

The discovery of multiple entry and trafficking routes within cells has changed the classical view of endocytosis: once thought to be a simple one-way mechanism to downmodulate plasma membrane (PM) signaling, it is now considered to be a network of intricate and interconnected highly regulated pathways, which allows the resolution of receptor-originated signals in space and time.1 In addition to the extensively studied clathrin-mediated endocytosis (CME) pathway, several other entry portals and routes (globally referred to as non-clathrin endocytosis, NCE) have emerged.2 Notably, there is increasing evidence that individual receptor species can be trafficked through different endocytic routes, frequently in a cell context-dependent fashion.
The epidermal growth factor receptor (EGFR) is a useful model to exemplify this concept.3,4 We demonstrated that the integrated output of CME and NCE directly controls the net levels of EGFR-dependent signaling and consequently the final cellular response. Internalization via CME targets receptors preponderantly to recycling and sustains the EGFR signaling capacity.3 In contrast, EGFR internalization via NCE is mostly associated with receptor degradation in the presence of high ligand concentrations; this mechanism thus safeguards cells against excessive signaling3 (Fig. 1, top).
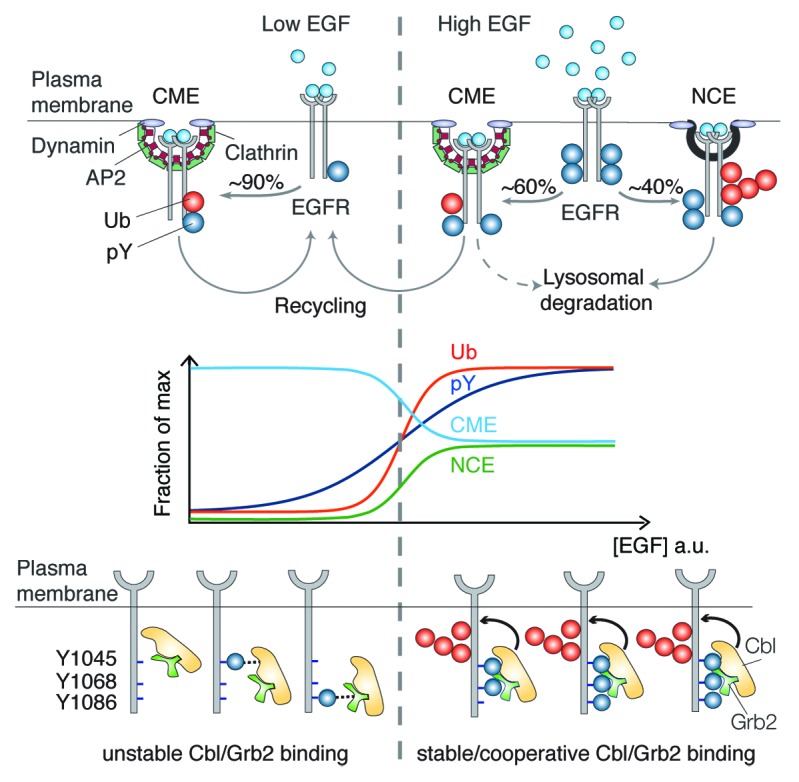
Figure 1. Top: pathways of EGFR internalization at low and high EGF concentration. Middle: schematic representation of EGFR ubiquitination, phosphorylation, and endocytic routes as a function of ligand concentration. Bottom: cooperativity mechanism responsible for the EGFR–Ub threshold. Three phosphotyrosines (pY) are critical for the cooperative recruitment of Cbl to active EGFR: pY1045 binds directly to Cbl, pY1068/pY1086 bind indirectly to Cbl:Grb2 complex.
So how does EGF dosage influence the selection of a particular endocytic pathway, and how are the different pathways coordinated? Our recent work5 has shown that the same range of EGF concentrations that activates EGFR-NCE also causes a sharp increase in EGFR ubiquitination (Fig. 1, middle). We succeeded in mechanistically linking EGFR ubiquitination to EGFR-NCE and demonstrated that this covalent modification functions as a “switch” that commits the EGFR to NCE. The molecular mechanism governing the switch is beautifully designed: upon EGF binding, the EGFR becomes multiply phosphorylated, a modification that permits the cooperative binding of the E3 ligase Cbl, in complex with Grb2, at 2 specific EGFR phosphorylation sites (Fig. 1, bottom). The higher the concentration of EGF, the greater the probability that EGFR is sufficiently phosphorylated at both sites to allow Cbl/Grb2 binding. Efficient recruitment of Cbl results in efficient EGFR ubiquitination and triggers its internalization via NCE. Thus, Cbl acts as an analogical-to-digital converter, allowing cells to react to a linear gradient of ligand with an (almost) all-or-nothing ubiquitination response that, in turn, regulates EGFR internalization via NCE and, ultimately, EGFR fate and signaling capacity.
