

Despite continuous advances in the knowledge of breast cancer pathophysiology, this type of neoplasia remains a leading cause of cancer-related death in women, with more than 450 000 deaths every year worldwide.1 Surgery, coupled with radiation and/or chemotherapy, remains the main approach for breast cancer treatment. However, another common therapeutic option is based on the use of the estrogen receptor (ER) antagonist, tamoxifen.2 Indeed, approximately 60–70% of early-stage breast cancers overexpress the ER, making their growth dependent on estrogens. Tamoxifen blocks estrogen signaling by competitively binding the ER, thus antagonizing its proliferative and pro-survival effects.
Breast cancers can be inherently drug-resistant or develop resistance after exposure to chemotherapeutic drugs, such as the anthracyline, doxorubicin.1 Resistance can also develop in patients receiving tamoxifen.2 Therefore, it is very important to understand how breast cancers become drug- and hormone-resistant, and whether or not their drug-resistance can be reversed. There also is a need for novel targeted therapies for breast cancer patients who develop resistance to traditional therapies.
GSK-3β is a serine/threonine kinase involved in several signal transduction cascades, including the PI3K/Akt/mTOR, Wnt/β-catenin, and MEK/ERK pathways.3 In particular, Akt and other kinases phosphorylate GSK-3β at Ser9. This phosphorylative event inactivates GSK-3β3. GSK-3β regulates cell cycle progression, differentiation, survival, embryogenesis, migration, and metabolism. However, aberrant GSK-3β activity has been linked with an increasing number of pathologies, including cancer, heart disease, immune system disorders, diabetes, atherosclerosis, and different neurological diseases.3
Although its role in cancer remains controversial, GSK-3β has also been implicated in breast cancer development and drug resistance.4,5 Using ER-positive MCF-7 breast cancer cells, Sokolosky and coworkers have performed a detailed analysis of the roles played by GSK-3β in causing resistance to doxorubicin or tamoxifen.6 They found that MCF-7 cells overexpressing a kinase-dead (KD) form of GSK-3β were more resistant to doxorubicin and tamoxifen compared with MCF-7 cells overexpressing either wild-type (WT) or constitutively active (CA) GSK-3β. Ectopic expression of GSK-3β(KD) also resulted in increased clonogenic activity of MCF-7 cells in comparison with either GSK-3β(WT) or GSK-3β(CA) overexpression.
Moreover, treatment of parental MCF-7 and MCF-7/GSK-3β(WT) cells with doxorubicin abrogated the phosphorylation of GSK-3β at Ser9. In contrast, Ser9 p-GSK-3β was still detectable in MCF-7/GSK-3β(KD) and MCF-7/GSK-3β(CA) cells. This indicated that one of the effects of doxorubicin on MCF-7 cells was suppression of Ser9 p-GSK-3β, which could then result in increased GSK-3β activity. Downregulation of Ser9 p-GSK-3β levels was detected despite increased Akt activity induced in these cell types by doxorubicin. This finding documented that the control of GSK-3β activity is complex and multi-factorial in MCF-7 cells, as there may be several alternate routes of GSK-3β inactivation that are independent of Akt,3 including downregulation of protein phosphatase activity or upregulation of other kinases (Fig. 1).
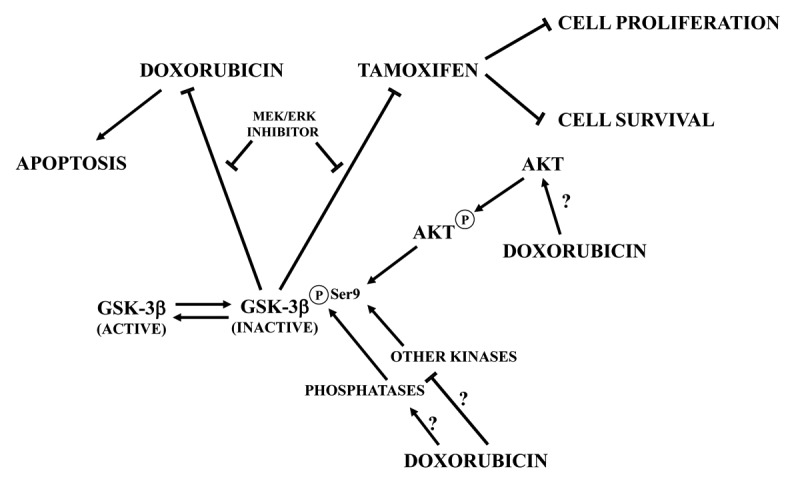
Figure 1. Schematic of the effects of GSK-3β inactivation on MCF-7 cell sensitivity to doxorubicin or tamoxifen. Arrows indicate activating events, whereas perpendicular lines indicate inhibitory events. At present, it is not clear how doxorubicin could dephosphorylate GSK-3β and activate Akt at the same time. Doxorubicin could either inhibit other protein kinases, targeting Ser9 p-GSK-3β, or stimulate protein phosphatases.
