

Abstract
Objective
Regulator of G-protein Signaling (RGS) proteins inhibit chemokine signaling by desensitizing G-protein coupled receptor signals. The mechanisms by which RGS13 promotes the generation of pathogenic autoantibodies in germinal centers (GC) were determined using BXD2-Rgs13−/− mice.
Methods
Confocal and light microscopy imaging was used to determine the location of cells that express RGS13 and activation-induced cytidine deaminase (AID) in the spleen and the number of plasmablasts. The levels of GC and plasma cell program transcripts in GC B cells were determined by quantitative real-time PCR. Differential IL-17-mediated expression of Rgs13 in GC versus non-GC B cells was analyzed using A20 versus 70Z/3 B cells.
Results
In spleens of BXD2 mice, RGS13 was mainly expressed by GC B cells and was stimulated by IL-17 but not IL-21. IL-17 upregulated Rgs13 in A20 GC but not 70Z/3 non-GC B cells. BXD2-Rgs13−/− mice exhibited smaller GCs, lower AID levels, suggesting lower somatic hypermutation and affinity maturation. There were, however, increased IgMbright plasmablasts, upregulation of plasma program genes Irf4, Blimp1, Xbp1 and pCREB target genes Fosb and Obf1, with down-regulation of GC program genes Aicda, Pax5 and Bach2 in GC B cells of BXD2-Rgs13−/− mice. BXD2-Rgs13−/− mice showed lower titers of IgG autoantibodies and IgG deposits in the glomeruli, suggesting reduced autoantibody pathogenicity.
Conclusion
RGS13 deficiency is associated with reduction in GC program genes and exit of less pathogenic IgM plasmablasts in BXD2 mice. Prolonged GC program, mediated by upregulation RGS13, enhanced AID expression and enabled generation of pathogenic autoantibodies in autoreactive GCs.
INTRODUCTION
The production of pathogenic autoantibodies in SLE (1) and RA (2, 3) has been extensively studied. Bootsma et al have shown that disease relapse in SLE was associated with a rise in IgG anti-dsDNA but not IgM anti-dsDNA (4). In addition, non-pathogenic autoantibodies have been extensively observed in the rheumatic diseases as elevated levels of anti-Ro or anti-CCP can be observed for many years before the onset of SLE (5) or rheumatoid arthritis (RA) (6, 7) and serum autoantibodies are not directly correlated with disease activity. However, the mechanisms of production of pathogenic vs. non-pathogenic autoantibodies are not known.
A key event for transition from non-pathogenic to pathogenic autoantibodies is somatic hypermutation (SHM) and class switch recombination (CSR) that regulated by activation-induced cytidine deaminase (AID). Diamond and colleagues (8) showed that IgM anti-dsDNA antibodies from peripheral blood from a patient with SLE can be derived from germline DNA that does not necessarily recognize self-antigens, but SHM enables anti-DNA recognition (1). Jiang et al (9) have shown that MRL-Faslpr/lpr mice that exhibit a heterozygous genotype for AID exhibit impaired SHM, affinity maturation, lower levels of specific anti-dsDNA and delayed development of renal disease, suggesting that AID levels impact the SHM and CSR and directly implicate affinity maturation of autoantibodies in autoimmunity. We have previously shown that BXD2 mice exhibit large, well-formed and numerous germinal centers (GCs) with high expression of AID in B cells (10–12). Importantly, BXD2 AID-dominant negative (AID-DN) Tg mice that express an AID with mutations in the catalytic domain and the PKA binding site exhibit decreased SHM, CSR, decreased development of autoantibodies and decreased autoimmune disease (13). Together these results indicate that upregulation of AID, leading to increased SHM and CSR is a crucial event to development of pathogenic autoantibodies.
Although AID plays a central role to promote development of pathogenic autoantibodies, the mechanism for the high expression of AID in autoreactive GCs remains unclear. There is, however, an extensive literature on the role of T cells to promote GC development (14, 15) and defects in GC selection has been shown to be operative in SLE (16, 17). IL-4, which is has been described to induce AID expression, does not appear to be upregulated in autoreactive T cells or in SLE (18, 19). Interestingly, although IL-21, the key cytokine produced by follicular T helper cells, has been shown to upregulate AID, a main function of IL-21 was shown to promote plasma B cell differentiation and it does not help B-cell SHM (20). BXD2 mice develop a lupus-like disease with high titers of high-affinity, class-switched autoantibodies and glomerulonephritis (10–12). We have previously shown that TH17 CD4 T cells in BXD2 mice are essential for development of large, numerous GCs that produce highly pathogenic autoantibodies (11). Further, IL-17 does not directly affect BCR or anti-CD40-induced B cell proliferative responses (21) and thus, IL-17-mediated development of autoreactive GC differs from the effects of IL-21 (20). Instead, IL-17 induces expression of regulator of G-protein signaling 13 (RGS13), which retards the B-cell chemotaxis response to CXCL12 and CXCL13. RGS13 is a critical GTPase accelerator (GTPase-activating protein) for Gα subunits that can control the magnitude and duration of the chemokine receptor signals (22, 23). Importantly, the CD4 T cell-B cell interaction promoted by IL-17 and upregulation of RGS13 was strongly needed for AID upregulation since B cells from BXD2-Il17ra−/− mice exhibited dramatically smaller GCs and reduced levels of AID (21).
To enable analysis of the mechanisms by which IL-17-mediated upregulation of RGS13 influences the generation of pathogenic autoantibodies, we generated BXD2-Rgs13−/− mice. We found that RGS13 expression occurred exclusively in GC B cells and its expression in BXD2 B cells was stimulated by IL-17 but not IL-21. Interestingly, although a deficiency of RGS13 does not completely block the formation of GC structure, it resulted in an increase in IgMhi plasmablasts as well as increased expression of plasma program genes and decreased expression of GC program genes. AID expression in situ was significantly attenuated in the GC B cells of BXD2-Rgs13−/− mice even though equal numbers of PNA+ B cells were found surrounding each CD4+ T cell in the spleen of BXD2-Rgs13−/− versus BXD2 mice. Collectively, these results propose a novel mechanism of generation of pathogenic autoantibodies as a result of IL-17-induced RGS13 which prolonged GC to plasma cell transition and promoted AID-mediated SHM and CSR.
