

Abstract
Oral diseases including periodontal disease and caries are some of the most prevalent infectious diseases in humans. Different microbial species cohabitate and form a polymicrobial biofilm called dental plaque in the oral cavity. Metagenomics using next generation sequencing technologies has produced bacterial profiles and genomic profiles to study the relationships between microbial diversity, genetic variation, and oral diseases. Several oral metagenomic studies have examined the oral microbiome of periodontal disease and caries. Gene annotations in these studies support the association of specific genes or metabolic pathways with oral health and with specific diseases. The roles of pathogenic species and functions of specific genes in oral disease development have been recognized by metagenomic analysis. A model is proposed in which three levels of interactions occur in the oral microbiome that determines oral health or disease.
Keywords: metagenomics, periodontal disease, caries, polymicrobial, biofilm, dental plaque, metabolic pathway, pathogen, oral microbiome, systems biology
Introduction
As development of DNA sequencing technologies have progressed, metagenomics has become a popular approach to microbial analysis. Metagenomics is a DNA sequencing approach in which a large amount of genomic DNA is randomly sheared and shotgun sequenced. Several recent studies have used a metagenomic approach to examine microbial roles in oral diseases. In this review we discuss hypotheses regarding the role of bacteria and recent metagenomic studies aimed at elucidating bacterial roles in oral diseases. We also discuss current and potential applications of metagenomics in oral microbiology.
Oral microorganisms are important for human health
Oral diseases, such as periodontal disease and dental caries are some of the most prevalent infectious diseases of humans. Oral diseases affect nearly all ages, all geographic locations, and all races of people worldwide.1,2 It is reported that up to 90% of the population are affected by periodontal disease.1,3 According to WHO reports, dental caries affects 60–90% of schoolchildren even in developed countries (http://www.who.int/oral_health/publications/report03/en/index.html). Oral diseases are associated with multiple microorganisms of different species in a polymicrobial milieu. The contiguous environment of the oral cavity supports a complex oral community.4-11 More than 700 species are estimated to occur in the human oral cavity and many of them are uncultivated bacteria.6,8
Periodontal disease arises from bacterial infection of periodontal tissues causing gingivitis and periodontitis. Gingivitis involves gum tissue but does not affect the underlying supporting structures of the teeth. In contrast, periodontitis is an inflammatory disease extending deep into the tissues, which causes loss of supporting connective tissue. Periodontitis can result in loss of connective tissue and bone support, and is a major cause of tooth loss in adults.3 Caries, or dental decay, is the ruin of tooth structure by oral bacterial acids produced from microbial fermentation of dietary debris.12 Three features of bacterial species are involved in caries development including biofilm formation, acid production, and acid tolerance.13 Many acidogenic and aciduric bacteria are involved in caries, including Streptococcus mutans and Streptococcus sobrinus, collectively known as mutans streptococci, as well as lactobacilli and other aciduric strains of non-mutans streptococci.14
Bacteria inhabit a biofilm (or plaque) in the oral environment. Biofilm is formed by the adherence of a group of microorganisms to surfaces of both hard and soft tissue in the oral cavity. The bacterial cells of a biofilm are attached to one another and are surrounded by extracellular polymeric substances including exopolysaccharides. Current evidence suggests that biofilm formation is an essential characteristic of these oral infectious diseases. Periodontal diseases can often be resolved by good oral hygiene to eliminate biofilm. Biofilms begin to develop on the teeth within a few hours in the absence of oral hygiene maintenance (such as tooth brushing), and gingivitis may result in 10 to 21 d. In cases of uncomplicated gingivitis, complete tooth cleaning treatment can return the gingival tissue (gums) to a healthy condition in about 1 wk.3 The initial step in biofilm development is adhesion of pioneering microorganisms to the tooth surface.15 Streptococci are the most recognized pioneering colonizers16—for example, S. sanguinis has been isolated from freshly cleaned tooth surfaces within 4 h.17 These pioneering streptococci provide signals and metabolites that attract subsequent bacterial species and enable them to colonize.15 The microorganisms that follow the pioneers interact with not only the pioneer colonizers but also with each other to form the mature biofilm. Some of these later microorganisms are pathogens that can instigate inflammation and cause infectious disease. For example, Aggregatibacter actinomycetemcomitans,18 Fusobacterium nucleatum,19 Porphyromonas gingivalis,20 Prevotella intermedia,21 Tannerella forsythia,22 and Treponema denticola23 are found in biofilms and appear to play a role in the development of periodontal disease.
Oral infectious pathogens have been associated not only with inflammation in the oral cavity but also with multiple systemic diseases24,25 such as infective endocarditis,26,27 bacterial pneumonia,28 diabetes,29 adverse pregnancy outcomes,30 rheumatoid arthritis,31 inflammatory bowel disease,32 and colon cancer.33
Roles of microorganisms in oral infection
The composition of oral microorganisms depends on multiple factors including lifestyle (e.g., diet, oral care habits), health (e.g., oral diseases, host immune responses, genetic susceptibility), and/or physical location in the oral cavity (tongue or tooth surfaces, as well as supragingival or subgingival sites).34
The original hypothesis put forth to explain the role of microorganisms in oral diseases was the non-specific plaque hypothesis, proposed by Schultz-Haudt et al.35 in 1954 and Macdonald et al.36 in 1956, which postulated that accumulation of any microorganisms (or non-specific species) in the biofilm played a role in causing inflammation. In the absence of tooth brushing, Theilade37 found that a complex biofilm, which contained more than 200 different species, was present on the gingival tissues of patients. The numbers of bacterial cells increased and the distribution of bacterial species shifted as destructive periodontitis developed, but no single species appeared in active sites or in inactive sites. A second hypothesis, proposed by Loesche38 in 1979, the specific plaque hypothesis, suggested that specific species were involved with periodontal disease. This hypothesis was based on the fact that multiple studies had identified particular bacterial species, which occurred in large numbers in samples from diseased tissues of patients. Specific immune responses against these bacteria were also found in the patients.38 A third hypothesis, proposed by Marsh in 1994 for explaining bacterial roles in caries and periodontal diseases, the ecological plaque hypothesis,39 postulated that both microorganisms and environmental factors were causative elements of periodontal disease. It was shown that a relatively balanced composition of bacterial species in biofilm (microbial homeostasis) could be significantly shifted, leading to disease, as a consequence of environmental changes. For example, changes in sugar intake, diet, or pH can change the balance of microbiota shifting the biofilm composition toward cariogenic species. In a low-pH environment, the proportions of potentially cariogenic species, such as mutans streptococci and lactobacilli, will increase. Dr Marsh proposed that oral diseases can be prevented not only by targeting the pathogenic species but also by interfering with the processes that drive the breakdown of homeostasis. A fourth hypothesis, which has been put forth recently, is the polymicrobial synergy and dysbiosis (PSD) hypothesis for explaining bacterial roles in periodontal diseases.40 This comprehensive hypothesis postulates that oral diseases are initiated by a synergistic and dysbiotic microbial community rather than by specific oral pathogens. Different members of polymicrobial communities, which in aggregate represent a specific genetic combination, work together to shape and stabilize a disease-provoking microbiota. Certain infectious bacterial species or “keystone pathogens”25 are thought to play important roles in modulating the host response and, through interactions with other pathogens, disrupting homeostasis resulting in dysbiosis, causing periodontal disease.
