

Abstract
Malignant mesothelioma (MM) is a very aggressive asbestos-related neoplasm of the serous membranes, whose incidence is increasing worldwide. Although the introduction of new drug combinations, such as cisplatin plus pemetrexed/gemcitabine, has determined an improvement in the patient quality of life, MM remains a universally fatal disease. The observation that key G1/S checkpoint regulators are often functionally inactivated in MM prompted us to test whether the use of G2/M checkpoint inhibitors, able to sensitize G1/S checkpoint-defective cancer cells to DNA-damaging agents, could be successful in MM. We treated six MM cell lines, representative of different histotypes (epithelioid, biphasic, and sarcomatoid), with cisplatin in combination with MK-1775, an inhibitor of the G2/M checkpoint kinase WEE1. We observed that MK-1775 enhanced the cisplatin cytotoxic effect in all MM cell lines, except the sarcomatoid cell line, which is representative of the most aggressive histotype. As expected, the enhancement in cisplatin toxicity was accompanied by a decrease in the inactive phosphorylated form of cyclin-dependent kinase 1 (CDK1), a key substrate of WEE1, which is indicative of G2/M checkpoint inactivation. Consistently, we also observed a decrease in G2/M accumulation and an increase in mitotic entry of DNA-damaged cells and apoptosis, probably due to the loss of the cell ability to arrest cell cycle in response to DNA damage, irrespectively of p53 mutational status. Notably, this treatment did not increase cisplatin cytotoxicity on normal cells, thus suggesting a possible use of MK-1775 in combination with cisplatin for a safe and efficient treatment of epithelioid and biphasic MM.
Keywords: WEE1, MK-1775, cisplatin, mesothelioma, CDK1, G2/M checkpoint, apoptosis
Introduction
Malignant mesothelioma (MM) is a very aggressive neoplasm that originates from the malignant transformation of mesothelium, a membrane covering the pleural, peritoneal and pericardial cavities. The most common MM type is that arising from the mesothelium lining the pleura, which accounts for approximately 70% of cases.1 MM is classified into three histotypes: epithelioid, biphasic, and sarcomatoid, the last one having the poorest prognosis.
The main risk factor for MM is exposure to asbestos.2,3 Indeed, the extensive use of this natural fibrous material during the twentieth century determined an increase in incidence of this cancer, which was originally extremely rare. Despite the ban on asbestos use in many developed countries, MM incidence is expected to further rise, with a predicted peak in 2020–2050, because of the long-latency time between the first exposure and diagnosis, which is typically longer than 30 y.
At present there is no known curative modality for MM. Indeed, the current therapeutic regimens have only limited effects on patients. A potentially curative surgical option for pleural MM, which aims at removing all gross disease, involves an extrapleural pneumonectomy.2,3 However, despite the combination with some form of adjuvant therapy, residual microscopic disease cannot be eradicated. Therefore, the occurrence of relapses together with the high risk of perioperative morbidity and mortality render the role of radical surgical resection very controversial. No radiotherapy or chemotherapy regimen has proved to be curative, although some regimens are valuable for palliation. In particular, the use of cisplatin in combination with pemetrexed/gemcitabine has determined a significant improvement in the patient quality of life. However, the prognosis remains extremely poor, with a median survival ranging between 9 and 17 mo from diagnosis.3 Therefore, there is an urgent need to develop new effective therapeutic strategies.
Over the last decades, intensive research has been devoted to the development of G2/M checkpoint inhibitors as novel therapeutic agents able to increase sensitivity to DNA-damaging anticancer drugs, such as chemotherapeutic agents and ionizing radiation.4-6 This novel therapeutic strategy is based on the observation that most human cancers rely on G2/M checkpoint rather than on G1/S checkpoint to detect and repair damaged DNA by stalling cell cycle. Indeed, the G1/S checkpoint is defective in most cancers, owing to the loss of key regulators of this checkpoint, such as retinoblastoma 1 (RB1) and p53 tumor suppressors. Therefore, tumor cells treated with a G2/M checkpoint inhibitor might lose the ability to arrest cell cycle in response to DNA damage and be forced to enter an aberrant and lethal mitosis. Conversely, non-neoplastic cells retain G1/S checkpoint activity and, therefore, are unaffected by these treatments.
Despite the cellular mechanisms underlying MM development are not yet clarified, many different molecular alterations have been described.7 In particular, a major genetic alteration, which occurs with a frequency of >70% in MM cell lines, is the homozygous deletion of the CDKN2A locus,8 which encodes both the p16 and p14 tumor suppressors through the use of an alternative first exon.9 p16 and p14 are crucial positive regulators of RB1 and p53, respectively, and the loss of the CDKN2A locus can result in the functional inactivation of both RB1 and p53. Moreover, the gene encoding p16 can also be inactivated in MM by methylation of its promoter.10 Another mechanism whereby RB1 and p53 pathways can be disrupted in MM involves the simian virus 40 (SV40), which has been implicated in MM pathogenesis.11 Indeed, the SV40 large T antigen can bind and inactivate both RB1 and p53 in MM cells.12
Therefore, despite the key G1/S checkpoint regulators, RB1 and p53, are rarely mutated in MM,7 the therapeutic approach based on the combination of DNA-damaging agents and G2/M checkpoint inhibitors could potentially be successful also in MM, in which these central G1/S checkpoint regulators are principally inactivated through indirect mechanisms, such as the altered expression/activity of their upstream regulators.