Much remains to be understood. First, while we have been able to shed some light on the role of NCE in EGFR homeostasis, we are still lacking a full molecular characterization of the components of the EGFR-NCE pathway. Our initial results show that this modality of EGF internalization does not fall into any of the previously described clathrin-independent pathways of internalization. Indeed, EGFR-NCE is independent of caveolin, Arf6, RhoA, CDC42, but dependent on cholesterol, dynamin, eps15, eps15R, and epsin1 (refs. 3–5 and our unpublished results). Furthermore, although it is actin-dependent, EGFR-NCE does not match with macropinocytosis, being Rac-independent while dependent on dynamin (our unpublished results). We are currently characterizing putative new components of this pathway, identified through mass-spectrometry in purified EGFR-containing vesicles internalized via NCE.
Second, while the EGFR-NCE pathway is present in various normal and cancer cell lines,5 its relevance to physiology and to cancer is yet to be demonstrated. This issue is linked to the surprisingly poor knowledge of the physiology of EGFR ligands (half a dozen). In the case of EGF, for instance, we know that the ligand is present in biological fluids in a range of concentrations spanning at least 2 orders of magnitude (reviewed in ref. 3). While this observation lends support to the idea that mechanisms that enable cells to cope with rather different ligand concentrations must exist, it is not clear why EGF concentrations are apparently “locally” controlled, nor have the target cells been defined in the various settings. Furthermore, in some instances, EGF might act as a membrane-bound juxtacrine stimulator, whose internalization might be subjected to different regulation.
Notwithstanding all the unknowns, it has been ascertained that aberrant EGFR signaling contributes to tumorigenesis. Our studies have now highlighted that the EGFR signaling output is the result of complex interactions of multiple EGF-dependent events, such as ligand concentration, EGFR number, endocytic route, and downstream intracellular fate. Therefore, the identification of the parameters that directly control the trigger point of the Ub/NCE threshold as a function of EGF dose, and, consequently, the selection of a particular endocytic route might help in the identification of potentially critical points of intervention for therapeutic purposes. Most likely, to obtain this knowledge and to actualize it into intervention tools, a higher level of “systems” understanding will be required. Thanks to the public availability of many quantitative studies, the EGFR system is amenable to such analytical approaches. In this framework, we are now analyzing the outcomes and predictions of a bottom-up model of EGFR activation/ubiquitination, built on the basis of our results5 and of the many pre-existing models of EGF activation (see for instance refs. 6 and 7). In parallel, there are important ongoing efforts aimed at engineering top-down models accounting for subsequent levels of receptor trafficking, for instance at the level of endosomal dynamics.8 The convergence of these approaches into unified models represents one the most challenging and promising avenues of investigation in the field.
Sigismund S, et al. EMBO J. 2013;32:2140–57. doi: 10.1038/emboj.2013.149.
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/27855
References
- 1.Sigismund S, et al. Physiol Rev. 2012;92:273–366. doi: 10.1152/physrev.00005.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Howes MT, et al. Curr Opin Cell Biol. 2010;22:519–27. doi: 10.1016/j.ceb.2010.04.001. [DOI] [PubMed] [Google Scholar]
- 3.Sigismund S, et al. Dev Cell. 2008;15:209–19. doi: 10.1016/j.devcel.2008.06.012. [DOI] [PubMed] [Google Scholar]
- 4.Sigismund S, et al. Proc Natl Acad Sci U S A. 2005;102:2760–5. doi: 10.1073/pnas.0409817102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sigismund S, et al. EMBO J. 2013;32:2140–57. doi: 10.1038/emboj.2013.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nguyen LK, et al. Cell Commun Signal. 2013;11:52. doi: 10.1186/1478-811X-11-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wiley HS, et al. Trends Cell Biol. 2003;13:43–50. doi: 10.1016/S0962-8924(02)00009-0. [DOI] [PubMed] [Google Scholar]
- 8.Foret L, et al. Curr Biol. 2012;22:1381–90. doi: 10.1016/j.cub.2012.06.021. [DOI] [PubMed] [Google Scholar]