Metagenomics for studying oral microorganisms
Current evidence suggests that the human is a “super-organism”. More than 1013 individual microorganisms co-exist with the 1012 cells in the human body.41 This collective of microorganisms makes up the human microbiome. Oral microbiota represents a major human microbiome that affects human health because it affects the microbiota at other sites, for example, those of the respiratory and gastrointestinal tracts. To identify and classify uncultivable oral microbial species, many molecular biological techniques have been applied in the past two decades, for example, Restriction Fragment Length Polymorphism (RFLP),42 Random Amplified Polymorphic DNA fingerprinting (RPAD),43 Denaturing Gradient Gel Electrophoresis (DGGE),44 Quantitative Real-time Polymerase Chain Reaction (qPCR),45 Microarray Chip,46 and Checkerboard Hybridization47 and others.48 Recently, the next generation sequence technologies (NGS)49 allow the analysis of a large number of microorganisms in different niches without bacterial culture. Several reviews have described results from studies based on NGS technologies in dissecting the microbiome in oral diseases.8,12,24,48,50,51 Many new species of microorganisms have been identified using DNA sequencing to analyze different environmental niches. For example, new species have been identified in samples from the ocean,52 from a low-complexity microbial biofilm community of acidic water,53 and from soil.54 New genes and metabolic pathways have been recognized through precise analysis of these nucleotide sequences.
NGS technologies have dramatically increased sequencing capabilities. There are currently four popular NGS technologies: Roche 454 pyrosequencing, Illumina HiSeq/MiSeq, ABI SOLiD, and Ion Torrent semiconductor sequencers. These NGS technologies have been applied in studies of the human microbiome including studies of the oral microbiome.55,56 Additional sequencing technologies are being developed that will further increase DNA sequencing capabilities (such as PacBio, microfluidic, or nanopore technologies)57
Two basic DNA sequencing approaches have been commonly applied to study uncultivated oral microbial communities—16S rRNA sequence analysis and metagenomics. The former involves sequencing of the conserved 16S rRNA gene. All bacterial 16S rRNA sequences contain conserved regions (most located in stem regions of the palindromic structure) and relatively variable regions (most located in loop regions of the palindromic structure). The conserved regions of 16S rRNA sequences are used to design PCR primers to amplify genes while the sequence differences in the variable regions are used in classification of different bacterial genera. Phylogenetic analysis of these 16S rRNA sequences produces bacterial profiles in oral samples. This method is relatively cost effective and has been used in many oral microbiome studies.6,9,11,58-64
Although 16S rRNA sequence analysis is in some ways straightforward, several aspects should be considered in analysis of polymicrobial profiles. First, the choice of primers is important for amplification of 16S rRNA genes. The primers should cover most (if not all) 16S rRNA genes of known bacterial species; this is accomplished by employing degenerate sequences. The amplicons should contain hypervariable regions of the 16S rRNA gene for classification. Different bacterial profiles can be found by using different primer sets.11 Second, the multiple rounds of PCR amplification that are required in 16S rRNA sequence analysis can affect the abundance of detected operational taxonomic units (OTU) and, therefore, can alter the apparent bacterial community structure.65 Third, this approach bases classification of bacteria to genera on the sequence of hypervariable regions of the 16S rRNA gene. However, it is sometimes difficult to separate closely related bacterial species or to build precise phylogenetic trees from these sequences due to the conservation of 16S rRNA genes and the high degree complexity of bacterial species in the samples.66 Finally, 16S rRNA sequence analysis cannot capture DNA segments and variations in bacterial genomes other than 16S rRNA genes. Many of the DNA sequence differences that are missed in 16S rRNA sequence analysis, such as differences that affect metabolic pathways, may be directly related to health or disease, for example, pathogenicity islands,67 horizontal gene transfer (HGT),68 or mobile genetic elements.69
Metagenomics is another DNA sequencing approach that is used to study uncultivated oral microbial communities. In this approach, whole genome shotgun (WGS) sequencing is used. Entire DNA samples are randomly sheared by a “shotgun” method and sequenced by either classical Sanger sequencing or NGS. The comprehensive sequences can then be analyzed to obtain either bacterial profiles based on 16S rRNA genes or genomic profiles based on whole genomes.54 For obtaining bacterial profiles by metagenomics, the DNA sequences are grouped (binned) by conserved gene sequences (such as 16S rRNA) that are represented in individual genomes of different microorganisms. Metagenomics requires less PCR amplification than 16S rRNA sequence analysis, and therefore, more accurately quantifies individual bacterial species in samples. For obtaining genomic profiles by metagenomics, the shotgun sequence reads are filtered for high-quality sequences, and contaminating human sequences are removed. The filtered sequences are then assembled, based on sequence overlaps, to form longer genomic sequence contigs. Coding sequences in the contigs are identified by computational algorithms. Whole-genome information in the bacterial samples is collected including protein coding regions, RNA genes, repeats, mobile elements, and plasmids. Genes are annotated via data mining and database searches using different algorithms.70 Abnormal genetic segments that are associated with health or disease conditions, such as pathogenicity islands, plasmids, and genes acquired from HGT can be identified.
Although most current studies employ 16S rRNA sequence analysis because of its low cost and lesser computational requirements, 16S rRNA sequence analysis only provides information on microbial taxa. Metagenomics provides more comprehensive information on the oral microbiome. Metagenomic analysis will become more common in oral microbiology research as improvements in sequencing technologies occur and new algorithms are developed for high-speed computational analysis. New sequencing technologies will produce longer sequence reads, high accuracy, shorter processing times, and significantly reduced costs. Many new computational algorithms will help speed the handling of sequence metadata, allow for rapid searching of multiple databases, efficient data mining, and the integration of different genomic information.
Recent Studies of the Oral Microbiome Using Metagenomics
Reference genomes and databases for oral metagenomics
The first complete sequence of a microbial genome was published in 1995.71 Numerous genomes, including numerous oral pathogens, have been sequenced since with support from the NIH. These completed reference genomes provide prodigious amounts of information for studying oral disease pathogens. All of these genomes are publically available, and can be downloaded from the National Center for Biotechnology Information (NCBI), Human Microbiome Project (HMP), or Human Oral Microbiome Database (HOMD). The current HMP contains a large number (~3000) of reference microbial genomes from different human body sites. The HOMD is a database commonly used for oral microbiology research. There are currently over 300 genomes of oral taxa available in HOMD. These microbial reference genomes allow precise bacterial classification and comparative genomic analysis in oral metagenomics. In addition to the updated set of complete genomes of oral bacterial species, the HOMD database also provides a 16S rRNA gene catalog for bacterial species classification in the human oral cavity.6,72 The 16S rRNA gene catalog in the database includes approximately 700 bacterial taxa. Both complete reference genomes and the 16S rRNA gene catalog in the database are essential for metagenomic analysis. The complete genomes provide comprehensive genetic information that can be used in the oral metagenomics for binning of DNA sequences, for annotation of metabolic pathways, and for identification of missed or acquired DNA segments; whereas, the 16S rRNA gene catalog provides information for classification of bacterial profiles.
More complete reference genome sequence projects are underway including the sequencing of genomes from many clinical isolates in comparative genomics and pan-genome studies. The “pan-genome” refers to all genes among the various strains of a bacterial species, and includes both the “core genome”, which is present in all strains, and the “disposable genome”, which is not present in all strains. It is hypothesized that these genome variations are key components permitting adaptive evolution of microorganisms.73 Comparisons of genomes of multiple clinical isolates have been used to identify virulence factors, to associate their geographic and epidemiological relationships, and to study bacterial evolution. For example, genomes of several clinical isolates of S. mutans were compared in a study of evolutionary selection of this caries-causing pathogen by associating their genetic variations with demographic history.74 All isolates of S. mutans analyzed shared a core genome of ~1500 genes. A large number of genes belonging to the disposable genome, which were not present in all isolates, were identified in the pan-genome. The pan-genome contained more than twice the number of genes in the core-genome. Comparison of the genomic sequences suggested that the S. mutans population increased dramatically starting about 10 000 y ago, which corresponds to the time when human agriculture started. Although most evolutionary selection was negative, 14 genes that were related to sugar metabolism and acid tolerance were under positive evolutionary selection. Additional oral pan-genome projects are underway; for example, genomes of clinical isolates of S. sobrinus are currently being sequenced at the J Craig Venter Institute, and genomes of clinical isolates of S. sanguinis have been sequenced at Baylor College of Medicine. The comparison of closely related isolates will be useful in identifying common ancestries, revealing differences in gene content, and elucidating the role of environmental stresses on genome evolution.