MATERIALS AND METHODS
Mice
Female homozygous C57BL/6J (B6), BXD2 recombinant inbred mice were obtained from The Jackson Laboratory; B6-Rgs13−/− mice were provided by Dr. Kirk Druey at NIH. BXD2-Rgs13−/− were generated by backcrossing B6-Rgs13−/− mice, with BXD2 mice for > seven generations. All mouse procedures were approved by the University of Alabama at Birmingham Institutional Animal Care and Use Committee.
Confocal Imaging Analysis
Spleens from mice were collected, embedded in Frozen Tissue Media (Fisher Scientific) and snap-frozen in 2-methylbutane. Frozen sections (8 µm thick) were fixed and processed as we previous described (21). The following primary antibodies conjugated to Alexa dyes were used to stain spleen tissues for confocal image analysis: biotin-PNA (Vector Laboratory) to Alexa 350–streptavidin (Invitrogen); anti-IgM to Alexa 555; anti-AID (mAID-2, eBioscience) to Alexa 555, anti-RGS13 (N1C3-2, GeneTex, Irvine, CA) to Alexa 647, and anti-CD4 (RM4-5, Invitrogen) to Alexa 488 or 647.
Confocal imaging intensity analysis was carried out using the 1.4 version of the Image J software developed by the U.S. National Institutes of Health and available on the Internet at http://rsb.info.nih.gov/nih-image/). Background intensity was subtracted for each image. The single color intensity plot was generated using the Image J interactive 3D surface plot module. The localization of RGS13 and AID staining was determined by analysis of the color intensity of each relative to the color intensity of PNA, IgM or CD4. The area of the marginal zone (MZ) was defined as the IgM+ area outside the marginal sinus (MS); the area of the FO as the IgM+ area inside the MS but excluding the mantle area; the mantle area as the IgM+ area between the GC and MZ (24).
Immunohistochemical (IHC) staining
IHC staining was carried out as described previously (13) using 4-µM sections of formalin-fixed tissues. Sections were incubated with HRP-linked anti-mouse IgM (Southern Biotech) and/or anti-mouse IgG (Southern Biotech) then treated with 3,3',5,5'-tetramethylbenzidine (Sigma) and counterstained with methyl green. Stained sections were viewed using an Olympus BX41 System Microscope. Quantitation of IgG+ glomeruli was carried out using the ImageJ program as described above.
Flow cytometry analysis and sorting
Flow cytometry was carried out on fluorescently-labeled single-cell suspensions as we described previously (13, 21). Unless specified, all fluorochrome conjugated antibodies were obtained from Biolegend (San Diego, CA). For the detection or isolation of GC B cells, cells were labeled with biotin-conjugated PNA (Vector Laboratories), APC-conjugated GL-7 (GL7 eBioscience), PE-anti-Fas (Jo2, BD-Pharmingen), APC-conjugated anti-mouse B220 (RA3-6B2), biotin-anti-CD138 (281-2), Alexa700- or pacific blue-anti-CD19 (clone 6D5), and Pacific blue- or Alexa647-conjugated streptavidin (SA).
For sorting of subsets of B cells, pan B cells were first enriched from single-cell spleen preparations using anti-CD19 MACS Beads (Miltenyi Biotech Inc, Auburn, CA). The FO (IgMloCD1dloCD21loCD23hi), MZ (IgMhiCD1dhiCD21hiCD23lo), and MZ-precursor (MZ-P) (IgMhiCD1dhiCD21hiCD23hi) subsets of B cells were sorted and gated based on the method that we described previously (25, 26).
Cytokine stimulation of B cells
Single-cell suspensions were prepared from the spleens of BXD2 mice. B cells were enriched by positive selection using magnetic anti-CD19 microbeads as described above. The purified B cells were rested at 4°C overnight and then cultured for 2 h at 37 °C/5% CO2 in RPMI 1640 medium (Invitrogen) supplemented with 2 mM L-glutamine, 25 mM HEPES, 100 U/ml of penicillin, 100 µg/ml of streptomycin, 5.5 × 10−5 M β-mercaptoethanol and 10% FCS in the presence and absence of 50 ng/ml of either recombinant mouse IL-17 or IL-21 (R&D Systems). GC B cell line A20 and the pre-GC B cell line 70Z/3 were obtained from ATCC (Manassas, VA).
ELISA
Serum levels of autoantibodies were determined by ELISA as we have described previously (13). BiP was purchased from Assay Designs, Inc. and all other autoantigens from Sigma-Aldrich. Urinary albumin was analyzed using a competitive Albuwell M ELISA kit (Exocell, Inc.) as described previously (13).
Anti-NP response analysis
Mice were immunized intraperitoneally with 50 µg of NP21–chicken gamma globulin (NP21-CGG; BioSearch Technologies) adsorbed to 1.3 mg alum (Sigma-Aldrich) in a total volume of 100 µl NP21-CGG alum/phosphate-buffered saline (PBS). Anti-NP antibodies in the serum were measured by ELISA in which the target antigens (Biosearch Technologies) were either NP7–bovine serum albumin (BSA)(a low hapten density with a molar ratio of NP to BSA of ~7) to detect high-affinity anti-NP antibodies; or NP33–BSA (a high hapten density with a molar ratio of NP to BSA of ~33) to detect both high and low-affinity anti-NP antibodies.
RNA Quantitation
Quantitative real-time-PCR (qRT-PCR) was carried out using the Bio-Rad IQ5 Thermocycler. cDNA was synthesized using the Maxima® First Strand cDNA Synthesis Kit (Fermentas Inc.). The real-time qPCR mixtures contained SYBR Green PCR Master Mix (Bio-Rad) and a pair of primer sequences as shown in Supplementary Table 1 for each gene.
Statistical analysis
All results are shown as the mean ± standard error of the mean (SEM). A two-tail t test was used when two groups were compared for statistical differences. ANOVA test was used when more than 2 groups were compared for statistical differences. P values less than 0.05 were considered significant.
RESULTS
RGS13 is expressed in GC B cells and is induced by IL-17 but not IL-21
The expression of RGS13 in autoimmune B cell subpopulations had not been examined previously. We found that RGS13 is expressed exclusively in GC B cells among splenic B cell populations (Fig. 1A, 1B). By confocal imaging of spleens from 3-mo-old BXD2 mice, we found high intensity staining of the RGS13 protein in cells in the GCs with only minimal staining of cells in the MZ, FO and mantle areas (Fig. 1A, 1B). Very minimal RGS13 expression could be detected in the spleen of age-matched BXD2-Rgs13−/− mice (Fig. 1C), although PNA+ GC cells were not completely absent (Fig. 1C).