Among the G2/M abrogators, WEE1 kinase inhibitors proved to be effective in sensitizing different tumor types to various anticancer therapeutics.13-23 WEE1 kinase is a crucial component of the G2/M DNA damage checkpoint, whose direct substrate is the key mitotic protein CDK1.24 WEE1 inhibitors prevent the WEE1-mediated phosphorylation and inactivation of CDK1 in response to DNA damage, thus resulting in G2/M checkpoint override and increased mitotic and cell death rates. To date, different small molecules targeting WEE1 have been used.13-23 In the present study, we tested MK-1775, an efficient and selective WEE1 inhibitor, which has entered phase I clinical trials in combination with gemcitabine, cisplatin, or carboplatin in patients with advanced solid tumors.25 In particular, we treated a panel of six MM cell lines, representative of the three different histotypes, with MK-1775 in combination with cisplatin, the chemotherapic agent currently used in MM therapy. We aimed to assess whether this strategy, which holds the potential for a rapid clinical translation, could be feasible for the treatment of MM. We observed that MK-1775 significantly enhanced the cisplatin cytotoxic effect in all cell lines, except the one representative of the most aggressive histotype, without enhancing toxicity in non-neoplastic cells. Moreover, we observed that the enhancement in cisplatin toxicity was accompanied by a reduction in CDK1 phosphorylation and a shift in the balance toward apoptosis rather than growth arrest.
Results
WEE1 inhibition selectively sensitizes MM cell lines to cisplatin
We treated six MM cell lines, representing the epithelioid (IST-MES1, IST-MES2, REN, and NCI-H2452), biphasic (MSTO-211H), and sarcomatoid (NCI-H2052) histotypes, with cisplatin at concentrations ranging from 2.5 to 15 µM, alone or in combination with the WEE1 inhibitor, MK-1775, at the concentration of 150 nM or 250 nM. We evaluated cell viability by MTS assay at 72 h after treatment (Fig. 1A). We observed that MK-1775 enhanced the cisplatin cytotoxic effect in a concentration-dependent manner in all cell lines except one (NCI-H2052), as also shown by the comparison between the half maximal inhibitory concentration (IC50) values of cisplatin alone and cisplatin in combination with MK-1775 (as in the table included in Fig. 1A).

Figure 1. Effect of co-treatments with cisplatin and MK-1775 on cell viability of MM cells and fibroblasts. (A) MTS analysis of cell viability. Results are reported as means of three experiments and expressed as percentages of cell viability calculated with respect to control cells treated with DMSO alone. The absorbance values of treated and control samples were subjected to one-way Anova with Dunnett post-test. Statistically significant differences between treated and control cells are indicated with *significant (P < 0.05) or **very significant (P < 0.01). In the table are reported the half maximal inhibitory concentration (IC50) values of cisplatin alone and cisplatin in combination with 150 nM or 250 nM MK-1775 on MM cell lines and fibroblasts. (B) Combination index (CI) values for cisplatin and MK-1775 in MM cell lines. CI = 1 indicates additivity, CI > 1 indicates antagonism, and CI < 1 indicates synergy.
MK-1775 alone showed a slight cytotoxicity on the majority of MM cell lines (data not shown) and a more significant antiproliferative activity on the IST-MES2 cells, in which cell viability was reduced to approximately 65% and 47% after treatment with 150 and 250 nM MK-1775, respectively (P < 0.05).
To rule out a possible increase in cisplatin cytotoxicity on non-neoplastic cells, the drug combination was also tested on primary human skin fibroblasts by MTS assay. We found that MK-1775 did not significantly enhance the cisplatin cytotoxic effects on fibroblasts, as evident by the IC50 values reported in Figure 1A.
To evaluate whether the effect of cisplatin and MK-1775 was synergic, we determined the combination index (CI) values through the Chou–Talalay method26 for the cell lines in which the drug combination was found to be effective. This analysis revealed a synergy between the two compounds (Fig. 1B).
MK-1775 increases cisplatin efficacy by decreasing G2/M accumulation and inducing mitotic entry and apoptosis
To study the mechanism whereby MK-1775 sensitizes MM cells to cisplatin, we first verified the inhibition of the enzymatic activity of WEE1 by evaluating the phosphorylation on Tyr15 of its substrate, CDK1, by western blotting 72 h after treatment with cisplatin in combination with 250 nM MK-1775. We used cisplatin concentrations corresponding to the IC50 values achieved by this chemotherapeutic agent when used in combination with 250 nM MK-1775 (as in the table included in Fig. 1A). As expected, we observed a decrease in phospho-CDK1 Tyr15 in all MM cell lines treated with the two drug combination with respect to cells treated with cisplatin alone (Fig. 2A). This result suggests that MK-1775, by preventing the WEE1-mediated phosphorylation and inactivation of CDK1, could be indeed able to abrogate the G2/M DNA damage checkpoint in MM cell lines.

Figure 2. Effect of co-treatments with cisplatin and MK-1775 on CDK1 activation and cell cycle progression. (A) Western blotting evaluation of phospho-CDK1 Tyr15 (pCDK1) and total CDK1 in MM cell lines treated with cisplatin and MK-1775 alone or in combination. DMSO alone was added to control cells. An anti-β-actin antibody was used for a loading control. (B) Representative cell cycle profiles, obtained by FACS analysis, showing the phase distribution of NCI-H2452 and MSTO-211H cell lines treated with cisplatin and/or MK-1775. DMSO alone was added to control cells. (C) Determination of percentage of mitotic cells by staining with acridine orange of NCI-H2452 and MSTO-211H cells treated with cisplatin and/or MK-1775 plus nocodazole. Control cells were treated with DMSO and nocodazole. The histograms show the means and standard deviations of three experiments. Statistically significant differences were evaluated by one-way Anova with Tukey post-test and are indicated with *significant (P < 0.05) and ***extremely significant (P < 0.001). Representative micrographs of MSTO-211H cells treated with cisplatin and/or MK-1775 plus nocodazole and stained with acridine orange are shown. Arrows point to examples of mitotic cells.
