to the interpretation of results from studies on the mechanism by which orotidine monophosphate decarboxylase (OMPDC) utilizes the 12 kcal/mol intrinsic binding energy of the phosphodianion of OMP in the stabilization of the vinyl carbanion intermediate of the enzyme-catalyzed decarboxylation reaction.[33, 63-65] The relatively large distance between the phosphodianion of OMP and the reactive orotate ring, compared with distance between the phosphodianion of GAP and the α–carbonyl hydrogen poses additional challenges for determining the mechanism by which binding interactions at the phosphodianion binding site are utilized in the stabilization of a transition state that forms the distant orotate binding site.
Scheme 3.
Acknowledgment
We acknowledge the National Institutes of Health Grant GM39754 for generous support of this work. This mini-review is based on a lecture given by JPR at the 12th Latin American Conference on Physical Organic Chemistry (CLAFQO-12), Foz do Iguaçu, PR, Brazil
REFERENCES
- 1.Pauling L. Nature. 1948;161:707. doi: 10.1038/161707a0. [DOI] [PubMed] [Google Scholar]
- 2.Pauling L. Chem. & Eng. News. 1946;24:1375. [Google Scholar]
- 3.Wolfenden R, Snider MJ. Acc. Chem. Res. 2001;34:938. doi: 10.1021/ar000058i. [DOI] [PubMed] [Google Scholar]
- 4.Jencks WP. Adv. Enzymology Relat. Areas Mol. Biol. 1975;43:219. doi: 10.1002/9780470122884.ch4. [DOI] [PubMed] [Google Scholar]
- 5.Amyes TL, Richard JP. Biochemistry. 2013;52:2021. doi: 10.1021/bi301491r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Amyes TL, Richard JP. In: Hydrogen-Transfer Reactions. Hynes JT, Klinman JP, Limbach H-H, Schowen RL, editors. Vol. 3. WILEY-VCH; Weinheim: 2007. p. 949. [Google Scholar]
- 7.Richard JP, Amyes TL. Cur. Opin. Chem. Biol. 2001;5:626. doi: 10.1016/s1367-5931(01)00258-7. [DOI] [PubMed] [Google Scholar]
- 8.Richard JP. Biochemistry. 2013;52:2009. doi: 10.1021/bi3017119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Richard JP, Amyes TL. Bioorg. Chem. 2004;32:354. doi: 10.1016/j.bioorg.2004.05.002. [DOI] [PubMed] [Google Scholar]
- 10.Warshel A. J. Biol. Chem. 1998;273:27035. doi: 10.1074/jbc.273.42.27035. [DOI] [PubMed] [Google Scholar]
- 11.Cannon WR, Benkovic SJ. J. Biol. Chem. 1998;273:26257. doi: 10.1074/jbc.273.41.26257. [DOI] [PubMed] [Google Scholar]
- 12.Bell RP. The Proton in Chemistry. Second Cornell University Press; Ithaca, New York: 1973. [Google Scholar]
- 13.Alhambra C, Gao J, Corchado JC, Villà J, Truhlar DG. J. Am. Chem. Soc. 1999;121:2253. [Google Scholar]
- 14.Truhlar DG, Gao J, Alhambra C, Garcia-Biloca M, Corchado J, Sanchez ML, Villa J. Acc. Chem. Res. 2002;35:341. doi: 10.1021/ar0100226. [DOI] [PubMed] [Google Scholar]
- 15.Hammond GS. J. Am. Chem. Soc. 1955;77:334. [Google Scholar]