Figure 1.

RGS13 is exclusively expressed by GC B cells. Confocal imaging photomicrographs of RGS13 expression (white) in the spleen of 3-mo-old (A, B) BXD2 or (C) control BXD2-Rgs13−/− mice. A, C, Frozen spleen sections were costained with PNA (blue), anti-IgM (red) and anti-CD4 (green)(Objective lens = 20×). Abbreviations used: MZ, marginal zone; FO, follicular area. Image-J analysis of the RGS13 color intensity relative to the color intensity of PNA and IgM by overlay of separate color projections. B, The individual 3D plots represent the section of a representative BXD2 spleen tissue examined by confocal microscopy, with the z-axis representing intensity. Intensity of RGS13 in specified areas is shown as the mean ± SEM. Four sections were evaluated with 3–5 randomly chosen follicles analyzed per section (*** p<0.005 between RGS13 in GC compared to RGS13 in other regions).
Analysis of GC, FO, MZ and MZ-P B-cell subpopulations by qRT-PCR indicated that Rgs13 transcripts were limited to the GC B cells and increased in BXD2 compared to B6 mice, with extremely low expression in the FO, MZ and MZ-P B cells (Fig. 2A).
Figure 2.

Induction of Rgs13 in GC B cells by IL-17. A, qRT-PCR analysis of Rgs13 expression in B cells sorted from the spleens of indicated strains (ND = not detectable; ** p<0.01 for the indicated comparisons). B, qRT-PCR analysis of Rgs13 after normalization to Gapdh, in purified BXD2 B cells 2 hrs after treatment with IL-17 or IL-21 (50 ng/ml) and Bcl6, after normalization to Gapdh, in purified BXD2 B cells 2 hrs after treatment with IL-21 (50 ng/ml)(Rt bar). Fold changes of the indicated genes relative to the untreated control (indicated by a gray line) are shown. C, qRT-PCR analysis of basal levels of Il17ra, after normalization to Gapdh, in A20 and 70Z/3 cells. (D) Gel electrophoresis (upper) and qRT-PCR analysis (lower) of Rgs13, after normalization to Gapdh, by A20 and 70Z/3 cells. Cells were stimulation with IL-17 at the indicated time points. Results are shown as the mean ± SEM (ND = not detectable; *** p< 0.005 between the indicated comparison or between basal versus stimulated cells)(N=4 for all panels).
To verify the GC T helper cytokine that can potentially stimulate Rgs13, 3-mo-old BXD2 B cells were stimulated with either IL-17 or IL-21 for 2 hrs, and the increase in Rgs13 expression in cytokine stimulated compared to unstimulated control (fold induction) was analyzed. The results showed that IL-17 induced the upregulation of Rgs13. In contrast, IL-21, which up-regulated Bcl-6, did not induce the expression of Rgs13 and even slightly downregulated its expression relative to unstimulated cells (Fig. 2B). To further determine that upregulation of Rgs13 is a GC B cell specific response to IL-17 stimulation, we analyzed the effect of IL-17 on the GC B cell line A20 and the pre-GC B cell line 70Z/3. Flow cytometry analysis revealed that A20 were predominantly a GC phenotype as indicated by Fas+ PNA+, whereas 70Z/3 cells were Fas−PNAlow (not shown). Interestingly, despite relatively lower expression of Il17r by A20 versus 70Z/3 B cells (Fig. 2C), there were higher levels of Rgs13 and higher induction of Rgs13 after 0.5-hour co-culture with IL-17 in the A20 cell line compared to no detectable upregulation of Rgs13 in the 70Z/3 cell line. Upregulation of Rgs13 in the A20 cell line was not transient, but was maintained in the presence of IL-17 for at least 24 hours (Fig. 2D).
Rgs13 deficiency suppressed autoantibody pathogenicity
A deficiency of RGS13 significantly reduced the manifestations of lupus characteristic of 6-mo-old BXD2-WT mice, including proteinuria (Fig. 3A) and the deposition of IgG-containing immune complexes in the glomeruli (Fig. 3B). At 6 mo of age, BXD2-WT mice had very high sera titers of class-switched IgG anti-DNA, anti-histone, and anti-BiP autoantibodies (10, 11). A deficiency of RGS13 resulted in a significant reduction in the titers of these IgG autoantibodies in 6-mo-old mice (Fig. 3C, left). The titers of these IgG autoantibodies were also elevated in the 1.5 mo-old BXD2-WT mice, compared to those detected in B6 mice (Fig. 3C, right). A deficiency of RGS13 is associated with reduction of IgG autoantibody titers in BXD2 mice at both ages (Fig. 3C). However, there was no significant difference in the titers of the IgM autoantibodies in the BXD2-WT and BXD2-Rgs13−/− mice at either 6 mo or 1.5 mo of age (Fig. 3D). Thus, a deficiency of the Rgs13 gene restricted age-related development of IgG pathogenic autoantibodies in the BXD2 mice but has a limited effect on the production of IgM autoantibodies.
Figure 3.

Kidney disease and production of pathogenic IgG autoantibodies are reduced in BXD2-Rgs13−/− mice. A, ELISA analysis of urinary albumin in 6-mo-old mice (N=6, ***p<0.005). B, Left: Representative photomicrographs of immunohistochemical staining of IgG antibody deposits on glomeruli. Objective lens was 20×. Box indicates area that was enlarged and shown in lower panels. Right: ImageJ quantitation of the intensity of IgG deposits in glomeruli of the indicated strain at 6-mo of age. Data are shown as mean ± SEM. At least 4 kidney sections were examined per mouse, and staining of at least 10 glomeruli quantified for each kidney section. C, D, ELISA analysis of serum levels of (C) IgG and (D) IgM autoantibodies in the indicated strains at 6.0-mo and 1.5-mo of age (mean ± SEM, N=6–10 mice/group; * p<0.05, ** p<0.01, *** p<0.005 for results for each strain in comparison with age-matched BXD2-WT mice).
