

Polyubiquitination is a post-translational modification process, which modulates a myriad of biological processes. Its dysregulation is associated with the development of human cancer and many other diseases, implicating the potential of targeting the ubiquitination system as a therapeutic strategy. Skp2, as an F-box protein of SCF E3 ubiquitin ligase, directly interacts with Skp1 and indirectly associates with Cullin1 and Rbx1 to bridge E2-conjugating enzyme with its protein substrate to execute its E3 ligase activity. Skp2 is proposed to be an oncoprotein because of its ability to trigger Lys 48 ubiquitination and destruction of p27, a cell cycle inhibitor.1 In line with this notion, Skp2 is overexpressed in multiple cancer types. As a result, many attempts have been made to develop small-molecule inhibitors to target Skp2-mediated p27 ubiquitination. Using high-throughput screening or in silico approaches, Chen et al. and Wu et al. identified small-molecule inhibitors that attenuate p27 ubiquitination and subsequent cell cycle progression.2,3 However, both groups did not further examine whether their small molecules displayed antitumor activity, and thus it is unclear whether these compounds could be used for cancer treatment. In addition, our previous finding illustrated that Skp2 orchestrates tumorigenesis through promoting Akt Lys-63 ubiquitination, membrane translocation, and activation upon Her2/Neu overexpression,4 highlighting the need for developing a specific Skp2 inhibitor that globally impairs Skp2 SCF E3 ligase activity.
Our recent work published in Cell reveals a specific Skp2 inhibitor, termed compound #25 (also named as SZL-P1–41), which physically binds to the F-box domain of Skp2 to prevent Skp1 association and Skp2 SCF complex formation.5 Consequently, compound #25, like Skp2 deficiency, augments p27-mediated apoptosis/senescence, while it impairs Akt-driven glycolysis. Such effects collectively restrain growth in multiple differentiated cancer cells. The efficacy of compound #25 is also validated but not limited to restrict tumor progression in lung and prostate cancer models.5 Strikingly, we uncover an unrecognized role of Skp2 targeting in diminishing cancer stem cell properties. Given that cancer stem cells are generally refractory to chemotherapy, the existence of cancer stem cells is regarded as one major cause of drug resistance and treatment failure. Indeed, combination of compound #25 and chemotherapeutic agents like doxorubicin and cyclophosphamide results in synergistic lethality. This underscores that compound #25 may be beneficial for enhancing the efficacy of chemotherapies currently used in clinic.
The anticancer activity of compound #25 demonstrated in our study proves that our rigorous in silico modeling platform is a powerful tool for drug discovery and development.5 It started with a diverse ligand data set of drug-like and lead-like compounds that were then subjected to a cascade of computational analyses for lead identification. The resulting hits from these studies were then individually inspected for a variety of computational parameters, and the best candidates were selected for further experimental evaluations. In particular, we investigated the mode by which the ligand occupied the Skp2/Skp1 interface, the ranking and predicted binding affinities, and the diversity of molecular scaffolds. The higher scrutiny at this stage ensured that the diversity of compounds was high, and that the compounds would strongly disrupt Skp2–Skp1 interactions. The validated computational hot spot analysis6 allowed us to predict that the residues Trp97 and Asp98 are essential for the compound #25 binding. Consistent with this notion, our mutagenesis assays and structure–activity relationship (SAR) analysis confirmed that such binding conveys the biological activities of compound #25 on impairing Skp2 SCF ligase activity and cancer cell growth. Further chemical modification of compound #25 will be performed to improve its binding affinity based on the information obtained from our modeling and SAR analysis.5 This will generate even more potent and specific Skp2 inhibitors.
