

Acute myeloid leukemia (AML) is a life-threatening stem cell neoplasm that arises from heterogeneous, complex, and dynamic genetic and epigenetic aberrations, resulting in a maturation arrest of neoplastic progenitor cells and consecutive expansion of malignant myeloblasts. To date, we know more than 100 driver mutations occurring in countless combinations, which poses a daunting challenge for the classification and management of AML patients and for the development of targeted therapies. Depending on the type of mutation, the cellular background, and the epigenetic context, driver mutations may converge functionally to deregulate a limited number of cellular processes, which may provide additional therapeutic opportunities.
One promising set of targets are epigenetic regulators maintaining aberrant chromatin states—a common hallmark of AML. The dependency on chromatin-associated genes has recently been probed systematically in an AML mouse model using an advanced RNAi screening method,1 which revealed the BET bromodomain containing protein 4 (BRD4) as the top sensitivity, and led to the testing of JQ1, a small-molecule BET bromodomain inhibitor, in AML. JQ1 treatment precisely recapitulated the potent anti-leukemic effects of RNAi-mediated BRD4 suppression, and showed strong effects in a broad range of AML contexts and subtypes, both in AML cell lines and primary patient-derived cells. In addition, this study revealed that BRD4 suppression directly triggers a breakdown of MYC transcription, implicating BRD4 inhibitors as an effective small-molecule approach to suppress MYC in cancer cells.
BRD4 functions as an epigenetic “reader” of acetylated histones that activates transcription through recruitment of pTEFb2 (Fig. 1) and other interaction partners. More recently, BRD4 has been implicated as a key factor driving oncogene expression through interaction with so-called “super enhancers”3. Promising anti-tumor activities of BET-bromodomain inhibitors have, meanwhile, been reported in a variety of cancer and leukemia types, including AML,1,4 multiple myeloma,5 lung adenocarcinoma,6 and others. Collectively, these studies illustrate that both MYC’s dependency on BRD4 as well as the general sensitivity to BRD4 suppression are extremely variable between different cancer contexts, with IC50 values ranging from 5 nM to >100 µM. However, this context-dependent requirement of BRD4 cannot be fully explained based on its known molecular functions, and so far no genetic, transcriptional, or chromatin marker has been identified to predict responsiveness to BRD4 inhibition. Moreover, in leukemia, where MYC transcription appears to be largely BRD4-dependent, the level of MYC suppression does not directly predict drug sensitivity, suggesting that other BRD4 target genes may play important roles in the response to BET bromodomain inhibition.1
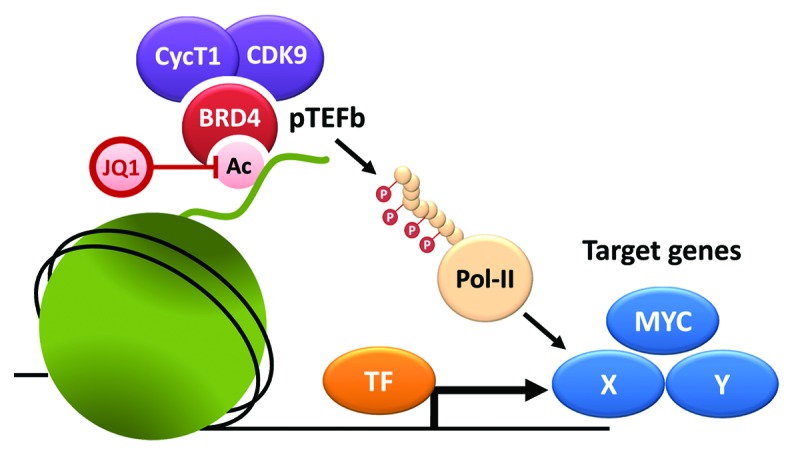
Figure 1. BRD4 recruits pTEFb to promote transcriptional elongation. BRD4 tightly binds acetylated histones via its BET bromodomains; JQ1 competes with this binding and displaces BRD4 from chromatin. One known effector mechanism of BRD4 is the recruitment of pTEFb, which, in turn, promotes transcriptional elongation of target genes through phosphorylation of RNA polymerase II (Pol-II). Inhibitory effects of JQ1 in cancer cells are associated with transcriptional suppression of specific BRD4 target genes, including MYC and others (X and Y). However, the vast context-dependency of these effects cannot be explained based on known BRD4 functions and may depend on direct or indirect interactions with transcription factors (TF).