So, to define further MK-1775 mechanism of action in MM cells, we focused on two MM cell lines representative of the histotypes in which the drug combination was effective: the epithelioid NCI-H2452 and the biphasic MSTO-211H cell lines. We treated the two cell lines with MK-1775 and/or cisplatin, as described above, and analyzed their cell cycle profile by FACS 72 h after treatment. As expected, we observed a decrease in G2/M accumulation of cells treated with the two drug combination with respect to cells treated with cisplatin alone (Fig. 2B). Moreover, the analysis showed an increase in the sub-G1 peak, which could be indicative of apoptosis, in cells co-treated with MK-1775 and cisplatin with respect to cells treated with cisplatin alone. These results support the hypothesis that MM cells treated with MK-1775 lose the ability to stall cell cycle in response to cisplatin-induced DNA damage and enter an aberrant and lethal mitosis.
We verified the ability of MK-1775 to force cisplatin-treated MM cells to enter mitosis by treating NCI-H2452 and MSTO-211H cells, growing on coverslips, with MK-1775 and/or cisplatin, as described above, plus nocodazole to “capture” cells entering mitosis. Eight hours after treatment we stained cells with acridine orange and observed a significant increase in mitotic cells, which were identified by their distinct morphology, in cell lines co-treated with MK-1775 and cisplatin with respect to cells treated with cisplatin alone (Fig. 2C).
To verify that the NCI-H2452 and MSTO-211H cells forced to enter mitosis following exposure to MK-1775 indeed harbor unrepaired DNA lesions, we evaluated by immunofluorescence the co-expression of phospho-histone H2AX (γ-H2AX) and phospho-histone H3 (pHH3), which are markers of DNA double-strand breaks (DSBs) and mitosis, respectively. Eight hours after treatment with MK-1775 and cisplatin plus nocodazole we observed that a very large fraction of cells staining positively for pHH3 were also positive for γ-H2AX (Fig. 3). Conversely, after treatment with cisplatin alone no double staining was detected. A low percentage of γ-H2AX positive cells was also observed after treatment with MK-1775 alone, consistent with the low cytotoxicity of this molecule, and these γ-H2AX positive cells were also positive for pHH3. These results show that MK-1775 causes the loss of MM cell ability to stall cell cycle in response to DSBs (induced by both cisplatin and, to a lower extent, also by MK-1775 itself) and forces cells to enter mitosis regardless of the presence of DNA damage. This premature entry into an aberrant mitosis could underlie an increase in apoptotic cell death.
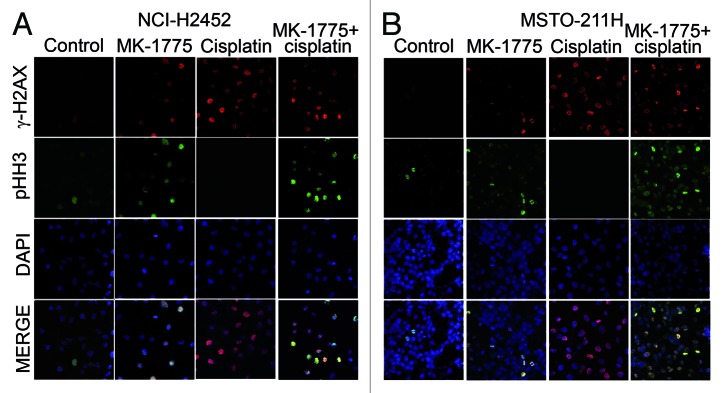
Figure 3. Detection of MM cells bearing damaged DNA that are forced to enter mitosis after treatment with cisplatin and MK-1775. Representative micrographs of NCI-H2452 (A) and MSTO (B) cells treated with cisplatin and/or MK-1775 plus nocodazole and stained by immunofluorescence with antibodies against γ-H2AX and pHH3 and by the fluorescent nuclear stain DAPI. Control cells were treated with DMSO and nocodazole.
To confirm the ability of MK-1775 to increase the cisplatin-induced apoptosis, we evaluated early and late apoptosis in both NCI-H2452 and MSTO-211H cell lines by cell staining with an annexin V-FITC antibody and propidium iodide (PI) and FACS analysis 72 h after treatment. These analyses showed an increase in apoptosis induction in MM cells co-treated with MK-1775 and cisplatin with respect to cells treated with cisplatin alone (Fig. 4A). We also analyzed caspase-3 activity in both NCI-H2452 and MSTO-211H cell lines 72 h after treatment. Consistently, we observed a significant increase in caspase-3 activity in cells treated with the two drug combination with respect to cells treated with cisplatin alone (Fig. 4B).

Figure 4. Apoptosis induction in MM cell lines treated with cisplatin and/or MK-1775. (A) A representative FACS analysis of apoptosis by cell staining with annexin V-FITC and propidium iodide (PI) of NCI-H2452 and MSTO-211H cells treated with cisplatin and/or MK-1775 or DMSO, as a control. The table reports the values relative to early apoptosis (annexin V-positive and PI-negative), late apoptosis (annexin V-positive and PI-positive) and total apoptosis. (B) Histograms showing caspase-3 activity in NCI-H2452 and MSTO-211H cell lines treated with cisplatin and/or MK-1775 or DMSO, as a control. Caspase-3 activity is expressed as pmol p-nitroaniline (pNA)/µg protein × time (h). The reported values represent the means and standard deviations of three experiments. Statistically significant differences were evaluated by one-way Anova with Tukey post-test and are indicated with *significant (P < 0.05), **very significant (P < 0.01) and ***extremely significant (P < 0.001).