Mounting evidence showed that the amplification of EGFR and activation of PI3K/Akt pathway are frequently observed in glioblastomas.7 As we previously showed that Skp2 is required for EGFR-driven Akt activation,4 it is intriguing to test whether compound #25 also targets glioblastomas and other caner types driven by oncogenic Akt activation. In addition to serving as a cancer therapeutic, compound #25 may have other clinical applications. Sakai and colleagues found that genetic ablation of Skp2 protects mice from development of obesity due to the reduction in the number of adipocytes, which is associated with p27 accumulation in white adipocyte tissues.8 Therefore, it is worthwhile to assess the efficacy of compound #25 on obesity and other metabolic disorders. To date, there are approximately 69 F-box proteins identified in the human genome. While compound #25 specifically targets Skp2 but not β-TrCP and Fbw7, further studies are required to determine whether compound #25 has off-target effects on other members of F-box protein family. Although in the last decade, targeting protein–protein interactions remains challenging, our study demonstrates that integrated in silico and experimental approaches prove to be feasible and cost-efficient in discovering specific small-molecule inhibitors for cancer therapy. (Fig. 1)
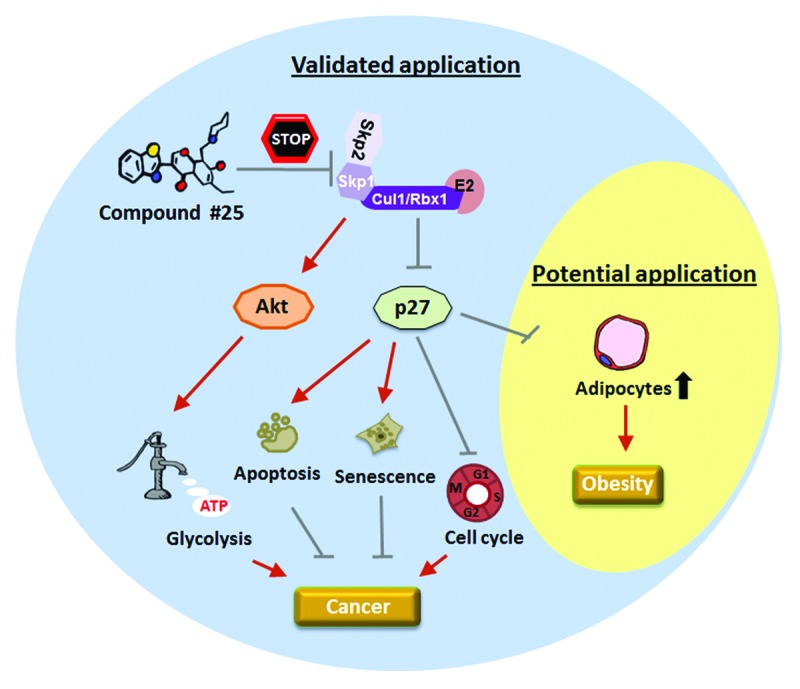
Figure 1. Potential applications of Skp2 inhibitor compound #25. The current study identifies a specific Skp2 inhibitor compound #25 that phenocopies the effect of Skp2 deficiency to target tumorigenesis by upregulation of p27 tumor suppressor and inactivation of Akt oncoprotein. Given that Skp2 loss reduces the number of adipocytes, possibly through p27 accumulation, compound #25 may also be used as a tool for obesity prevention.
Chan CH, et al. Cell. 2013;154:556–68. doi: 10.1016/j.cell.2013.06.048.
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/27853
References
- 1.Hershko DD. Cancer. 2008;112:1415–24. doi: 10.1002/cncr.23317. [DOI] [PubMed] [Google Scholar]
- 2.Chen Q, et al. Blood. 2008;111:4690–9. doi: 10.1182/blood-2007-09-112904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wu L, et al. Chem Biol. 2012;19:1515–24. doi: 10.1016/j.chembiol.2012.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chan CH, et al. Cell. 2012;149:1098–111. doi: 10.1016/j.cell.2012.02.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chan CH, et al. Cell. 2013;154:556–68. doi: 10.1016/j.cell.2013.06.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Morrow JK, et al. Curr Pharm Des. 2012;18:1255–65. doi: 10.2174/138161212799436412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huang PH, et al. Sci Signal. 2009;2:re6. doi: 10.1126/scisignal.287re6. [DOI] [PubMed] [Google Scholar]
- 8.Sakai T, et al. J Biol Chem. 2007;282:2038–46. doi: 10.1074/jbc.M608144200. [DOI] [PubMed] [Google Scholar]