In AML, BET bromodomain inhibitors show potent anti-leukemic effects, with IC50 values in the submicromolar range in a wide variety of subtypes—way beyond MLL-rearranged leukemia, where BRD4 was initially identified as a candidate target.1,4 Importantly, susceptible contexts include AML cell lines harboring unfavorable aberrations such as TP53 mutations and complex-aberrant karyotypes, as well as samples from patients with chemotherapy-refractory or relapsed AML.1,7 Despite these encouraging results, the curative potential of anti-leukemic agents depends on their ability to attack and eradicate leukemia-initiating cells, also known as leukemic stem cells (LSC).8 Recent data suggest that BRD4 is expressed in the CD34+/CD38− compartment of AML blasts.7 Interestingly, leukemic blasts and LSC, in contrast to normal bone marrow progenitor cells, consistently show strong cytoplasmic BRD4 expression, although the mechanistic relevance of this finding remains elusive. Importantly, JQ1 was found to induce growth arrest and apoptosis in CD34+/CD38+ and CD34+/CD38+ AML cells, and these effects were again independent of the AML subtype or disease stage.7 Together, these ex vivo data suggest that BRD4 may serve as a leukemia stem cell target that is highly expressed in the LSC compartment of most AML patients.
Over the past 2 y, BET bromodomain inhibition has emerged as an entirely novel concept for treatment of AML and other cancer types. Despite very promising pre-clinical findings, evaluating the efficacy and safety of this approach, as well as its ability to eradicate LSCs in vivo, requires further pre-clinical and clinical investigation. The observation that JQ1 synergizes with ARA-C,7 the standard chemotherapeutic agent in current clinical AML therapy, should encourage combinatorial therapy trials. One major challenge for the further development and clinical testing of BET bromodomain inhibitors is the lack of reliable biomarkers to predict sensitivity to these compounds—a problem that is common to many emerging chromatin-targeted therapeutics. In contrast to direct small-molecule inhibitors of driving oncoproteins, where specific DNA mutations serve as reliable biomarkers, the establishment of diagnostic tests guiding the clinical use of BET bromodomain inhibitors will require a better mechanistic understanding about factors determining drug sensitivity and resistance. In the case of BRD4, these may involve multiple effector functions and target genes, which may vary between different cancer contexts. Despite these challenges and remaining questions, the discovery of BRD4 as a candidate target in AML highlights the great potential of systematically exploring non-oncogene addictions in chromatin regulatory networks and beyond.
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/27859
References
- 1.Zuber J, et al. Nature. 2011;478:524–8. doi: 10.1038/nature10334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yang Z, et al. Mol Cell. 2005;19:535–45. doi: 10.1016/j.molcel.2005.06.029. [DOI] [PubMed] [Google Scholar]
- 3.Lovén J, et al. Cell. 2013;153:320–34. doi: 10.1016/j.cell.2013.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dawson MA, et al. Nature. 2011;478:529–33. doi: 10.1038/nature10509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Delmore JE, et al. Cell. 2011;146:904–17. doi: 10.1016/j.cell.2011.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lockwood WW, et al. Proc Natl Acad Sci U S A. 2012;109:19408–13. doi: 10.1073/pnas.1216363109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Herrmann H, et al. Oncotarget. 2012;3:1588–99. doi: 10.18632/oncotarget.733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Valent P, et al. Nat Rev Cancer. 2012;12:767–75. doi: 10.1038/nrc3368. [DOI] [PubMed] [Google Scholar]