Metagenomics provides insight of bacterial profiles
Metagenomic approaches have been applied to analysis of bacterial profiles in the oral microbiome. Metagenomic analysis permits longer, even full-length, 16S rRNA genes to be assembled from sequences and used for classification of bacterial species. The longer 16S rRNA genes obtained from these assembled sequences through metagenomic analysis permit precise taxonomic analysis of bacterial species compared with the much shorter amplicons of specific regions (~400 bp) of 16S rRNA genes obtained in 16S rRNA sequence analysis. In addition, the ability to use all loci in the classification of bacterial species by metagenomics provides for more precise taxonomy. One caveat regarding metagenomic analysis is that sequences have been obtained for far fewer bacterial isolates than are available from 16S rRNA sequence analysis. Thus, because of the limited number of bacterial isolates and the limited number of patient samples, metagenomic analysis may have less power to associate bacterial species with diseases.
Bacterial profiles have been examined in several metagenomics studies. Xie and colleagues examined biofilm samples from a caries-free, periodontally healthy, subject by metagenomics, and identified 12 well characterized phyla, including members of the TM-7 and BRC1 clades from a total of 860 megabases (Mb) of sequence.75 Both pathogens and opportunistic pathogens were found in the samples supporting the ecological plaque hypothesis of oral diseases.75
In a comparison of 15 subgingival plaque samples from two periodontitis patients and three healthy individuals using metagenomics, Liu and colleagues10 found that the disease samples shared a similar bacterial species cluster that was different from the completely healthy samples suggesting that the disease state occupied a narrow region within the space of possible configurations of the oral microbiome. They observed a shift in the oral bacterial composition from a gram-positive dominated community in the healthy subject to a gram-negative dominated community in periodontal disease.10 The shift in bacterial species from gram-positive to gram-negative confirmed previous findings using different molecular biological methods.39 Liu and colleagues also observed higher bacterial diversity in the diseased samples than in the healthy samples, which confirmed results obtained using 16S rRNA sequence analysis.59
In a metagenomic analysis, Wang and colleagues analyzed 16 periodontal samples including 5 swab samples from three healthy, plaque-free subjects and two periodontal patients and 11 dental plaque samples from six healthy subjects and five periodontal patients. These samples represented four periodontal groups: swab of periodontal disease group (H-1), plaque of periodontal disease group (H-2), swab of healthy periodontal tissue group (Z), and plaque of healthy periodontal tissue group (PZ).76 They found a strong correlation between bacterial community structure and disease status, and identified numerous novel microbial inhabitants. They also examined FimA type, an important biofilm gene involved in interactions of P. gingivalis with other microorganisms. They found that the most prevalent P. gingivalis FimA was type II, which is consistent with previous studies.77 They observed that Prevotella species were widely present in both healthy and disease samples. Although some species, such as P. nigrescens (ATCC 33563), P. sp. oral taxon 472, and P. sp. oral taxon 317, were found to be more prevalent in the healthy group, a wider Prevotella taxa was observed in plaques of the periodontal disease group (H-2) than in the healthy group (PZ). These results indicate that a precise classification of bacterial taxa, for example to the species level or better, is necessary to understand the association of the microbiome with oral health conditions.
Belda-Ferre and colleagues employed a metagenomic approach to compare bacterial profiles in 6 supragingival dental plaque samples (from six subjects—two healthy, two with a low number of active caries, and two with a high number of active caries) and in two cavity samples from which sufficient DNA for sequencing was obtained. They found that the array of bacterial species in the caries samples differed from the array of bacteria found in healthy individuals.78 Bacilli and Gammaproteobacteria were more common in the healthy samples—whereas, some typically anaerobic taxa like Clostridiales and Bacteroidetes were more common in the caries samples. It is interesting that higher levels of Streptococcus sanguinis and Aggregatibacter sp. were found in the healthy samples—whereas, higher levels of Streptococcus gordonii and Leptotrichia buccalis were found in the caries samples. They also found that strains of Veillonella parvula were present in all samples but were more abundant in the caries samples. The bacterial profile analysis indicated that tooth cavities were almost complete absent of S. mutans, the originally identified etiological agent, but by a complex community formed of numerous bacterial species including the most common genera, Veillonella, Corynebacterium, or Leptotrichia.78 They proposed that S. mutans might initiate caries and interact with other bacterial species and other species could colonize the niche leading to disease development. For example, it was previously reported the synergistic effect of S. mutans and Veillonella alcalescens in which the mixture of both species produced more acid than either one of them separately.79 The analysis also found that diversity of the bacterial community diminished as caries advanced. Individuals who have never suffered from dental caries do not have mutans streptococci but have high recruitment of other species such as S. sanguinis.78 This finding is consistent with a previously reported analysis showing reduced bacterial diversity concomitant with caries development.80 The reduction in bacterial diversity in caries development may result not only from the harsh acidic environmental but may also be caused by antagonistic factors, such as the mutacins produced by acid tolerant S. mutans.81
Metagenomics identifies specific metabolic pathways and genes associated with oral disease
To compare metabolism in bacterial communities between healthy and periodontal disease samples, Liu and colleagues examined metabolic pathways by metagenomics.10 Sequence reads were assembled into contigs, which were then annotated and assigned to metabolic functions using the Kyoto Encyclopedia of Genes and Genomes (KEGG) database.10 The metabolic pathway analysis indicated different metabolic profiles between healthy and disease samples. The periodontal disease samples were enriched in metabolic pathway genes associated with “a parasitic lifestyle”, such as for utilization of degraded nutrients from lysed host cells and other bacterial cells, genes associated with an intracellular lifestyle, such as for fatty acid metabolism, and genes associated with an anaerobic environment lifestyle, such as for ferredoxin oxidation, acetyl-CoA degradation, and energy-coupling factor class transporters.10 Several pathways were found to be enriched in the healthy samples including fatty acid biosynthesis, aspartate metabolism, and glycerol-3-phosphate metabolism. Certain fatty acids have been reported to have a protective role in periodontal health.82,83 An interesting finding in the analysis by Liu et al. was enrichment for genes involved in homoserine metabolism in healthy samples. Homoserine is a signaling molecule for quorum sensing (a bacterial system of stimulus and response correlated with environmental population density).This result suggests that vigorous metabolic activity for quorum sensing occurs within the healthy microbiome.10 Wang and colleagues also compared metabolic pathway genes between healthy samples and periodontal disease samples. In the subgingival plaques of chronic periodontal patients, they observed under-representation of genes for carbohydrate metabolism, amino acid metabolism, energy metabolism, lipid metabolism, membrane transport, and signal transduction but over-representation of genes for glycan biosynthesis and cell motility.76
Liu and colleagues found that a number of genes for virulence factors, such as type IV secretion systems and the lipid-A of lipopolysaccharide (LPS) biosynthesis, were enriched in the periodontal disease samples.10 LPS (endotoxin) is a major constituent of the cell wall of gram-negative bacteria and a well-known virulence factor involved in oral disease.84 Similarly, Wang and colleagues also found a number of virulence factor genes in periodontal samples including genes for LPS and collagenase PrtC protein. Wang and colleagues also observed significant differences in occurrence of several genes between healthy tissues and periodontal disease samples; these included genes for membrane transport, signal transduction, and cell motility.76 For example, they found that almost all genes involved in bacterial chemotaxis were significantly overrepresented in the supragingival biofilm of periodontal disease samples. They also found a high number of flagellar assembly-related genes in periodontal disease samples. They proposed that the presence of these genes indicated that active motility and colonization were occurring in the periodontal microbiome.