What is even more interesting, MCF-7/GSK-3β(KD) cells displayed an elevated sensitivity to the mTORC1 inhibitor, rapamycin, compared with MCF-7/GSK-3β(WT) or MCF-7/GSK-3β(CA) cells, while concurrent MEK/ERK inhibition alleviated doxorubicin and tamoxifen resistance in MCF-7/GSK-3β(KD) cells (Fig. 1). Overall, these results demonstrated that GSK-3β is a key regulatory molecule in sensitivity of breast cancer cells to chemo-, hormonal, and targeted therapy (Fig. 1). Moreover, they fit well with the conclusions of an immunohistochemical study performed on paraffin-embedded samples from 72 consecutive invasive mammary carcinomas, which documented how higher Ser9 p-GSK-3β levels correlated with a worse clinical outcome in ER-positive cases.7 GSK-3β-controlled drug resistance may be mediated by the BH3-only member of the Bcl-2 family, Mcl-1. Indeed, the expression levels of this anti-apoptotic protein inversely correlated with GSK-3β activity (assessed by immunostochemical staining for p-GSK-3β) in breast cancer specimens, and active GSK-3β was found to be required for proteasome-mediated Mcl-1 degradation.8
In conclusion, the findings by Sokolosky et al.6 unequivocally demonstrated that loss of GSK-3β kinase activity could dramatically increase the drug and hormonal resistance of breast cancer cells. However, this may confer an Achilles’ heel by sensitizing cancer cells to targeted therapy with small-molecule kinase inhibitors. Although further studies are needed to determine the clinical relevance of the complex interactions between GSK-3β, mTORC1, and MEK/ERK, it could be envisaged that mTORC1 and MEK/ERK inhibitors should be able to potentiate the effects of chemo- and hormonal therapy, thereby presenting an attractive treatment route for overcoming GSK-3β-mediated drug-resistance in breast cancer.
Sokolosky M, et al. Cell Cycle. 2014;13:820–33. doi: 10.4161/cc.27728.
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/28091
References
- 1.McCubrey JA, et al. Adv Enzyme Regul. 2010;50:285–307. doi: 10.1016/j.advenzreg.2009.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Steelman LS, et al. Cell Cycle. 2011;10:3003–15. doi: 10.4161/cc.10.17.17119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Phukan S, et al. Br J Pharmacol. 2010;160:1–19. doi: 10.1111/j.1476-5381.2010.00661.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Farago M, et al. Cancer Res. 2005;65:5792–801. doi: 10.1158/0008-5472.CAN-05-1021. [DOI] [PubMed] [Google Scholar]
- 5.Dong J, et al. Cancer Res. 2005;65:1961–72. doi: 10.1158/0008-5472.CAN-04-2501. [DOI] [PubMed] [Google Scholar]
- 6.Sokolosky M, et al. Cell Cycle. doi: 10.4161/cc.27728. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Armanious H, et al. Hum Pathol. 2010;41:1657–63. doi: 10.1016/j.humpath.2010.04.015. [DOI] [PubMed] [Google Scholar]
- 8.Ding Q, et al. Cancer Res. 2007;67:4564–71. doi: 10.1158/0008-5472.CAN-06-1788. [DOI] [PubMed] [Google Scholar]