- 16.Herschlag D, Natarajan A. Biochemistry. 2013;52:2050. doi: 10.1021/bi4000113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schwans JP, Sunden F, Gonzalez A, Tsai Y, Herschlag D. J. Am. Chem. Soc. 2011;133:20052. doi: 10.1021/ja208050t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Go MK, Koudelka A, Amyes TL, Richard JP. Biochemistry. 2010;49:5377. doi: 10.1021/bi100538b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Go MK, Amyes TL, Richard JP. J. Am. Chem. Soc. 2010;132:13525. doi: 10.1021/ja106104h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Malabanan MM, Go MK, Amyes TL, Richard JP. Biochemistry. 2011;50:5767. doi: 10.1021/bi2005416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Amyes TL, Richard JP. Biochemistry. 2007;46:5841. doi: 10.1021/bi700409b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Richard JP. Biochemistry. 2012;51:2652. doi: 10.1021/bi300195b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Malabanan MM, Amyes TL, Richard JP. Cur. Opin. Struct. Biol. 2010;20:702. doi: 10.1016/j.sbi.2010.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Knowles JR. Philos. Trans. Roy. Soc., London Ser. B. 1991;332:115. doi: 10.1098/rstb.1991.0039. [DOI] [PubMed] [Google Scholar]
- 25.Knowles JR. Nature. 1991;350:121. doi: 10.1038/350121a0. [DOI] [PubMed] [Google Scholar]
- 26.Knowles JR, Albery WJ. Acc. Chem. Res. 1977;10:105. [Google Scholar]
- 27.Rieder SV, Rose IA. J. Biol. Chem. 1959;234:1007. [PubMed] [Google Scholar]
- 28.O'Donoghue AC, Amyes TL, Richard JP. Biochemistry. 2005;44:2622. doi: 10.1021/bi047953k. [DOI] [PubMed] [Google Scholar]
- 29.O'Donoghue AC, Amyes TL, Richard JP. Biochemistry. 2005;44:2610. doi: 10.1021/bi047954c. [DOI] [PubMed] [Google Scholar]
- 30.Amyes TL, O'Donoghue AC, Richard JP. J. Am. Chem. Soc. 2001;123:11325. doi: 10.1021/ja016754a. [DOI] [PubMed] [Google Scholar]
- 31.Richard JP. J. Am. Chem. Soc. 1984;106:4926. [Google Scholar]
- 32.Tsang W-Y, Amyes TL, Richard JP. Biochemistry. 2008;47:4575. doi: 10.1021/bi8001743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Amyes TL, Richard JP, Tait JJ. J. Am. Chem. Soc. 2005;127:15708. doi: 10.1021/ja055493s. [DOI] [PubMed] [Google Scholar]
- 34.Ray WJ, Jr., Long JW, Owens JD. Biochemistry. 1976;15:4006. doi: 10.1021/bi00663a015. [DOI] [PubMed] [Google Scholar]
- 35.Go MK, Amyes TL, Richard JP. Biochemistry. 2009;48:5769. doi: 10.1021/bi900636c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhai X. 2013 Unpublished results. [Google Scholar]
- 37.Jencks WP. Proc. Natl. Acad. Sci. 1981;78:4046. doi: 10.1073/pnas.78.7.4046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lolis E, Petsko GA. Biochemistry. 1990;29:6619. doi: 10.1021/bi00480a010. [DOI] [PubMed] [Google Scholar]
- 39.Alber T, Banner DW, Bloomer AC, Petsko GA, Phillips D, Rivers PS, Wilson IA. Philos. Trans. R. Soc. London, Ser. B. 1981;293:159. doi: 10.1098/rstb.1981.0069. [DOI] [PubMed] [Google Scholar]
- 40.Jogl G, Rozovsky S, McDermott AE, Tong L. Proc. Natl. Acad. Sci. 2003;100:50. doi: 10.1073/pnas.0233793100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kursula I, Wierenga RK. J. Biol. Chem. 2003;278:9544. doi: 10.1074/jbc.M211389200. [DOI] [PubMed] [Google Scholar]
- 42.Davenport RC, Bash PA, Seaton BA, Karplus M, Petsko GA, Ringe D. Biochemistry. 1991;30:5821. doi: 10.1021/bi00238a002. [DOI] [PubMed] [Google Scholar]
- 43.Alahuhta M, Wierenga RK. Prot.: Strut., Func., Bioinf. 2010;78:1878. [Google Scholar]