Belda-Ferre and colleagues found more genes for mixed-acid fermentation and DNA uptake (or competence) in samples from active caries78; whereas, they found more genes for antibacterial peptides, periplasmic stress response genes, capsular, and extracellular polysaccharides, and bacitracin stress response genes in samples from healthy periodontal tissues.78 Above metagenomic studies suggest that particular metabolic pathways and specific genes are associated with oral diseases although further in-depth studies are required to confirm the conclusions. Some of these genes may become new diagnostic markers for clinical analysis.
Xie and colleagues found many resistance genes for antibiotics and toxic compounds (representing 2.8% of the total predicted genes coding for proteins) in oral biofilm samples.75 Among these were antibiotic resistance genes for the major classes of antibiotics and for different resistant mechanisms, such as β-lactams, aminoglycosides, fluoroquinolones, and the peptide antibiotic bacitracin, but also genes for general multidrug or heavy-metal resistance functions, such as efflux pumps. Liu and colleagues also found a number of genes related to antibiotic and metal resistance (mercury and cobalt-zinc-cadmium).10 The occurrence of these various resistance genes identified in the oral cavity implies that the oral microbiota is an important gene pool for antibiotic resistance in the human microbiome.
Metagenomics links bacterial species with metabolic pathways
Belda-Ferre and colleagues compared both bacterial profiles and genomic profiles between caries and healthy samples in an effort to elucidate specific functions played by individual bacterial species in caries.78 They examined gene categories by Clusters of Orthologous Groups (COG), and found that gene categories were not equally distributed. Specific taxonomic groups were associated with specific functions. For example, more genes for defense mechanisms, signal transduction, and carbohydrate metabolism were found in Bacilli, while more genes for cell motility were found in Clostridia. The different distributions of functional genes may explain the different bacterial species associated with healthy tissues (Bacilli) or with caries (Clostridia). They suggested that the dental biofilm from individuals who have never suffered from caries may represent a genetic reservoir of new anti-caries compounds and probiotics.78 One well-known example in the oral cavity is the competition between S. sanguinis and S. mutans,85 in which S. sanguinis produces H2O2, which inhibits S. mutans, while S. mutans produces mutacins, which inhibit S. sanguinis.86,87
By deeper sequence analysis, Belda-Ferre and colleagues found that genomic sequences of V. parvula strains differed between samples from healthy periodontal tissues and samples from caries.78 The Veillonella strains in the caries samples contained a DNA region in a genomic island that included CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats)-associated protein genes, hypothetical protein genes, a gene encoding protein for DNA uptake, and a gene encoding amidophosphoribosyltransferase. However, the Veillonella strains in the two healthy samples did not contain this region.78 This result suggests that these different genomic regions may be related to caries.
Metagenomics for Research of the Oral Microbiome
Advanced technologies can be used for studying the oral microbiome
Metagenomics produce a large amount of genomic information for oral microbiome. Several advanced technologies can be used in this investigation. Figure 1 indicates the relationship of metagenomics to different “omics” methods. The central dogma of molecular biology is DNA to RNA to protein (and to metabolites). Based on genomic information from metagenomics, multiple “omics” approaches will collect massive data and provide different molecule profiles for systems biology analysis. Systems biology is an approach to integrate comprehensive interactions of biological systems and study all components as a whole to discover emergent properties. Several advanced “omics” technologies have been used to investigate molecular activities in microbiome. For example, metaproteomics was used to examine a pooled salivary supernatant from six healthy subjects.88 In the future, we anticipate more advanced “omics” technologies will be used to provide complementary biologic data of metagenomics for studying the oral microbiome. Ultimately, by integrating different data sets using systems biology, the precise effects of the microbiome on the host will be ascertained (Fig. 1).
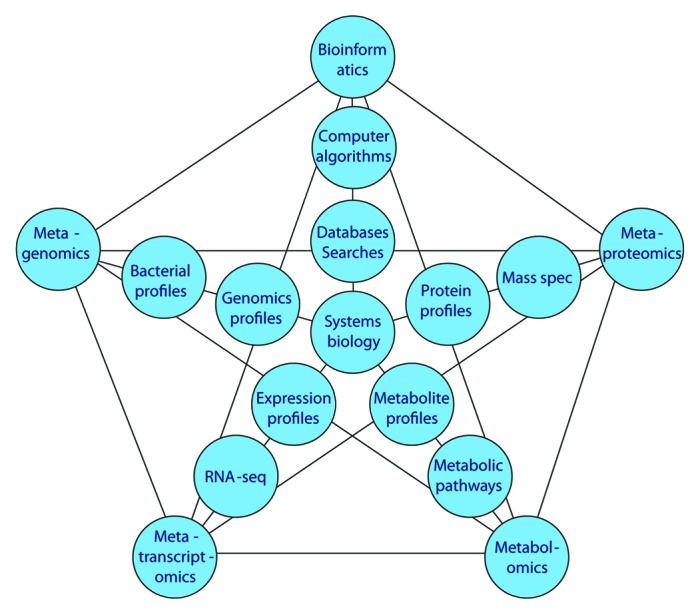
Figure 1. Metagenomics and other “omics” relationships. Current “omics” approaches to metadata analysis and their relationships to oral microbiology research.
Key points in associating oral health and disease by metagenomics
There are several key points that affect oral metagenomic analysis. The first is DNA sequencing.49 Although many improvements have occurred in NGS, current sequencing projects for oral metagenomics remain costly and time consuming. Metagenomics requires much higher sequence coverage than 16S rRNA sequence analysis; whole bacterial genomes average 1.5 to 9.5 Mb compared with 1.5 kb of 16S rRNA genes.89 There is a tradeoff between the length of sequences and the cost. Current sequence reads of NGS are short compared with reads from the Sanger sequencing method. Longer sequence reads will be useful in sequence assembly, data mining, and gene annotation. This key point also includes the preparation of DNA samples for oral metagenomics. DNA preparation has potential to introduce multiple biases that may affect the final results. Representative DNA should be isolated from equally broken cells of different bacterial species. DNA preparation procedures for bacteria, eukaryotes, archaea, and viruses are different. High quality and sufficient quantity of DNA samples are required for obtaining high coverage required for metagenomics. In currently reported metagenomic studies, from 50% to 90% of sequences represent human contaminants even though precautionary steps have been taken.56 It is important to reduce contaminating host DNA, and efforts have been made in this regard.90
A second key point is the collection of patient samples and the study design for oral metagenomics. The oral microbiome varies by location and differs significantly among different locations such as tooth surface, tongue, hard palate, supragingival, or subgingival sites. Human activities and behaviors, such as smoking, diet, brushing or flossing of teeth, also alter the oral microbiome. A detailed protocol for sample collection and preparation for dental metagenomics has been presented by Mullany and colleagues.91 The sample collection procedures depend on the research goals. For example, caries biofilm samples should be precisely collected from tooth sites without contamination from subgingival sites—whereas, periodontitis samples should be collected carefully to reduce human DNA contamination. Sample collection devices may also result in different microbiome compositions, e.g., cotton swab or loop-like device for supragingival samples. Many current reports are based on a small number of samples that may not adequately describe the different environments within the oral cavity. Thus, some of the differences between diseased and healthy tissues previously reported may represent other differences within the oral cavity, rather than differences between diseased and healthy tissue.