Discussion
In recent years there has been great concern about the asbestos-related disease MM because of its increasing incidence worldwide.2,3 Indeed, the extensive use of asbestos during the twentieth century, together with its long-latency time (longer than 30 y), determined a rise in incidence, which is expected to peak over the period 2020–2050.
Several efforts have been devoted to design new effective therapeutic strategies for MM, which led to some developments in the management of this disease. In particular, the recent introduction of new drug combinations, such as cisplatin plus pemetrexed/gemcitabine, has shown a significant improvement in the patient quality of life.2,3 Nevertheless, MM remains a universally fatal disease.
Understanding the molecular events underlying the development, progression and resistance to therapy of MM is essential to design new therapeutic strategies and to increase the chance of recovery of patients for whom there is currently no specific curative modality. Many different molecular alterations have been described, among which the functional inactivation of RB1 and p53 proteins.7 Indeed, despite these tumor suppressors are rarely mutated in MM, their pathways can be disrupted by the altered expression of upstream regulators, such as p16 and p14. Moreover, the SV40 large T antigen, which has been implicated in MM pathogenesis, can bind and inactivate both p53 and RB1.
The observation that key G1/S checkpoint regulators can be functionally inactivated in MM suggests that patients with MM might benefit from the new therapeutic approach based on the combination of DNA-damaging agents and G2/M checkpoint inhibitors. Indeed, these G2/M checkpoint abrogators proved to be effective in sensitizing G1/S checkpoint-defective cancer cells to various anticancer therapeutics, by preventing cells to arrest cell cycle in response to DNA damage and thus forcing them to enter mitosis and undergo cell death.13-19 Although this approach proved to be principally effective on cells bearing p53 mutations, it has been suggested that it could be successful also in tumors, such as MM, in which p53 is indirectly inactivated by other mechanisms.6
In the present study, we treated six MM cell lines, representative of the three different histotypes (epithelioid, biphasic, and sarcomatoid), with cisplatin, the chemotherapic agent currently used in MM therapy, in combination with MK-1775, an efficient and selective inhibitor of the G2/M checkpoint kinase WEE1, which has entered phase I clinical trials in combination with several chemotherapic agents in patients with different solid tumors.25
We observed that MK-1775 significantly enhanced the cisplatin cytotoxic effect in all MM cell lines, except the sarcomatoid cell line NCI-H2052, which is representative of the most aggressive histotype. This treatment was effective both in p53 wild-type cell lines, namely IST-MES1 (Sanger Institute, Catalogue of Somatic Mutations in Cancer) and MSTO-211H,27 and p53 mutated cell lines, namely NCI-H2452,27 REN,28 and IST-MES2.29 However, in all the p53 wild-type cell lines examined in this study (IST-MES1, NCI-H2052,27 and MSTO-211H) the gene encoding p14, a positive p53 regulator able to inhibit the MDM2-dependent degradation of p53,30 was previously found to be mutated (Sanger Institute, Catalogue of Somatic Mutations in Cancer). Therefore, p53 might be functionally inactive in these p53 wild-type cells lines. These observations, together with some previous studies showing the effectiveness of this type of treatment regardless of the p53 mutational status,20-22 support the hypothesis that this approach can be effective not only in tumors in which the G1/S checkpoint regulators are directly inactivated by mutations but also in tumors in which these regulators are inactivated by other defects in their pathway. Thus, the ineffectiveness of the treatment on the NCI-H2052 cell line seems to be correlated with the well-known intrinsic resistance to apoptosis,31-33 which characterize this cell line and the very aggressive sarcomatoid histotype, rather than to the presence of a wild-type p53.27
We also observed a slight cytotoxicity of MK-1775 alone on the majority of MM cell lines and a more significant antiproliferative activity on the IST-MES2 cell line. This observation is consistent with previous studies showing the effectiveness of WEE1 inhibitors as single agents on some tumor cell types.34-36 A possible explanation could be that WEE1 inhibition might itself cause DNA damage and, consequently, cell death, by allowing cells with DNA damage to enter an aberrant mitosis, as previously suggested.36 Support for this hypothesis comes from a recent study showing a novel WEE1 function in controlling DNA replication and maintaining genomic stability.37
To rule out a possible MK-1775-induced increase in cisplatin cytotoxicity on non-neoplastic cells, we also tested the drug combination on primary human skin fibroblasts. Consistent with previous studies showing the selective action of MK-1775 toward cancer cells,18,20 we found that MK-1775 did not significantly enhance the cisplatin cytotoxic effect on fibroblasts. This observation further supports the hypothesis that non-neoplastic cells are unaffected by this type of treatment because normal cells retain their G1/S checkpoint activity and are protected from premature entry into mitosis.6
To study the mechanism underlying the MK-1775-induced sensitization to cisplatin in MM cells, we first verified the inhibition of the enzymatic activity of WEE1 by evaluating the phosphorylation of its substrate, the key mitotic protein CDK1. We observed a decrease in the inactive phosphorylated form of CDK1 (phospho-CDK1 Tyr15) in all MM cell lines treated with the two drug combination with respect to cells treated with cisplatin alone, which can be indicative of a G2/M checkpoint inactivation. Consistently, we also observed a decrease in G2/M accumulation and an increase in mitotic entry of cells harboring unrepaired DNA lesions, which ultimately led to an increase in apoptosis in two MM cell lines (NCI-H2452 and MSTO-211H) representative of the epithelioid and biphasic histotypes.