- 44.Malabanan MM, Koudelka AP, Amyes TL, Richard JP. J. Am. Chem. Soc. 2012;134:10286. doi: 10.1021/ja303695u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Malabanan MM, Amyes TL, Richard JP. J. Am. Chem. Soc. 2011;133:16428. doi: 10.1021/ja208019p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wierenga RK. Cell. Mol. Life Sci. 2010;67:3961. doi: 10.1007/s00018-010-0473-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kursula I, Salin M, Sun J, Norledge BV, Haapalainen AM, Sampson NS, Wierenga RK. Protein Engineering, Design & Selection. 2004;17:375. doi: 10.1093/protein/gzh048. [DOI] [PubMed] [Google Scholar]
- 48.Desamero R, Rozovsky S, Zhadin N, McDermott A, Callender R. Biochemistry. 2003;42:2941. doi: 10.1021/bi026994i. [DOI] [PubMed] [Google Scholar]
- 49.Rozovsky S, Jogl G, Tong L, McDermott AE. J. Mol. Biol. 2001;310:271. doi: 10.1006/jmbi.2001.4673. [DOI] [PubMed] [Google Scholar]
- 50.Williams JC, McDermott AE. Biochemistry. 1995;34:8309. doi: 10.1021/bi00026a012. [DOI] [PubMed] [Google Scholar]
- 51.Wang Y, Berlow RB, Loria JP. Biochemistry. 2009;48:4548. doi: 10.1021/bi9002887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Berlow RB, Igumenova TI, Loria JP. Biochemistry. 2007;46:6001. doi: 10.1021/bi700344v. [DOI] [PubMed] [Google Scholar]
- 53.Wolfenden R. Acc. Chem. Res. 1972;5:10. doi: 10.1021/ar000058i. [DOI] [PubMed] [Google Scholar]
- 54.Wolfenden RV. Biochemistry. 1970;9:3404. doi: 10.1021/bi00819a018. [DOI] [PubMed] [Google Scholar]
- 55.Campbell ID, Jones RB, Kiener PA, Waley SG. Biochem. J. 1979;179:607. doi: 10.1042/bj1790607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Campbell ID, Jones RB, Kiener PA, Richards E, Waley SC, Wolfenden R. Biochem. Biophys. Res. Commun. 1978;83:347. doi: 10.1016/0006-291x(78)90438-2. [DOI] [PubMed] [Google Scholar]
- 57.Hartman FC, LaMuraglia GM, Tomozawa Y, Wolfenden R. Biochemistry. 1975;14:5274. doi: 10.1021/bi00695a007. [DOI] [PubMed] [Google Scholar]
- 58.Richard JP. Biochemistry. 1998;37:4305. doi: 10.1021/bi972655r. [DOI] [PubMed] [Google Scholar]
- 59.Malabanan MM, Nitsch-Velasquez L, Amyes TL, Richard JP. J. Am. Chem. Soc. 2013;135:5978. doi: 10.1021/ja401504w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Richard JP, Huber RE, Heo C, Amyes TL, Lin S. Biochemistry. 1996;35:12387. doi: 10.1021/bi961029b. [DOI] [PubMed] [Google Scholar]
- 61.Casteleijn MG, Alahuhta M, Groebel K, El-Sayed I, Augustyns K, Lambeir AM, Neubauer P, Wierenga RK. Biochemistry. 2006;45:15483. doi: 10.1021/bi061683j. [DOI] [PubMed] [Google Scholar]
- 62.Zhai X, Amyes TL, Loria JP, Wierenga RK, Richard JP. Biochemistry. 2013;52 doi: 10.1021/bi401019h. Submitted for Publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Amyes TL, Ming SA, Goldman LM, Wood BM, Desai BJ, Gerlt JA, Richard JP. Biochemistry. 2012;51:4630. doi: 10.1021/bi300585e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Goryanova B, Amyes TL, Gerlt JA, Richard JP. J. Am. Chem. Soc. 2011;133:6545. doi: 10.1021/ja201734z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Amyes TL, Wood BM, Chan K, Gerlt JA, Richard JP. J. Am. Chem. Soc. 2008;130:1574. doi: 10.1021/ja710384t. [DOI] [PMC free article] [PubMed] [Google Scholar]