A third key point is handling large data sets and performing computational analyses.92 A much larger sequence data set has to be handled in metagenomic analysis than in 16S rRNA sequence analysis. Dramatic increases are expected in the amounts of sequence data to be produced and analyzed in the near future resulting from the development of new sequencing technologies. Larger data sets require more storage capacity and higher transfer speed. The amount of computational analysis required will similarly increase. New algorithms for computational analysis have been and will be developed. One of the obstacles in the computational analyses is to properly assigning new sequences or new genes that are absent in current databases.
The final key point is to associate metagenomics with disease. As indicated in Figure 2, different interactions will affect the composition of the oral microbiome. In addition to issues noted above including numbers of human subjects collected, variations in sample processing, or collection location, there is the fact that new genes identified in metagenomic analysis cannot necessarily be annotated to metabolic pathways. All of these variations present a challenge for reliably associating metagenomic data with oral diseases. Well-designed experiments, sufficient numbers of patient samples, adequate sequence coverage, reliable annotations of gene functions and metabolic pathways, and powerful statistical analysis are essential.
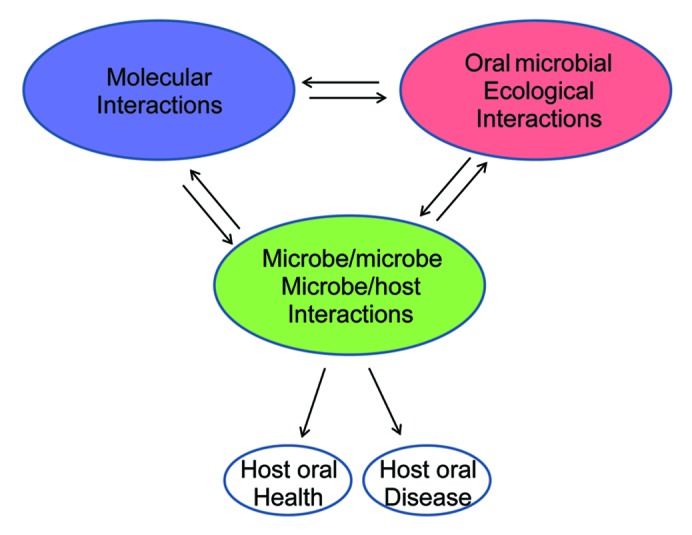
Figure 2. Interactions of the oral microbiome and its relationships with oral health and disease. Different interactions involved in the oral microbiome. Changes in microbial diversity depend on different interactions. Molecular interactions are interactions of bacterial intercellular molecules (DNAs, RNAs, proteins, and metabolites). Microbe/microbe, microbe/host interactions are biological interactions among different organisms in the oral cavity. Microbial ecological interactions are the relationships between microorganisms and the oral environment.
Opportunities in oral metagenomics
The prospects for important contributions from oral metagenomics toward understanding oral disease are many. First, metagenomics promises to provide reliable bacterial profiles unencumbered by the biases inherent in culture-dependent microbiologic methods or PCR-dependent molecular biologic methods. Bacterial profiles will help to identify “keystone” microorganisms in oral diseases. The healthy bacterial profile may be used to build new therapeutic treatments or to manipulate the oral microbiome by introducing probiotic microorganisms or by selectively inhibiting “keystone” pathogens. Second, many reference genomes will be assembled or completed. Multiple genomes can be compared with study microbial genome evolution. The reference genomes of new oral microorganisms will provide chances to study the roles of these microorganisms precisely in the oral cavity. Third, metagenomics will identify new genes or genetic variations (e.g., pathogenicity islands, HGT) that may be associated with oral diseases. Disease-related genetic variations may be used to develop new diagnostic markers for clinical applications. Finally, bacterial profiles can be associated with genomic profiles through metagenomics to understand which microorganism(s) and which genes or metabolic pathways play keystone functions in dysbiosis of oral disease.
Conclusion
Metagenomics is in an early stage of application to the oral microbiome. However, both bacterial profiles and genomic profiles can be examined and compared in metagenomics to study relationships between microbial diversity, genetic variations, and oral diseases. Current data suggest deep complexity of the oral microbiome. We propose a model in which three levels of interactions are involved in the oral community and, consequently, determine oral health or disease (Fig. 2). The first level includes the molecular interactions among biological molecules, such as DNA, RNA, protein, and metabolites. These molecules interact with each other intracellularly or intercellularly in bacterial cells, and determine microbial growth. The second level includes biological interactions between microbes and microbes and host, such as neutralism, amensalism, antagonism, mutualism, commensalism, or parasitism. The different biological interactions shape the composition of the oral microbiome. The third level includes the relationship of microorganisms with the oral environment. For example, oral environmental changes, such as high-sugar diet, low-pH, smoking, and fluoride use, may affect oral microbial diversity. These three levels of interactions work together to determine the microbiome and to affect pathogen capacities. Oral health or disease is a consequence of the effects of the microbiome on the host. A systems biology approach is required to explain the complex interactions of microbiome and host. The advances in metagenomics will lead the way for understanding oral disease.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors thank Drs Todd Kitten and Xiuchun Ge for discussions and suggestions. We have tried to present a comprehensive review of the field, and we apologize to all colleagues whose work we may have overlooked. Research in the author laboratory is supported by grants R01DE018138 (P.X.) and R01DE023078 (P.X.) from the National Institutes of Health.
Glossary
Abbreviations:
- NGS
next generation sequence
- OTU
operational taxonomic units
- PSD
polymicrobial synergy and dysbiosis hypothesis
- PCR
polymerase chain reaction
- HGT
horizontal gene transfer
- WGS
whole genome shotgun
- NCBI
National Center for Biotechnology Information
- HMP
Human Microbiome Project
- HOMD
Human Oral Microbiome Database
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- LPS
lipopolysaccharide
- COG
Clusters of Orthologous Groups
- Mb
mega base
Footnotes
Previously published online: www.landesbioscience.com/journals/virulence/article/28532
References
- 1.Horz HP, Conrads G. Diagnosis and anti-infective therapy of periodontitis. Expert Rev Anti Infect Ther. 2007;5:703–15. doi: 10.1586/14787210.5.4.703. [DOI] [PubMed] [Google Scholar]
- 2.Selwitz RH, Ismail AI, Pitts NB. Dental caries. Lancet. 2007;369:51–9. doi: 10.1016/S0140-6736(07)60031-2. [DOI] [PubMed] [Google Scholar]
- 3.Pihlstrom BL, Michalowicz BS, Johnson NW. Periodontal diseases. Lancet. 2005;366:1809–20. doi: 10.1016/S0140-6736(05)67728-8. [DOI] [PubMed] [Google Scholar]
- 4.Konopka A. What is microbial community ecology? ISME J. 2009;3:1223–30. doi: 10.1038/ismej.2009.88. [DOI] [PubMed] [Google Scholar]