Rgs13 deficiency inhibited GC progression but enhanced IgM plasmablasts
We next determined if the lower titers of autoantibody production in these mice is associated with reduction in GC formation and plasma cell development. Consistent with our previous observations (13, 21), we found significantly higher numbers of GC B cells of 1.5 mo-old and 6 mo-old BXD2-WT mice as compared to age-matched B6 mice, with the frequency being 8-fold higher in the 6-mo old mice (Fig. 4A, right). A deficiency of RGS13 was associated with a significant reduction in the frequency of GC B cells in the 6-mo-old BXD2 mice (Fig. 4A, right), but the frequency of GC B cells were only slightly lower in the 1.5 mo-old BXD2-Rgs13−/− as compared to BXD2-WT mice (Fig. 4A, left).
Figure 4.
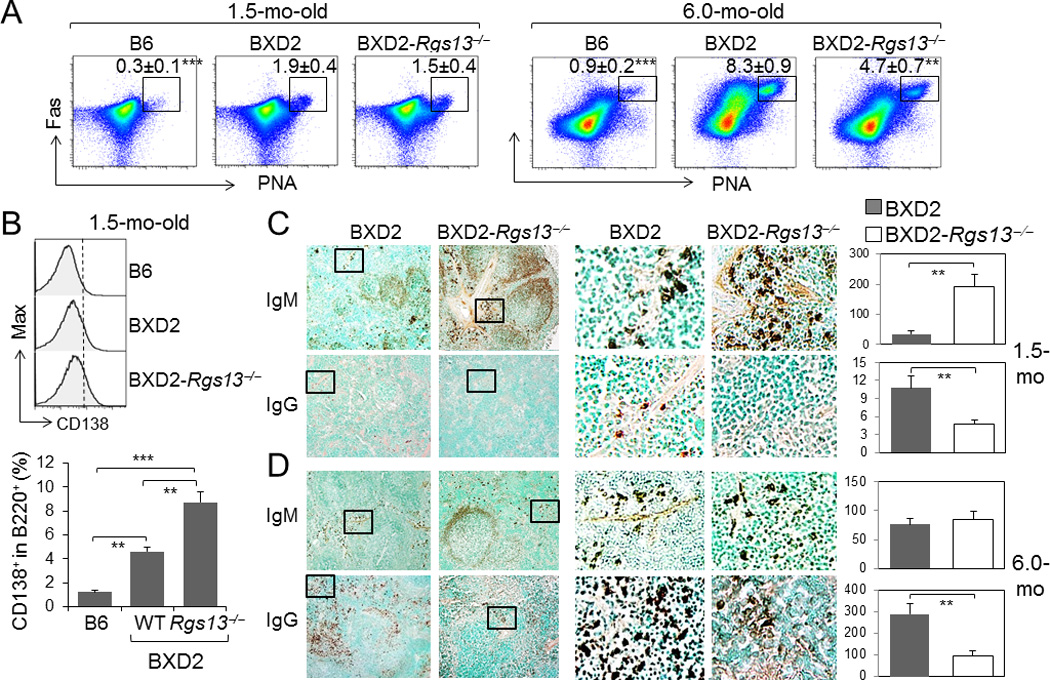
A deficiency of RGS13 in BXD2 mice obstructs age-related increase of GCs but enhances the numbers of IgM plasma cells in young mice. A, FACS analysis of the percent of PNA+Fas+ CD19+ GC B cells in the spleens of 1.5- and 6-mo-old mice (mean ± SEM of the percent of gated population within B cells, N=4; ** p<0.01, *** p<0.005 for the indicated strain in comparison with BXD2-WT mice). B, FACS analysis of the percent of CD138+ (Syndecan-1) B220+ plasmablasts in B6, BXD2, and BXD2-Rgs13−/− mice, at 1.5-mo-old. C, D, Left, Low power (10× objective lens) and Middle, zoomed view: Representative photomicrographs of immunohistochemical staining of IgMbright and IgGbright cells in the spleens of (C) 1.5-mo-old and (D) 6-mo-old mice. Right, Quantitation of the number of IgMbright and IgGbright cells per 10× objective lens microscopic view (mean ± SEM. N=4 mice per group). At least 3 randomly chosen areas were counted per section (** p<0.01 or *** p<0.005 between the indicated comparisons, for Panels B - D).
The development of plasmablasts within the spleen can be quantified via enumerating the percent of CD138+ B220+ B cells. There was a significant increase in CD138 plasmablasts in the 1.5-mo-old BXD2 mice compared to B6 mice (Fig. 4B). Surprisingly, there was a significantly increased percentage of CD138+ plasmablasts in BXD2-Rgs13−/− mice, compared to BXD2 mice (Fig. 4B). The presence of plasmablasts was further quantitated by enumerating IgMbright or IgGbright plasmablasts B cells exiting through bridging channels as they migrate into the perifollicular areas (27). Consistent with the flow cytometry findings, at 1.5 mo of age, there were significantly higher numbers of IgMbright plasmablasts in the spleens of the BXD2-Rgs13−/− mice than the BXD2-WT mice (Fig. 4C, upper left and right). A high power view of the represented area further shows higher numbers of IgMbright plasmablasts located in bridging channels and outside of the marginal zone in the BXD2-Rgs13−/− mice, compared to BXD2 mice (Fig. 4C, upper middle). At this age, very few IgGbright cells were observed in the spleens but higher numbers of IgGbright cells were seen in the BXD2-WT mice than the BXD2-Rgs13−/− mice (Fig. 4C, lower). Together these results confirm that in 1.5-mo-old BXD2-Rgs13−/− mice, there is a significant increase in the number of plasmablasts, especially IgM plasmablasts, compared to BXD2 mice.
At 6 months of age, there was no significant difference in the numbers of IgMbright plasmablasts in the spleens of these strains (Fig. 4D, upper). In contrast, the numbers of IgGbright plasmablasts in the spleens of BXD2-WT mice were markedly higher than in the BXD2-Rgs13−/− mice (Fig. 4D, lower).
Decreased GC but increased plasma cell program genes in Rgs13−/− GC B cells
The above results suggested the possibility that a deficiency of RGS13 enhances the transition of GC B cells to plasma cells. To determine if RGS13 deficiency favors plasma program over GC program, we sorted GC B cells from 3-mo-old mice, and quantified the expression of plasma cell program genes and GC program genes by qRT-PCR. In terms of GC program genes, there was significantly lower expression of Bach2, Pax5 and activation-induced cytidine deaminase (Aid or Aicda), but not Bcl6, in GC B cells from BXD2-Rgs13−/− mice as compared to GC B cells from BXD2-WT mice (Fig. 5A).
Figure 5.