A similar approach, using doxorubicin in combination with a siRNA against another key G2/M checkpoint regulator, the checkpoint kinase 1 (CHK1), was recently tested on NCI-H2452 and MSTO-211H MM cell lines.38 Consistent with our data, the authors observed a decrease in cells in G2/M phase and an increase in cell death in NCI-H2452 cell line co-treated with the two agents with respect to cells treated with doxorubicin alone. Conversely, the CHK1-siRNA did not sensitize MSTO-211H cells to doxorubicin. This discrepancy with our data, which showed the effectiveness of this type of approach also on MSTO-211H cells, could be explained by a difference in efficiency between the different types of agents used in the two studies.
In conclusion, we observed that MK-1775 sensitized epithelioid and biphasic MM cell lines to cisplatin, regardless of the p53 mutational status. The enhancement in cisplatin toxicity was accompanied by a reduction in the inactive phosphorylated form of CDK1 and a shift in the balance toward apoptosis rather than growth arrest. Moreover, we observed that this treatment selectively affected cancer cells without enhancing cisplatin cytotoxicity on normal cells. Therefore, our findings suggest a possible use of MK-1775 in combination with cisplatin for a safe and efficient treatment of epithelioid and biphasic MM. Although this two-drug combination has yet to be assessed in vivo in animal models of mesothelioma, this therapeutic approach has the potential to be rapidly translated to the clinic considering that MK-1775 is already being tested in clinical trials and patients with MM urgently need new therapeutic strategies.
Materials and Methods
Cell culture
NCI-H2052, MSTO-211H, and NCI-H2452 mesothelioma cell lines were purchased from American Type Culture Collection (ATCC). IST-MES1 and IST-MES2 mesothelioma cell lines were obtained from National Institute for Cancer Research, Genova, Italy and REN mesothelioma cells were kindly provided by Giovanni Gaudino (University of Hawaii Cancer Center, Manoa). Primary human skin fibroblasts were a kind gift of Michele Fimiani, Giancarlo Mariotti, and Stefania Mei (University of Siena, Italy).39 Cells were maintained in RPMI-1640 (NCI-H2052, MSTO-211H, and NCI-H2452) and DMEM (REN, IST-MES1, IST-MES2, and primary human skin fibroblasts) containing 10% fetal bovine serum and 2 mM l-glutamine. All cell lines were cultured at 37 °C in a humidified atmosphere containing 5% CO2.
Co-treatments with cisplatin and MK-1775 and MTS assay
MK-1775 (Axon Medchem) and cisplatin (Calbiochem) were dissolved in DMSO (Sigma-Aldrich) to concentrations of 20 mM and 3.3 mM, respectively, and then diluted in culture medium before use.
MM cell lines and primary human skin fibroblasts were seeded in 96 well plates and after 24 h were treated with MK-1775, at the concentrations of 150 or 250 nM, and cisplatin, at concentrations ranging from 2.5 to 15 µM, used either individually or in combination. Control cells were treated with DMSO at the maximum amount used to deliver the molecules. DMSO had no toxic effect on the cell lines.40 Seventy-two hours after treatment, cell viability was evaluated by MTS assay (CellTiter 96® AQueous One Solution Cell Proliferation Assay, Promega), following the manufacturer instructions, through spectrophotometric reading at two different wavelengths (540 and 630 nm).
IC50 values were calculated using GraphPad Prism version 5.01 for Windows, GraphPad Software. To calculate these values, the cisplatin concentration range was also extended to values higher than 15 μM (up to 50 μM), when necessary. CI values were calculated using the CalcuSyn software (Biosoft).
Protein extraction and western blotting
MM cell lines were seeded in 100 mm diameter petri dishes and, 24 h after seeding, treated with 250 nM ΜK-1775 and cisplatin, used either individually or in combination. We used cisplatin concentrations corresponding to the IC50 values achieved by this chemotherapeutic agent when used in combination with 250 nM MK-1775. Control cells were treated with DMSO alone. Seventy-two hours after treatment, cells were harvested on ice in lysis buffer consisting of 50 mM TRIS-HCl pH 7.5, 50 mM EDTA pH 8, 150 mM NaCl, 1% NP40, 2 mM NaOV, 10 mM NaF, 0.3 mM PMSF, a protease inhibitor cocktail (Roche), and the phosphatase inhibitor cocktail 3 (Sigma-Aldrich).
Equal amounts of proteins (70 µg) per sample were electrophoresed and blotted onto PVDF membranes (Thermo Scientific), which were then blocked in 3% BSA and incubated with antibodies against phospho-CDK1 Tyr15, CDK1 (Cell Signaling), and β-actin (Santa Cruz Biotechnology). After incubation with horseradish peroxidase-conjugated secondary antibodies, signals were detected using the Supersignal West Pico Chemiluminescent Substrate (Pierce) and autoradiography films (Pierce).
Cytofluorimetric analyses of cell cycle profile and apoptosis
NCI-H2452 and MSTO-211H cells were seeded in 100 mm diameter petri dishes and, 24 h after seeding, treated with ΜK-1775 and/or cisplatin or DMSO, as described above. Seventy-two hours after treatment, cells were harvested, washed with PBS and fixed in 70% ice-cold ethanol. Nuclei were stained with 5 µg/ml PI plus 20 µg/ml RNase (Sigma-Aldrich) at 4 °C overnight in the dark and analyzed with a BD FACSCanto II flow cytometer (Becton Dickinson). Data were analyzed using a BD FACSDiva software (Becton Dickinson).
Apoptosis was evaluated by cell staining with an annexin V-FITC antibody and PI (Annexin V-FITC kit, Miltenyi Biotec Inc.) and FACS analysis 72 h after treatment, according to the manufacturer instructions.