- 5.Aas JA, Paster BJ, Stokes LN, Olsen I, Dewhirst FE. Defining the normal bacterial flora of the oral cavity. J Clin Microbiol. 2005;43:5721–32. doi: 10.1128/JCM.43.11.5721-5732.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dewhirst FE, Chen T, Izard J, Paster BJ, Tanner AC, Yu WH, Lakshmanan A, Wade WG. The human oral microbiome. J Bacteriol. 2010;192:5002–17. doi: 10.1128/JB.00542-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Duran-Pinedo AE, Paster B, Teles R, Frias-Lopez J. Correlation network analysis applied to complex biofilm communities. PLoS One. 2011;6:e28438. doi: 10.1371/journal.pone.0028438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jenkinson HF. Beyond the oral microbiome. Environ Microbiol. 2011;13:3077–87. doi: 10.1111/j.1462-2920.2011.02573.x. [DOI] [PubMed] [Google Scholar]
- 9.McLean JS, Fansler SJ, Majors PD, McAteer K, Allen LZ, Shirtliff ME, Lux R, Shi W. Identifying low pH active and lactate-utilizing taxa within oral microbiome communities from healthy children using stable isotope probing techniques. PLoS One. 2012;7:e32219. doi: 10.1371/journal.pone.0032219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu B, Faller LL, Klitgord N, Mazumdar V, Ghodsi M, Sommer DD, Gibbons TR, Treangen TJ, Chang YC, Li S, et al. Deep sequencing of the oral microbiome reveals signatures of periodontal disease. PLoS One. 2012;7:e37919. doi: 10.1371/journal.pone.0037919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ge X, Rodriguez R, Trinh M, Gunsolley J, Xu P. Oral microbiome of deep and shallow dental pockets in chronic periodontitis. PLoS One. 2013;8:e65520. doi: 10.1371/journal.pone.0065520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wade WG. The oral microbiome in health and disease. Pharmacol Res. 2013;69:137–43. doi: 10.1016/j.phrs.2012.11.006. [DOI] [PubMed] [Google Scholar]
- 13.Peterson SN, Snesrud E, Schork NJ, Bretz WA. Dental caries pathogenicity: a genomic and metagenomic perspective. Int Dent J. 2011;61(Suppl 1):11–22. doi: 10.1111/j.1875-595X.2011.00025.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Takahashi N, Nyvad B. The role of bacteria in the caries process: ecological perspectives. J Dent Res. 2011;90:294–303. doi: 10.1177/0022034510379602. [DOI] [PubMed] [Google Scholar]
- 15.Jenkinson HF, Lamont RJ. Oral microbial communities in sickness and in health. Trends Microbiol. 2005;13:589–95. doi: 10.1016/j.tim.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 16.Rickard AH, Gilbert P, High NJ, Kolenbrander PE, Handley PS. Bacterial coaggregation: an integral process in the development of multi-species biofilms. Trends Microbiol. 2003;11:94–100. doi: 10.1016/S0966-842X(02)00034-3. [DOI] [PubMed] [Google Scholar]
- 17.Nyvad B, Kilian M. Microbiology of the early colonization of human enamel and root surfaces in vivo. Scand J Dent Res. 1987;95:369–80. doi: 10.1111/j.1600-0722.1987.tb01627.x. [DOI] [PubMed] [Google Scholar]
- 18.Fives-Taylor PM, Meyer DH, Mintz KP, Brissette C. Virulence factors of Actinobacillus actinomycetemcomitans. Periodontol 2000. 1999;20:136–67. doi: 10.1111/j.1600-0757.1999.tb00161.x. [DOI] [PubMed] [Google Scholar]
- 19.Témoin S, Wu KL, Wu V, Shoham M, Han YW. Signal peptide of FadA adhesin from Fusobacterium nucleatum plays a novel structural role by modulating the filament’s length and width. FEBS Lett. 2012;586:1–6. doi: 10.1016/j.febslet.2011.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lamont RJ, Jenkinson HF. Subgingival colonization by Porphyromonas gingivalis. Oral Microbiol Immunol. 2000;15:341–9. doi: 10.1034/j.1399-302x.2000.150601.x. [DOI] [PubMed] [Google Scholar]
- 21.Malm S, Jusko M, Eick S, Potempa J, Riesbeck K, Blom AM. Acquisition of complement inhibitor serine protease factor I and its cofactors C4b-binding protein and factor H by Prevotella intermedia. PLoS One. 2012;7:e34852. doi: 10.1371/journal.pone.0034852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Settem RP, Honma K, Stafford GP, Sharma A. Protein-linked glycans in periodontal bacteria: prevalence and role at the immune interface. Front Microbiol. 2013;4:310. doi: 10.3389/fmicb.2013.00310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bian J, Liu X, Cheng YQ, Li C. Inactivation of cyclic Di-GMP binding protein TDE0214 affects the motility, biofilm formation, and virulence of Treponema denticola. J Bacteriol. 2013;195:3897–905. doi: 10.1128/JB.00610-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Han YW, Wang X. Mobile microbiome: oral bacteria in extra-oral infections and inflammation. J Dent Res. 2013;92:485–91. doi: 10.1177/0022034513487559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hajishengallis G, Darveau RP, Curtis MA. The keystone-pathogen hypothesis. Nat Rev Microbiol. 2012;10:717–25. doi: 10.1038/nrmicro2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Paik S, Senty L, Das S, Noe JC, Munro CL, Kitten T. Identification of virulence determinants for endocarditis in Streptococcus sanguinis by signature-tagged mutagenesis. Infect Immun. 2005;73:6064–74. doi: 10.1128/IAI.73.9.6064-6074.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xu P, Alves JM, Kitten T, Brown A, Chen Z, Ozaki LS, Manque P, Ge X, Serrano MG, Puiu D, et al. Genome of the opportunistic pathogen Streptococcus sanguinis. J Bacteriol. 2007;189:3166–75. doi: 10.1128/JB.01808-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Paju S, Scannapieco FA. Oral biofilms, periodontitis, and pulmonary infections. Oral Dis. 2007;13:508–12. doi: 10.1111/j.1601-0825.2007.01410a.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moodley A, Wood NH, Shangase SL. The relationship between periodontitis and diabetes: a brief review. SADJ. 2013;68:260–, 262-4. [PubMed] [Google Scholar]
- 30.Han YW. Oral health and adverse pregnancy outcomes - what’s next? J Dent Res. 2011;90:289–93. doi: 10.1177/0022034510381905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yeoh N, Burton JP, Suppiah P, Reid G, Stebbings S. The role of the microbiome in rheumatic diseases. Curr Rheumatol Rep. 2013;15:314. doi: 10.1007/s11926-012-0314-y. [DOI] [PubMed] [Google Scholar]
- 32.Ismail Y, Mahendran V, Octavia S, Day AS, Riordan SM, Grimm MC, Lan R, Lemberg D, Tran TA, Zhang L. Investigation of the enteric pathogenic potential of oral Campylobacter concisus strains isolated from patients with inflammatory bowel disease. PLoS One. 2012;7:e38217. doi: 10.1371/journal.pone.0038217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Castellarin M, Warren RL, Freeman JD, Dreolini L, Krzywinski M, Strauss J, Barnes R, Watson P, Allen-Vercoe E, Moore RA, et al. Fusobacterium nucleatum infection is prevalent in human colorectal carcinoma. Genome Res. 2012;22:299–306. doi: 10.1101/gr.126516.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Palmer RJ., Jr. Composition and development of oral bacterial communities. Periodontol 2000. 2014;64:20–39. doi: 10.1111/j.1600-0757.2012.00453.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schultz-Haudt S, Bruce MA, Bibby BG. Bacterial factors in nonspecific gingivitis. J Dent Res. 1954;33:454–8. doi: 10.1177/00220345540330040301. [DOI] [PubMed] [Google Scholar]
- 36.MacDonald JB, Sutton RM, Knoll ML, Madlener EM, Grainger RM. The pathogenic components of an experimental fusospirochetal infection. J Infect Dis. 1956;98:15–20. doi: 10.1093/infdis/98.1.15. [DOI] [PubMed] [Google Scholar]