Decreased GC program and increased plasma program gene expression in the absence of RGS13. qRT-PCR analysis of (A) GC program-related, (B) Plasma program-related, and (C) pCREB targeted or POU family transcription factor genes (CD19+PNA+Fas+) sorted from the spleens of mice (N=2 mice per group with 3 independent measurements. * p<0.05, ** p<0.01, *** p<0.005 between BXD2 and BXD2-Rgs13−/−).
Notably, a deficiency of RGS13 resulted in significant upregulation of plasma cell program genes Blimp1, Irf4, and Xbp1 in the GC B cells of 3 mo of age (Fig. 5B). The B-cell-restricted Oct-2 is a POU family transcription factor, which recruits the coactivator OCA-B/Bob-1/OBF-1, and plays an important role for Ig gene transcription and plasma differentiation (28, 29). Xie et al (30) recently showed that RGS13 acts as a nuclear repressor of CREB and can suppress the downstream target genes of CREB phosphorylation including Fosb and Obf1. We also found that the expression of the genes encoding of Fosb and Obf1 was significantly upregulated in GC B cells of BXD2-Rgs13−/− as compared to BXD2-WT GC B cells (Fig. 5C). In contrast, expression of the gene encoding Oct-2 was not altered by a deficiency of RGS13 (Fig. 5C).
RGS13 deficiency reduced AID and SHM in vivo
It has been established that the expression of AID is associated with close contact and prolonged interactions between cytokine secreting CD4+ T cells and GC B cells in vivo (31). The lower expression of AID in GC B cells detected in the BXD2-Rgs13−/− mice was further verified using confocal microscopy. Regions of the GC that exhibited equivalent numbers of CD4 T cells and PNA+ GC B cells were examined to determine the relative number of AID+PNA+ GC B cells that are in contact with each CD4 T cell (Fig. 6A, left and middle high-power panels). In the RGS13 deficient spleen, although there were equivalent numbers of PNA+ B cells (red) in contact with CD4+ T cells (white), there was a significant decrease in the number of B cells in close proximity to T cells that also express AID (green), as compared to BXD2-WT spleens (Fig. 6A). These results suggest that in the absence of RGS13, PNA+ B cells that are in proximity with CD4+ T cells exhibited a lower ability to upregulate AID.
Figure 6.

A deficiency of RGS13 in BXD2 mice limits AID expression and antibody affinity maturation. A, Confocal immunofluorescent assessment of position of PNA+ or AID+ B cells and CD4 T cells in the GCs. Yellow rectangle gated areas were enlarged to reveal the contact between CD4 T cells (white) and PNA+ GC B cells (red) or between CD4 T cells (white) and AID+ B cells (green). Bar graph showing the average numbers of PNA+ GC B cells or AID+ GC B cells surrounding each CD4 T cell in the GCs. At least 3 GCs were analyzed per section with 4 sections per strain (** p<0.01, as compared to BXD2-WT mice). B, C, ELISA analysis of the serum (B) high affinity anti-NP and (C) high+low affinity anti-NP IgG1 and IgG2b antibody responses at the indicated days after immunization of mice with NP21-CGG (N=4 mice per group; * p<0.05; ** p<0.01; *** p<0.005 as compared to BXD2-WT mice). All Data are shown as mean ± SEM.
To determine if altered GC and plasma cell program genes in the BXD2-WT mice is important for a T-dependent antibody affinity maturation GC response, we compared the responses of BXD2-WT and BXD2-Rgs13−/− mice to immunization with NP21-CGG, a T-dependent antigen. Affinity maturation requires SHM of the V186.2/V3 genes of the J558 family (32, 33). From day 14 to day 28, the development of the high affinity anti-NP7, (Fig. 6B) and high + low affinity anti-NP33 (Fig. 6C) IgG1 and IgG2b antibodies was significantly higher in BXD2-WT mice than BXD2-Rgs13−/− mice, suggesting a reduction in GC-mediated SHM in the BXD2-Rgs13−/− mice. These results together suggest that while there was spontaneous release of low affinity antibodies, the generation of high-affinity T-dependent antibodies was markedly reduced in BXD2-Rgs13−/− mice as compared to the BXD2-WT counterparts.
DISCUSSION
The present results identify RGS13 as a key regulator in the GCs to prolong the GC program kinetics. In the presence of intact RGS13 in the GC of BXD2 mice, there was an increase in the number of GC B cells that interacted with CD4 T cells in a manner that enabled the high levels of AID in spontaneous GC B cells in BXD2 mice. Although large numbers of IgMbright plasmablasts were found in young BXD2-Rgs13−/− mice, autoantibodies produced from these mice exhibited lower pathogenicity as immune complexes and renal disease development were attenuated in older BXD2-Rgs13−/− mice. The results obtained by the NP21-CGG immunization in BXD2-Rgs13−/− mice further support the role of RGS13 in enabling SHM and the generation of high affinity antibody responses.
Upregulation of AID has been shown to be an essential step in murine models of SLE, humans with SLE (1, 9) and in RA (34), to promoting SHM, CSR and development of highly pathogenic autoantibodies. Upregulation of AID in autoimmunity may requires close T-B interactions in addition to factors that promote either GC development (TFH) or B cell survival and tolerance loss (such as BAFF). In BXD2 mice, we previously showed that inhibition of CD28/CD86 interaction within Ad-CTLA4Ig had a prolonged and profound effect on inhibition of Aicda and autoimmune disease (10). IL-4 (35), CD40L (36) and IL-21 (37–39) can all upregulate AID and promote CSR and SHM, as recently reviewed (40). IL-4+ TH-2 T cells were almost undetectable in the spleen of BXD2 mice (21). Importantly, the present study shows that IL-17, but not IL-21, upregulates Rgs13 in BXD2 B cells. Furthermore, a deficiency of RGS13 results in a significant increase in several plasma genes including Blimp1, Irf4, and Xbp1 but attenuates the expression of GC program genes Aicda, Pax5 and Bach2. The results suggest that IL-17-induced Rgs13 favors the GC program genes and delays the transition of GC B cells to plasma cells.