Mitosis analysis by nocodazole treatment and acridine orange staining
NCI-H2452 and MSTO-211H cells were grown on coverslips and treated with MK-1775 and/or cisplatin, as described above, plus 50 ng/ml of nocodazole (Sigma-Aldrich) to capture cells entering mitosis. Control cells were treated with DMSO and nocodazole. Eight hours after treatment, cells were washed twice in PBS, fixed in 4% paraformaldehyde for 20 min at room temperature and stained with 40 µg/ml acridine orange (Sigma-Aldrich) in PBS for 5 min at room temperature. Cells were analyzed by fluorescence microscopy using 490 nm band-pass blue excitation filters and a 515 nm long-pass barrier filter. Mitotic cells were identified by their morphology and counted in randomly selected areas.
Immunofluorescence
NCI-H2452 and MSTO-211H cells were grown on coverslips and treated with MK-1775 and/or cisplatin plus nocodazole, as described above. Control cells were treated with DMSO and nocodazole. Eight hours after treatment, cells were fixed in 3% paraformaldehyde for 10 min and permeabilized by 0.5% triton-X 100 for 10 min. Samples were then blocked in 1% BSA for 10 min and then incubated with anti-γ-H2AX (Upstate) and anti-ppH3-FITC conjugated (Cell Signaling) antibodies at 37 °C for 1 h. After washing, samples were incubated with a Cy3-conjugated secondary antibody (Alexa Fluor®, Life Technologies) at 37 °C for 45 min. The coverslips were mounted using the ProLong Gold Antifade Reagent with DAPI (Life Technologies). Images were obtained using a Zeiss Axiovert 100 M confocal microscope.
Caspase-3 enzymatic activity
NCI-H2452 and MSTO-211H cells were seeded and treated for 72 h with ΜK-1775 and/or cisplatin or DMSO, as described above. Caspase-3 enzymatic activity was evaluated in cellular lysates using the Colorimetric Caspase-3 Assay Kit (Sigma-Aldrich) following the manufacturer instructions. This assay is based on the hydrolysis of the peptide substrate acetyl-Asp-Glu-Val-Asp p-nitroanilide by caspase-3, resulting in the release of the p-nitroaniline (pNA) moiety. The absorbance of pNA was measured spectrophotometrically at 405 nm through a microplate reader. pNA pmols were determined by a calibration curve prepared with defined pNA solutions. Caspase-3 activity is expressed as pmol pNA/µg protein × time (hours).
Statistical analysis
Statistical analyses were performed using one-way Anova with Dunnett post-test, to compare all data vs. control, or Tukey post-test, to compare all pairs of data; P < 0.05 was considered to be statistically significant.
Disclosure of Potential Conflicts of Interest
No potential conflict of interest was disclosed.
Acknowledgments
This study was supported by the Sbarro Health Research Organization (http://www.shro.org), Human Health Foundation (http://www.hhfonlus.org), Fondazione T. and L. de Beaumont Bonelli (http://www.giuliotarro.it) and the Commonwealth of Pennsylvania. The authors wish to thank Giovanni Gaudino (University of Hawaii Cancer Center, Manoa) for providing REN cells; Michele Fimiani, Giancarlo Mariotti, and Stefania Mei (University of Siena, Italy) for providing primary human skin fibroblasts; Laura Pisapia and Pasquale Barba (Consiglio Nazionale delle Ricerche-Institute of Genetics and Biophysics, Naples, Italy) for FACS analyses; and Carmelina Antonella Iannuzzi for technical help.
Glossary
Abbreviations:
- CDK1
cyclin-dependent kinase 1
- CHK1
checkpoint kinase 1
- CI
combination index
- DSBs
double-strand breaks
- γ-H2AX
phosphorylated histone H2AX
- IC50
half maximal inhibitory concentration
- MM
malignant mesothelioma
- pHH3
phospho-histone H3
- PI
propidium iodide
- pNA
p-nitroaniline
- RB1
retinoblastoma 1
- SV40
simian virus 40
Footnotes
Previously published online: www.landesbioscience.com/journals/cbt/article/27623
References
- 1.Bridda A, Padoan I, Mencarelli R, Frego M. Peritoneal mesothelioma: a review. MedGenMed. 2007;9:32. [PMC free article] [PubMed] [Google Scholar]
- 2.Robinson BW, Musk AW, Lake RA. Malignant mesothelioma. Lancet. 2005;366:397–408. doi: 10.1016/S0140-6736(05)67025-0. [DOI] [PubMed] [Google Scholar]
- 3.Tsao AS, Wistuba I, Roth JA, Kindler HL. Malignant pleural mesothelioma. J Clin Oncol. 2009;27:2081–90. doi: 10.1200/JCO.2008.19.8523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhou BB, Bartek J. Targeting the checkpoint kinases: chemosensitization versus chemoprotection. Nat Rev Cancer. 2004;4:216–25. doi: 10.1038/nrc1296. [DOI] [PubMed] [Google Scholar]
- 5.Kawabe T. G2 checkpoint abrogators as anticancer drugs. Mol Cancer Ther. 2004;3:513–9. [PubMed] [Google Scholar]