- 37.Theilade E. The non-specific theory in microbial etiology of inflammatory periodontal diseases. J Clin Periodontol. 1986;13:905–11. doi: 10.1111/j.1600-051X.1986.tb01425.x. [DOI] [PubMed] [Google Scholar]
- 38.Loesche WJ. Clinical and microbiological aspects of chemotherapeutic agents used according to the specific plaque hypothesis. J Dent Res. 1979;58:2404–12. doi: 10.1177/00220345790580120905. [DOI] [PubMed] [Google Scholar]
- 39.Marsh PD. Microbial ecology of dental plaque and its significance in health and disease. Adv Dent Res. 1994;8:263–71. doi: 10.1177/08959374940080022001. [DOI] [PubMed] [Google Scholar]
- 40.Hajishengallis G, Lamont RJ. Beyond the red complex and into more complexity: the polymicrobial synergy and dysbiosis (PSD) model of periodontal disease etiology. Mol Oral Microbiol. 2012;27:409–19. doi: 10.1111/j.2041-1014.2012.00663.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bäckhed F, Ley RE, Sonnenburg JL, Peterson DA, Gordon JI. Host-bacterial mutualism in the human intestine. Science. 2005;307:1915–20. doi: 10.1126/science.1104816. [DOI] [PubMed] [Google Scholar]
- 42.Savett DA, Progulske-Fox A. Restriction fragment length polymorphism analysis of two hemagglutinin loci, serotyping and agglutinating activity of Porphyromonas gingivalis isolates. Oral Microbiol Immunol. 1995;10:1–7. doi: 10.1111/j.1399-302X.1995.tb00110.x. [DOI] [PubMed] [Google Scholar]
- 43.Ménard C, Mouton C. Clonal diversity of the taxon Porphyromonas gingivalis assessed by random amplified polymorphic DNA fingerprinting. Infect Immun. 1995;63:2522–31. doi: 10.1128/iai.63.7.2522-2531.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zijnge V, Harmsen HJ, Kleinfelder JW, van der Rest ME, Degener JE, Welling GW. Denaturing gradient gel electrophoresis analysis to study bacterial community structure in pockets of periodontitis patients. Oral Microbiol Immunol. 2003;18:59–65. doi: 10.1034/j.1399-302X.2003.180110.x. [DOI] [PubMed] [Google Scholar]
- 45.Blome B, Braun A, Sobarzo V, Jepsen S. Molecular identification and quantification of bacteria from endodontic infections using real-time polymerase chain reaction. Oral Microbiol Immunol. 2008;23:384–90. doi: 10.1111/j.1399-302X.2008.00440.x. [DOI] [PubMed] [Google Scholar]
- 46.Colombo AP, Boches SK, Cotton SL, Goodson JM, Kent R, Haffajee AD, Socransky SS, Hasturk H, Van Dyke TE, Dewhirst F, et al. Comparisons of subgingival microbial profiles of refractory periodontitis, severe periodontitis, and periodontal health using the human oral microbe identification microarray. J Periodontol. 2009;80:1421–32. doi: 10.1902/jop.2009.090185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sakamoto M, Umeda M, Benno Y. Molecular analysis of human oral microbiota. J Periodontal Res. 2005;40:277–85. doi: 10.1111/j.1600-0765.2005.00793.x. [DOI] [PubMed] [Google Scholar]
- 48.Nyvad B, Crielaard W, Mira A, Takahashi N, Beighton D. Dental caries from a molecular microbiological perspective. Caries Res. 2013;47:89–102. doi: 10.1159/000345367. [DOI] [PubMed] [Google Scholar]
- 49.Metzker ML. Sequencing technologies - the next generation. Nat Rev Genet. 2010;11:31–46. doi: 10.1038/nrg2626. [DOI] [PubMed] [Google Scholar]
- 50.Zaura E. Next-generation sequencing approaches to understanding the oral microbiome. Adv Dent Res. 2012;24:81–5. doi: 10.1177/0022034512449466. [DOI] [PubMed] [Google Scholar]
- 51.Burne RA, Zeng L, Ahn SJ, Palmer SR, Liu Y, Lefebure T, Stanhope MJ, Nascimento MM. Progress dissecting the oral microbiome in caries and health. Adv Dent Res. 2012;24:77–80. doi: 10.1177/0022034512449462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Venter JC, Remington K, Heidelberg JF, Halpern AL, Rusch D, Eisen JA, Wu D, Paulsen I, Nelson KE, Nelson W, et al. Environmental genome shotgun sequencing of the Sargasso Sea. Science. 2004;304:66–74. doi: 10.1126/science.1093857. [DOI] [PubMed] [Google Scholar]
- 53.Tyson GW, Chapman J, Hugenholtz P, Allen EE, Ram RJ, Richardson PM, Solovyev VV, Rubin EM, Rokhsar DS, Banfield JF. Community structure and metabolism through reconstruction of microbial genomes from the environment. Nature. 2004;428:37–43. doi: 10.1038/nature02340. [DOI] [PubMed] [Google Scholar]
- 54.Tringe SG, von Mering C, Kobayashi A, Salamov AA, Chen K, Chang HW, Podar M, Short JM, Mathur EJ, Detter JC, et al. Comparative metagenomics of microbial communities. Science. 2005;308:554–7. doi: 10.1126/science.1107851. [DOI] [PubMed] [Google Scholar]
- 55.Human Microbiome Project Consortium Structure, function and diversity of the healthy human microbiome. Nature. 2012;486:207–14. doi: 10.1038/nature11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Human Microbiome Project Consortium A framework for human microbiome research. Nature. 2012;486:215–21. doi: 10.1038/nature11209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dark MJ. Whole-genome sequencing in bacteriology: state of the art. Infect Drug Resist. 2013;6:115–23. doi: 10.2147/IDR.S35710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dewhirst FE, Klein EA, Thompson EC, Blanton JM, Chen T, Milella L, Buckley CM, Davis IJ, Bennett ML, Marshall-Jones ZV. The canine oral microbiome. PLoS One. 2012;7:e36067. doi: 10.1371/journal.pone.0036067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Griffen AL, Beall CJ, Campbell JH, Firestone ND, Kumar PS, Yang ZK, Podar M, Leys EJ. Distinct and complex bacterial profiles in human periodontitis and health revealed by 16S pyrosequencing. ISME J. 2012;6:1176–85. doi: 10.1038/ismej.2011.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jiang W, Zhang J, Chen H. Pyrosequencing analysis of oral microbiota in children with severe early childhood dental caries. Curr Microbiol. 2013;67:537–42. doi: 10.1007/s00284-013-0393-7. [DOI] [PubMed] [Google Scholar]
- 61.Simón-Soro A, Tomás I, Cabrera-Rubio R, Catalan MD, Nyvad B, Mira A. Microbial geography of the oral cavity. J Dent Res. 2013;92:616–21. doi: 10.1177/0022034513488119. [DOI] [PubMed] [Google Scholar]
- 62.Jiang B, Liang X, Chen Y, Ma T, Liu L, Li J, Jiang R, Chen T, Zhang X, Li S. Integrating next-generation sequencing and traditional tongue diagnosis to determine tongue coating microbiome. Sci Rep. 2012;2:936. doi: 10.1038/srep00936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kraneveld EA, Buijs MJ, Bonder MJ, Visser M, Keijser BJ, Crielaard W, Zaura E. The relation between oral Candida load and bacterial microbiome profiles in Dutch older adults. PLoS One. 2012;7:e42770. doi: 10.1371/journal.pone.0042770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hsiao WW, Li KL, Liu Z, Jones C, Fraser-Liggett CM, Fouad AF. Microbial transformation from normal oral microbiota to acute endodontic infections. BMC Genomics. 2012;13:345. doi: 10.1186/1471-2164-13-345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pinto AJ, Raskin L. PCR biases distort bacterial and archaeal community structure in pyrosequencing datasets. PLoS One. 2012;7:e43093. doi: 10.1371/journal.pone.0043093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Konstantinidis KT, Tiedje JM. Prokaryotic taxonomy and phylogeny in the genomic era: advancements and challenges ahead. Curr Opin Microbiol. 2007;10:504–9. doi: 10.1016/j.mib.2007.08.006. [DOI] [PubMed] [Google Scholar]