How extension of the GC program facilitated the formation of pathogenic autoantibodies in autoimmune BXD2 mice? A model of delay-driven diversity in which antibody affinity maturation processes, including SHM and CSR, can be controlled at the point in which GC B cells undergo the transition to plasma cell differentiation has been proposed recently by Igarashi and colleagues (41, 42). In this model, regulatory transcription factors that prolong the GC gene-expression program and thereby delay the transition to the plasma gene regulatory program promote antibody maturation and the generation of plasma cells that produce antibodies that have undergone CSR and exhibit high levels of SHM (41). This extension of the delay-driven diversity model to regulation of pathogenic autoantibody production suggests a previously unsuspected susceptibility to autoimmune disease in which temporal differences in relative expression of specific genes regulating GC to plasma B cell transition can affect disease severity. We have observed that, Fosb, a target of pCREB and can positively regulate Blimp1 expression for terminal differentiation of activated B cells to plasma cells (28, 43), was significantly upregulated in GC B cells of BXD2-Rgs13−/− mice. Consistently, Obf1, another target of pCREB (44) and can directly bind to XBP-1 to regulate plasma differentiation (29), was also significantly upregulated in GC B cells of BXD2-Rgs13−/− mice. In contrast, Oct2, which is not a target of pCREB, was not upregulated as a result of RGS13 deficiency. Together, these results suggest that one likely mechanism by which IL-17 induced RGS13 can enhance AID-mediated autoantibody SHM and CSR is via repression of the pCREB-related plasma cell transition, leading to a prolonged GC program kinetics of the GC B cells.
One question is if RGS13 directly affects GC to plasma cell transition or if it acts through prolonged T-B interaction to affect AID induction. RGS13 protein was first defined as molecules that modulate the chemokine signaling event, i.e., CXCR5 chemotaxis in response to CXCL13, and RGS13 and can act through desensitization of GPCR to enable retention and close contact of GC B cells and CD4 T cells (23). It has been reported that the retention of B cells play a key role in shaping a GC response (45) and is needed either to promote the interaction of CD4+ T cells with B cells, which facilitates recycling of B cells into the GC (46) or promotion of the interactions of FDC with B cells to enhance antigen encounter (47). The present results suggest that the expression of RGS13 at least regulates successful contacts between GC B cells and CD4+ T cells that can lead to the upregulation of AID in autoantigen triggered spontaneous GC B cells.
In summary, in addition to providing a framework for the understanding of IL-17-mediated promotion of GC formation and autoantibody production, the current studies identify new mechanisms that may promote the development and severity of autoimmune disease. The results imply that factors such as IL-17 and protein kinase A (48) that can directly regulate expression of RGS13 can also act through this gene to manipulate the size of GCs and the extent of affinity maturation and CSR of the antibodies produced. We therefore propose that upregulation of RGS13 can have a positive effect on AID by delaying the transition of the GC B cell from the GC program to the plasma cell program, and this in turn results in increased CSR, SHM, and production of pathogenic autoantibodies. This effect is distinct from factors that promote GC development, such as IL-21 from TFH, and factors that promote tolerance loss in GC B cell survival such as BAFF, and provide insights into a unique, and distinct step used for development of pathogenic autoantibodies.
Supplementary Material
ACKNOWLDGEMENTS
We thank Dr. Fiona Hunter for review of the manuscript and Ms. Karen Beeching for excellent secretarial assistance. Flow cytometry and confocal imaging data acquisition were carried out at the UAB Comprehensive Flow Cytometry Core (P30 AR048311 and P30 AI027767) and UAB Analytic Imaging and Immunoreagents Core (P30 AR048311), respectively. This work is supported by grants from VA Merit Review Grant (1I01BX000600-01), NIH/NIAID (1RO1 AI 071110 and 1RO1 AI083705), American College of Rheumatology (ACR)-Within-Our-Reach, Rheumatology Research Foundation, Alliance for Lupus Research, Arthritis Foundation, Lupus Research Institute, and, in part, by the Intramural Research Program of the NIAID, NIH.
Footnotes
All authors claim to have no financial interests which could create a potential conflict of interest or the appearance of a conflict of interest with regard to the work.
AUTHOR CONTRIBUTIONS
H-C. H. and J.D.M. designed experiments, analyzed data, and wrote the manuscript. J.H.W. carried out all confocal imaging analysis and was involved in manuscript preparation. J.S.N., S.X., PA.Y. Q.W., J.L., B.L., and Y.D. performed ELISA, cell sorting, flow cytometry, paraffin imaging, gene expression analysis. B.L. paired, bred, and genotyped all mice used for this study. K.M.D. assisted generation of BXD2-Rgs13−/− mouse, gene expression data interpretation, and was involved in manuscript preparation.
REFERENCES
- 1.Schroeder K, Herrmann M, Winkler TH. The role of somatic hypermutation in the generation of pathogenic antibodies in SLE. Autoimmunity. 2012 doi: 10.3109/08916934.2012.748751. [DOI] [PubMed] [Google Scholar]
- 2.Holers VM. Are anti-cyclic citrullinated peptide antibodies pathogenic in rheumatoid arthritis? Nat Clin Pract Rheumatol. 2006;2(8):400–401. doi: 10.1038/ncprheum0247. [DOI] [PubMed] [Google Scholar]
- 3.Snir O, Widhe M, Hermansson M, von Spee C, Lindberg J, Hensen S, et al. Antibodies to several citrullinated antigens are enriched in the joints of rheumatoid arthritis patients. Arthritis Rheum. 2010;62(1):44–52. doi: 10.1002/art.25036. [DOI] [PubMed] [Google Scholar]