- 6.Levesque AA, Eastman A. p53-based cancer therapies: Is defective p53 the Achilles heel of the tumor? Carcinogenesis. 2007;28:13–20. doi: 10.1093/carcin/bgl214. [DOI] [PubMed] [Google Scholar]
- 7.Lee AY, Raz DJ, He B, Jablons DM. Update on the molecular biology of malignant mesothelioma. Cancer. 2007;109:1454–61. doi: 10.1002/cncr.22552. [DOI] [PubMed] [Google Scholar]
- 8.Prins JB, Williamson KA, Kamp MM, Van Hezik EJ, Van der Kwast TH, Hagemeijer A, Versnel MA. The gene for the cyclin-dependent-kinase-4 inhibitor, CDKN2A, is preferentially deleted in malignant mesothelioma. Int J Cancer. 1998;75:649–53. doi: 10.1002/(SICI)1097-0215(19980209)75:4<649::AID-IJC25>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 9.Sharpless NE, DePinho RA. The INK4A/ARF locus and its two gene products. Curr Opin Genet Dev. 1999;9:22–30. doi: 10.1016/S0959-437X(99)80004-5. [DOI] [PubMed] [Google Scholar]
- 10.Wong L, Zhou J, Anderson D, Kratzke RA. Inactivation of p16INK4a expression in malignant mesothelioma by methylation. Lung Cancer. 2002;38:131–6. doi: 10.1016/S0169-5002(02)00178-2. [DOI] [PubMed] [Google Scholar]
- 11.Gazdar AF, Butel JS, Carbone M. SV40 and human tumours: myth, association or causality? Nat Rev Cancer. 2002;2:957–64. doi: 10.1038/nrc947. [DOI] [PubMed] [Google Scholar]
- 12.Testa JR, Giordano A. SV40 and cell cycle perturbations in malignant mesothelioma. Semin Cancer Biol. 2001;11:31–8. doi: 10.1006/scbi.2000.0344. [DOI] [PubMed] [Google Scholar]
- 13.Wang Y, Li J, Booher RN, Kraker A, Lawrence T, Leopold WR, Sun Y. Radiosensitization of p53 mutant cells by PD0166285, a novel G(2) checkpoint abrogator. Cancer Res. 2001;61:8211–7. [PubMed] [Google Scholar]
- 14.Wang Y, Decker SJ, Sebolt-Leopold J. Knockdown of Chk1, Wee1 and Myt1 by RNA interference abrogates G2 checkpoint and induces apoptosis. Cancer Biol Ther. 2004;3:305–13. doi: 10.4161/cbt.3.3.697. [DOI] [PubMed] [Google Scholar]
- 15.Hirai H, Iwasawa Y, Okada M, Arai T, Nishibata T, Kobayashi M, Kimura T, Kaneko N, Ohtani J, Yamanaka K, et al. Small-molecule inhibition of Wee1 kinase by MK-1775 selectively sensitizes p53-deficient tumor cells to DNA-damaging agents. Mol Cancer Ther. 2009;8:2992–3000. doi: 10.1158/1535-7163.MCT-09-0463. [DOI] [PubMed] [Google Scholar]
- 16.Hirai H, Arai T, Okada M, Nishibata T, Kobayashi M, Sakai N, Imagaki K, Ohtani J, Sakai T, Yoshizumi T, et al. MK-1775, a small molecule Wee1 inhibitor, enhances anti-tumor efficacy of various DNA-damaging agents, including 5-fluorouracil. Cancer Biol Ther. 2010;9:514–22. doi: 10.4161/cbt.9.7.11115. [DOI] [PubMed] [Google Scholar]
- 17.Indovina P, Giordano A. Targeting the checkpoint kinase WEE1: selective sensitization of cancer cells to DNA-damaging drugs. Cancer Biol Ther. 2010;9:523–5. doi: 10.4161/cbt.9.7.11276. [DOI] [PubMed] [Google Scholar]
- 18.Bridges KA, Hirai H, Buser CA, Brooks C, Liu H, Buchholz TA, Molkentine JM, Mason KA, Meyn RE. MK-1775, a novel Wee1 kinase inhibitor, radiosensitizes p53-defective human tumor cells. Clin Cancer Res. 2011;17:5638–48. doi: 10.1158/1078-0432.CCR-11-0650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rajeshkumar NV, De Oliveira E, Ottenhof N, Watters J, Brooks D, Demuth T, Shumway SD, Mizuarai S, Hirai H, Maitra A, et al. MK-1775, a potent Wee1 inhibitor, synergizes with gemcitabine to achieve tumor regressions, selectively in p53-deficient pancreatic cancer xenografts. Clin Cancer Res. 2011;17:2799–806. doi: 10.1158/1078-0432.CCR-10-2580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sarcar B, Kahali S, Prabhu AH, Shumway SD, Xu Y, Demuth T, Chinnaiyan P. Targeting radiation-induced G(2) checkpoint activation with the Wee-1 inhibitor MK-1775 in glioblastoma cell lines. Mol Cancer Ther. 2011;10:2405–14. doi: 10.1158/1535-7163.MCT-11-0469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.PosthumaDeBoer J, Würdinger T, Graat HC, van Beusechem VW, Helder MN, van Royen BJ, Kaspers GJ. WEE1 inhibition sensitizes osteosarcoma to radiotherapy. BMC Cancer. 2011;11:156. doi: 10.1186/1471-2407-11-156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mir SE, De Witt Hamer PC, Krawczyk PM, Balaj L, Claes A, Niers JM, Van Tilborg AA, Zwinderman AH, Geerts D, Kaspers GJ, et al. In silico analysis of kinase expression identifies WEE1 as a gatekeeper against mitotic catastrophe in glioblastoma. Cancer Cell. 2010;18:244–57. doi: 10.1016/j.ccr.2010.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cozzi M, Giorgi F, Marcelli E, Pentimalli F, Forte IM, Schenone S, D’Urso V, De Falco G, Botta M, Giordano A, et al. Antitumor activity of new pyrazolo[3,4-d]pyrimidine SRC kinase inhibitors in Burkitt lymphoma cell lines and its enhancement by WEE1 inhibition. Cell Cycle. 2012;11:1029–39. doi: 10.4161/cc.11.5.19519. [DOI] [PubMed] [Google Scholar]