- 67.Faruque SM, Mekalanos JJ. Pathogenicity islands and phages in Vibrio cholerae evolution. Trends Microbiol. 2003;11:505–10. doi: 10.1016/j.tim.2003.09.003. [DOI] [PubMed] [Google Scholar]
- 68.Lesic B, Carniel E. Horizontal transfer of the high-pathogenicity island of Yersinia pseudotuberculosis. J Bacteriol. 2005;187:3352–8. doi: 10.1128/JB.187.10.3352-3358.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Malachowa N, DeLeo FR. Mobile genetic elements of Staphylococcus aureus. Cell Mol Life Sci. 2010;67:3057–71. doi: 10.1007/s00018-010-0389-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Thomas T, Gilbert J, Meyer F. Metagenomics - a guide from sampling to data analysis. Microb Inform Exp. 2012;2:3. doi: 10.1186/2042-5783-2-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Fleischmann RD, Adams MD, White O, Clayton RA, Kirkness EF, Kerlavage AR, Bult CJ, Tomb JF, Dougherty BA, Merrick JM, et al. Whole-genome random sequencing and assembly of Haemophilus influenzae Rd. Science. 1995;269:496–512. doi: 10.1126/science.7542800. [see comments] [DOI] [PubMed] [Google Scholar]
- 72.Chen T, Yu WH, Izard J, Baranova OV, Lakshmanan A, Dewhirst FE. The Human Oral Microbiome Database: a web accessible resource for investigating oral microbe taxonomic and genomic information. Database (Oxford) 2010;2010:baq013. doi: 10.1093/database/baq013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tettelin H, Masignani V, Cieslewicz MJ, Donati C, Medini D, Ward NL, Angiuoli SV, Crabtree J, Jones AL, Durkin AS, et al. Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: implications for the microbial “pan-genome”. Proc Natl Acad Sci U S A. 2005;102:13950–5. doi: 10.1073/pnas.0506758102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Cornejo OE, Lefébure T, Bitar PD, Lang P, Richards VP, Eilertson K, Do T, Beighton D, Zeng L, Ahn SJ, et al. Evolutionary and population genomics of the cavity causing bacteria Streptococcus mutans. Mol Biol Evol. 2013;30:881–93. doi: 10.1093/molbev/mss278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Xie G, Chain PS, Lo CC, Liu KL, Gans J, Merritt J, Qi F. Community and gene composition of a human dental plaque microbiota obtained by metagenomic sequencing. Mol Oral Microbiol. 2010;25:391–405. doi: 10.1111/j.2041-1014.2010.00587.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wang J, Qi J, Zhao H, He S, Zhang Y, Wei S, Zhao F. Metagenomic sequencing reveals microbiota and its functional potential associated with periodontal disease. Sci Rep. 2013;3:1843. doi: 10.1038/srep01843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yoshino T, Laine ML, van Winkelhoff AJ, Dahlén G. Genotype variation and capsular serotypes of Porphyromonas gingivalis from chronic periodontitis and periodontal abscesses. FEMS Microbiol Lett. 2007;270:75–81. doi: 10.1111/j.1574-6968.2007.00651.x. [DOI] [PubMed] [Google Scholar]
- 78.Belda-Ferre P, Alcaraz LD, Cabrera-Rubio R, Romero H, Simón-Soro A, Pignatelli M, Mira A. The oral metagenome in health and disease. ISME J. 2012;6:46–56. doi: 10.1038/ismej.2011.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Noorda WD, Purdell-Lewis DJ, van Montfort AM, Weerkamp AH. Monobacterial and mixed bacterial plaques of Streptococcus mutans and Veillonella alcalescens in an artificial mouth: development, metabolism, and effect on human dental enamel. Caries Res. 1988;22:342–7. doi: 10.1159/000261134. [DOI] [PubMed] [Google Scholar]
- 80.Gross EL, Leys EJ, Gasparovich SR, Firestone ND, Schwartzbaum JA, Janies DA, Asnani K, Griffen AL. Bacterial 16S sequence analysis of severe caries in young permanent teeth. J Clin Microbiol. 2010;48:4121–8. doi: 10.1128/JCM.01232-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Merritt J, Qi F. The mutacins of Streptococcus mutans: regulation and ecology. Mol Oral Microbiol. 2012;27:57–69. doi: 10.1111/j.2041-1014.2011.00634.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Campan P, Planchand PO, Duran D. Pilot study on n-3 polyunsaturated fatty acids in the treatment of human experimental gingivitis. J Clin Periodontol. 1997;24:907–13. doi: 10.1111/j.1600-051X.1997.tb01210.x. [DOI] [PubMed] [Google Scholar]
- 83.Kesavalu L, Bakthavatchalu V, Rahman MM, Su J, Raghu B, Dawson D, Fernandes G, Ebersole JL. Omega-3 fatty acid regulates inflammatory cytokine/mediator messenger RNA expression in Porphyromonas gingivalis-induced experimental periodontal disease. Oral Microbiol Immunol. 2007;22:232–9. doi: 10.1111/j.1399-302X.2007.00346.x. [DOI] [PubMed] [Google Scholar]
- 84.Jain S, Darveau RP. Contribution of Porphyromonas gingivalis lipopolysaccharide to periodontitis. Periodontol 2000. 2010;54:53–70. doi: 10.1111/j.1600-0757.2009.00333.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Caufield PW, Dasanayake AP, Li Y, Pan Y, Hsu J, Hardin JM. Natural history of Streptococcus sanguinis in the oral cavity of infants: evidence for a discrete window of infectivity. Infect Immun. 2000;68:4018–23. doi: 10.1128/IAI.68.7.4018-4023.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kreth J, Merritt J, Shi W, Qi F. Competition and coexistence between Streptococcus mutans and Streptococcus sanguinis in the dental biofilm. J Bacteriol. 2005;187:7193–203. doi: 10.1128/JB.187.21.7193-7203.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Chen L, Ge X, Dou Y, Wang X, Patel JR, Xu P. Identification of hydrogen peroxide production-related genes in Streptococcus sanguinis and their functional relationship with pyruvate oxidase. Microbiology. 2011;157:13–20. doi: 10.1099/mic.0.039669-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Jagtap P, McGowan T, Bandhakavi S, Tu ZJ, Seymour S, Griffin TJ, Rudney JD. Deep metaproteomic analysis of human salivary supernatant. Proteomics. 2012;12:992–1001. doi: 10.1002/pmic.201100503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Raes J, Korbel JO, Lercher MJ, von Mering C, Bork P. Prediction of effective genome size in metagenomic samples. Genome Biol. 2007;8:R10. doi: 10.1186/gb-2007-8-1-r10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hunter SJ, Easton S, Booth V, Henderson B, Wade WG, Ward JM. Selective removal of human DNA from metagenomic DNA samples extracted from dental plaque. J Basic Microbiol. 2011;51:442–6. doi: 10.1002/jobm.201000372. [DOI] [PubMed] [Google Scholar]
- 91.Mullany P, Hunter S, Allan E. Metagenomics of dental biofilms. Adv Appl Microbiol. 2008;64:125–36. doi: 10.1016/S0065-2164(08)00404-8. [DOI] [PubMed] [Google Scholar]
- 92.Segata N, Boernigen D, Tickle TL, Morgan XC, Garrett WS, Huttenhower C. Computational meta’omics for microbial community studies. Mol Syst Biol. 2013;9:666. doi: 10.1038/msb.2013.22. [DOI] [PMC free article] [PubMed] [Google Scholar]