- 4.Bootsma H, Spronk PE, Ter Borg EJ, Hummel EJ, de Boer G, Limburg PC, et al. The predictive value of fluctuations in IgM and IgG class anti-dsDNA antibodies for relapses in systemic lupus erythematosus. A prospective long-term observation. Ann Rheum Dis. 1997;56(11):661–666. doi: 10.1136/ard.56.11.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Arbuckle MR, McClain MT, Rubertone MV, Scofield RH, Dennis GJ, James JA, et al. Development of autoantibodies before the clinical onset of systemic lupus erythematosus. N Engl J Med. 2003;349(16):1526–1533. doi: 10.1056/NEJMoa021933. [DOI] [PubMed] [Google Scholar]
- 6.Kokkonen H, Mullazehi M, Berglin E, Hallmans G, Wadell G, Ronnelid J, et al. Antibodies of IgG, IgA and IgM isotypes against cyclic citrullinated peptide precede the development of rheumatoid arthritis. Arthritis Res Ther. 2011;13(1):R13. doi: 10.1186/ar3237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rantapaa-Dahlqvist S, de Jong BA, Berglin E, Hallmans G, Wadell G, Stenlund H, et al. Antibodies against cyclic citrullinated peptide and IgA rheumatoid factor predict the development of rheumatoid arthritis. Arthritis Rheum. 2003;48(10):2741–2749. doi: 10.1002/art.11223. [DOI] [PubMed] [Google Scholar]
- 8.Zhang J, Jacobi AM, Wang T, Diamond B. Pathogenic autoantibodies in systemic lupus erythematosus are derived from both self-reactive and non-self-reactive B cells. Mol Med. 2008;14(11–12):675–681. doi: 10.2119/2008-00066.Zhang. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jiang C, Zhao ML, Diaz M. Activation-induced deaminase heterozygous MRL/lpr mice are delayed in the production of high-affinity pathogenic antibodies and in the development of lupus nephritis. Immunology. 2009;126(1):102–113. doi: 10.1111/j.1365-2567.2008.02882.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hsu HC, Wu Y, Yang P, Wu Q, Job G, Chen J, et al. Overexpression of activation-induced cytidine deaminase in B cells is associated with production of highly pathogenic autoantibodies. J Immunol. 2007;178(8):5357–5365. doi: 10.4049/jimmunol.178.8.5357. [DOI] [PubMed] [Google Scholar]
- 11.Hsu HC, Zhou T, Kim H, Barnes S, Yang P, Wu Q, et al. Production of a novel class of polyreactive pathogenic autoantibodies in BXD2 mice causes glomerulonephritis and arthritis. Arthritis Rheum. 2006;54(1):343–355. doi: 10.1002/art.21550. [DOI] [PubMed] [Google Scholar]
- 12.Mountz JD, Yang P, Wu Q, Zhou J, Tousson A, Fitzgerald A, et al. Genetic segregation of spontaneous erosive arthritis and generalized autoimmune disease in the BXD2 recombinant inbred strain of mice. Scand J Immunol. 2005;61(2):128–138. doi: 10.1111/j.0300-9475.2005.01548.x. [DOI] [PubMed] [Google Scholar]
- 13.Hsu HC, Yang P, Wu Q, Wang JH, Job G, Guentert T, et al. Inhibition of the catalytic function of activation-induced cytidine deaminase promotes apoptosis of germinal center B cells in BXD2 mice. Arthritis Rheum. 2011;63(7):2038–2048. doi: 10.1002/art.30257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mietzner B, Tsuiji M, Scheid J, Velinzon K, Tiller T, Abraham K, et al. Autoreactive IgG memory antibodies in patients with systemic lupus erythematosus arise from nonreactive and polyreactive precursors. Proc Natl Acad Sci U S A. 2008;105(28):9727–9732. doi: 10.1073/pnas.0803644105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shlomchik MJ, Aucoin AH, Pisetsky DS, Weigert MG. Structure and function of anti-DNA autoantibodies derived from a single autoimmune mouse. Proc Natl Acad Sci U S A. 1987;84(24):9150–9154. doi: 10.1073/pnas.84.24.9150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cappione A, 3rd, Anolik JH, Pugh-Bernard A, Barnard J, Dutcher P, Silverman G, et al. Germinal center exclusion of autoreactive B cells is defective in human systemic lupus erythematosus. J Clin Invest. 2005;115(11):3205–3216. doi: 10.1172/JCI24179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Grammer AC, Slota R, Fischer R, Gur H, Girschick H, Yarboro C, et al. Abnormal germinal center reactions in systemic lupus erythematosus demonstrated by blockade of CD154-CD40 interactions. J Clin Invest. 2003;112(10):1506–1520. doi: 10.1172/JCI19301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Horwitz DA, Wang H, Gray JD. Cytokine gene profile in circulating blood mononuclear cells from patients with systemic lupus erythematosus: increased interleukin-2 but not interleukin-4 mRNA. Lupus. 1994;3(5):423–428. doi: 10.1177/096120339400300511. [DOI] [PubMed] [Google Scholar]
- 19.Sugimoto K, Morimoto S, Kaneko H, Nozawa K, Tokano Y, Takasaki Y, et al. Decreased IL-4 producing CD4+ T cells in patients with active systemic lupus erythematosus-relation to IL-12R expression. Autoimmunity. 2002;35(6):381–387. doi: 10.1080/0891693021000008535. [DOI] [PubMed] [Google Scholar]
- 20.Ettinger R, Sims GP, Fairhurst AM, Robbins R, da Silva YS, Spolski R, et al. IL-21 induces differentiation of human naive and memory B cells into antibody-secreting plasma cells. J Immunol. 2005;175(12):7867–7879. doi: 10.4049/jimmunol.175.12.7867. [DOI] [PubMed] [Google Scholar]
- 21.Hsu HC, Yang P, Wang J, Wu Q, Myers R, Chen J, et al. Interleukin 17-producing T helper cells and interleukin 17 orchestrate autoreactive germinal center development in autoimmune BXD2 mice. Nat Immunol. 2008;9(2):166–175. doi: 10.1038/ni1552. [DOI] [PubMed] [Google Scholar]
- 22.Johnson EN, Druey KM. Functional characterization of the G protein regulator RGS13. J Biol Chem. 2002;277(19):16768–16774. doi: 10.1074/jbc.M200751200. [DOI] [PubMed] [Google Scholar]
- 23.Shi GX, Harrison K, Wilson GL, Moratz C, Kehrl JH. RGS13 regulates germinal center B lymphocytes responsiveness to CXC chemokine ligand (CXCL)12 and CXCL13. J Immunol. 2002;169(5):2507–2515. doi: 10.4049/jimmunol.169.5.2507. [DOI] [PubMed] [Google Scholar]
- 24.Bertoni F, Ponzoni M. The cellular origin of mantle cell lymphoma. Int J Biochem Cell Biol. 2007;39(10):1747–1753. doi: 10.1016/j.biocel.2007.04.026. [DOI] [PubMed] [Google Scholar]