- 24.O’Connell MJ, Raleigh JM, Verkade HM, Nurse P. Chk1 is a wee1 kinase in the G2 DNA damage checkpoint inhibiting cdc2 by Y15 phosphorylation. EMBO J. 1997;16:545–54. doi: 10.1093/emboj/16.3.545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Leijen S, Schellens JH, Shapiro G, Pavlick R, Tibes T, Demuth J, Viscusi J, Cheng JD, Xu Y, Oza AM. A phase I pharmacological and pharmacodynamics study of MK-1775, a Wee1 tyrosine kinase inhibitor, in monotherapy and combination with gemcitabine, cisplatin, or carboplatin in patients with advanced solid tumors. J Clin Oncol. 2010;28:15s. [Google Scholar]
- 26.Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 27.Manfredi JJ, Dong J, Liu WJ, Resnick-Silverman L, Qiao R, Chahinian P, Saric M, Gibbs AR, Phillips JI, Murray J, et al. Evidence against a role for SV40 in human mesothelioma. Cancer Res. 2005;65:2602–9. doi: 10.1158/0008-5472.CAN-04-2461. [DOI] [PubMed] [Google Scholar]
- 28.Pietruska JR, Kane AB. SV40 oncoproteins enhance asbestos-induced DNA double-strand breaks and abrogate senescence in murine mesothelial cells. Cancer Res. 2007;67:3637–45. doi: 10.1158/0008-5472.CAN-05-3727. [DOI] [PubMed] [Google Scholar]
- 29.Di Marzo D, Forte IM, Indovina P, Di Gennaro E, Rizzo V, Giorgi F, Mattioli E, Iannuzzi CA, Budillon A, Giordano A, et al. Pharmacological targeting of p53 through RITA is an effective antitumoral strategy for malignant pleural mesothelioma. Cell Cycle. 2014;13:652–65. doi: 10.4161/cc.27546. [DOI] [PubMed] [Google Scholar]
- 30.Brooks CL, Gu W. p53 ubiquitination: Mdm2 and beyond. Mol Cell. 2006;21:307–15. doi: 10.1016/j.molcel.2006.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Villanova F, Procopio A, Rippo MR. Malignant mesothelioma resistance to apoptosis: recent discoveries and their implication for effective therapeutic strategies. Curr Med Chem. 2008;15:631–41. doi: 10.2174/092986708783885273. [DOI] [PubMed] [Google Scholar]
- 32.Nilsonne G, Sun X, Nyström C, Rundlöf AK, Potamitou Fernandes A, Björnstedt M, Dobra K. Selenite induces apoptosis in sarcomatoid malignant mesothelioma cells through oxidative stress. Free Radic Biol Med. 2006;41:874–85. doi: 10.1016/j.freeradbiomed.2006.04.031. [DOI] [PubMed] [Google Scholar]
- 33.Aung W, Hasegawa S, Furukawa T, Saga T. Potential role of ferritin heavy chain in oxidative stress and apoptosis in human mesothelial and mesothelioma cells: implications for asbestos-induced oncogenesis. Carcinogenesis. 2007;28:2047–52. doi: 10.1093/carcin/bgm090. [DOI] [PubMed] [Google Scholar]
- 34.Hashimoto O, Shinkawa M, Torimura T, Nakamura T, Selvendiran K, Sakamoto M, Koga H, Ueno T, Sata M. Cell cycle regulation by the Wee1 inhibitor PD0166285, pyrido [2,3-d] pyimidine, in the B16 mouse melanoma cell line. BMC Cancer. 2006;6:292. doi: 10.1186/1471-2407-6-292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Murrow LM, Garimella SV, Jones TL, Caplen NJ, Lipkowitz S. Identification of WEE1 as a potential molecular target in cancer cells by RNAi screening of the human tyrosine kinome. Breast Cancer Res Treat. 2010;122:347–57. doi: 10.1007/s10549-009-0571-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kreahling JM, Gemmer JY, Reed D, Letson D, Bui M, Altiok S. MK1775, a selective Wee1 inhibitor, shows single-agent antitumor activity against sarcoma cells. Mol Cancer Ther. 2012;11:174–82. doi: 10.1158/1535-7163.MCT-11-0529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Domínguez-Kelly R, Martín Y, Koundrioukoff S, Tanenbaum ME, Smits VA, Medema RH, Debatisse M, Freire R. Wee1 controls genomic stability during replication by regulating the Mus81-Eme1 endonuclease. J Cell Biol. 2011;194:567–79. doi: 10.1083/jcb.201101047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Romagnoli S, Fasoli E, Vaira V, Falleni M, Pellegrini C, Catania A, Roncalli M, Marchetti A, Santambrogio L, Coggi G, et al. Identification of potential therapeutic targets in malignant mesothelioma using cell-cycle gene expression analysis. Am J Pathol. 2009;174:762–70. doi: 10.2353/ajpath.2009.080721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pianigiani E, Ierardi F, Mazzanti B, Saccardi R, Cuciti C, Fimiani M. Human de-epidermized dermis as a stem cell carrier. Transplant Proc. 2010;42:2244–6. doi: 10.1016/j.transproceed.2010.05.040. [DOI] [PubMed] [Google Scholar]
- 40.Indovina P, Giorgi F, Rizzo V, Khadang B, Schenone S, Di Marzo D, Forte IM, Tomei V, Mattioli E, D’Urso V, et al. New pyrazolo[3,4-d]pyrimidine SRC inhibitors induce apoptosis in mesothelioma cell lines through p27 nuclear stabilization. Oncogene. 2012;31:929–38. doi: 10.1038/onc.2011.286. [DOI] [PubMed] [Google Scholar]