- 25.Wang JH, Li J, Wu Q, Yang P, Pawar RD, Xie S, et al. Marginal zone precursor B cells as cellular agents for type I IFN-promoted antigen transport in autoimmunity. J Immunol. 2010;184(1):442–451. doi: 10.4049/jimmunol.0900870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang JH, Wu Q, Yang P, Li H, Li J, Mountz JD, et al. Type I interferon-dependent CD86(high) marginal zone precursor B cells are potent T cell costimulators in mice. Arthritis Rheum. 2011;63(4):1054–1064. doi: 10.1002/art.30231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hargreaves DC, Hyman PL, Lu TT, Ngo VN, Bidgol A, Suzuki G, et al. A coordinated change in chemokine responsiveness guides plasma cell movements. J Exp Med. 2001;194(1):45–56. doi: 10.1084/jem.194.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ohkubo Y, Arima M, Arguni E, Okada S, Yamashita K, Asari S, et al. A role for c-fos/activator protein 1 in B lymphocyte terminal differentiation. J Immunol. 2005;174(12):7703–7710. doi: 10.4049/jimmunol.174.12.7703. [DOI] [PubMed] [Google Scholar]
- 29.Shen Y, Hendershot LM. Identification of ERdj3 and OBF-1/BOB-1/OCA-B as direct targets of XBP-1 during plasma cell differentiation. J Immunol. 2007;179(5):2969–2978. doi: 10.4049/jimmunol.179.5.2969. [DOI] [PubMed] [Google Scholar]
- 30.Xie Z, Geiger TR, Johnson EN, Nyborg JK, Druey KM. RGS13 acts as a nuclear repressor of CREB. Mol Cell. 2008;31(5):660–670. doi: 10.1016/j.molcel.2008.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Reinhardt RL, Liang HE, Locksley RM. Cytokine-secreting follicular T cells shape the antibody repertoire. Nat Immunol. 2009;10(4):385–393. doi: 10.1038/ni.1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Maizels N, Bothwell A. The T-cell-independent immune response to the hapten NP uses a large repertoire of heavy chain genes. Cell. 1985;43(3 Pt 2):715–720. doi: 10.1016/0092-8674(85)90244-2. [DOI] [PubMed] [Google Scholar]
- 33.Cumano A, Rajewsky K. Clonal recruitment and somatic mutation in the generation of immunological memory to the hapten NP. EMBO J. 1986;5(10):2459–2468. doi: 10.1002/j.1460-2075.1986.tb04522.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xu X, Hsu HC, Chen J, Grizzle WE, Chatham WW, Stockard CR, et al. Increased expression of activation-induced cytidine deaminase is associated with anti-CCP and rheumatoid factor in rheumatoid arthritis. Scand J Immunol. 2009;70(3):309–316. doi: 10.1111/j.1365-3083.2009.02302.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nambu Y, Sugai M, Gonda H, Lee CG, Katakai T, Agata Y, et al. Transcription-coupled events associating with immunoglobulin switch region chromatin. Science. 2003;302(5653):2137–2140. doi: 10.1126/science.1092481. [DOI] [PubMed] [Google Scholar]
- 36.Tran TH, Nakata M, Suzuki K, Begum NA, Shinkura R, Fagarasan S, et al. B cell-specific and stimulation-responsive enhancers derepress Aicda by overcoming the effects of silencers. Nat Immunol. 2010;11(2):148–154. doi: 10.1038/ni.1829. [DOI] [PubMed] [Google Scholar]
- 37.Kobayashi S, Haruo N, Sugane K, Ochs HD, Agematsu K. Interleukin-21 stimulates B-cell immunoglobulin E synthesis in human beings concomitantly with activation-induced cytidine deaminase expression and differentiation into plasma cells. Hum Immunol. 2009;70(1):35–40. doi: 10.1016/j.humimm.2008.10.021. [DOI] [PubMed] [Google Scholar]
- 38.Pene J, Gauchat JF, Lecart S, Drouet E, Guglielmi P, Boulay V, et al. Cutting edge: IL-21 is a switch factor for the production of IgG1 and IgG3 by human B cells. J Immunol. 2004;172(9):5154–5157. doi: 10.4049/jimmunol.172.9.5154. [DOI] [PubMed] [Google Scholar]
- 39.Simard N, Konforte D, Tran AH, Esufali J, Leonard WJ, Paige CJ. Analysis of the role of IL-21 in development of murine B cell progenitors in the bone marrow. J Immunol. 2011;186(9):5244–5253. doi: 10.4049/jimmunol.1004040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zan H, Casali P. Regulation of Aicda expression and AID activity. Autoimmunity. 2012 doi: 10.3109/08916934.2012.749244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Muto A, Ochiai K, Kimura Y, Itoh-Nakadai A, Calame KL, Ikebe D, et al. Bach2 represses plasma cell gene regulatory network in B cells to promote antibody class switch. EMBO J. 2010;29(23):4048–4061. doi: 10.1038/emboj.2010.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Watanabe-Matsui M, Muto A, Matsui T, Itoh-Nakadai A, Nakajima O, Murayama K, et al. Heme regulates B-cell differentiation, antibody class switch, and heme oxygenase-1 expression in B cells as a ligand of Bach2. Blood. 2011;117(20):5438–5448. doi: 10.1182/blood-2010-07-296483. [DOI] [PubMed] [Google Scholar]
- 43.Corcoran LM, Hasbold J, Dietrich W, Hawkins E, Kallies A, Nutt SL, et al. Differential requirement for OBF-1 during antibody-secreting cell differentiation. J Exp Med. 2005;201(9):1385–1396. doi: 10.1084/jem.20042325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Qin XF, Reichlin A, Luo Y, Roeder RG, Nussenzweig MC. OCA-B integrates B cell antigen receptor-, CD40L- and IL 4-mediated signals for the germinal center pathway of B cell development. EMBO J. 1998;17(17):5066–5075. doi: 10.1093/emboj/17.17.5066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang X, Cho B, Suzuki K, Xu Y, Green JA, An J, et al. Follicular dendritic cells help establish follicle identity and promote B cell retention in germinal centers. J Exp Med. 2011;208(12):2497–2510. doi: 10.1084/jem.20111449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.McHeyzer-Williams LJ, McHeyzer-Williams MG. Antigen-specific memory B cell development. Annu Rev Immunol. 2005;23:487–513. doi: 10.1146/annurev.immunol.23.021704.115732. [DOI] [PubMed] [Google Scholar]
- 47.Suzuki K, Grigorova I, Phan TG, Kelly LM, Cyster JG. Visualizing B cell capture of cognate antigen from follicular dendritic cells. J Exp Med. 2009;206(7):1485–1493. doi: 10.1084/jem.20090209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xie Z, Yang Z, Druey KM. Phosphorylation of RGS13 by the cyclic AMP-dependent protein kinase inhibits RGS13 degradation. J Mol Cell Biol. 2010;2(6):357–365. doi: 10.1093/jmcb/mjq031. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.