

Abstract
Centromere protein A (CENP-A) is a homolog of histone H3 that epigenetically marks the heterochromatin of chromosomes. CENP-A is a critical component of the cell cycle machinery that is necessary for proper assembly of the mitotic spindle. However, the role of CENP-A in the heart and cardiac progenitor cells (CPCs) has not been previously studied. This study shows that CENP-A is expressed in CPCs and declines with age. Silencing CENP-A results in a decreased CPC growth rate, reduced cell number in phase G2/M of the cell cycle, and increased senescence associated β-galactosidase activity. Lineage commitment is not affected by CENP-A silencing, suggesting that cell cycle arrest induced by loss of CENP-A is a consequence of senescence and not differentiation. CENP-A knockdown does not exacerbate cell death in undifferentiated CPCs, but increases apoptosis upon lineage commitment. Taken together, these results indicate that CPCs maintain relatively high levels of CENP-A early in life, which is necessary for sustaining proliferation, inhibiting senescence, and promoting survival following differentiation of CPCs.
Keywords: CENP-A, cardiac progenitor cell, heart, cell cycle, senescence
Introduction
The past 10 y have witnessed a dramatic increase in knowledge regarding the existence of cardiac progenitor cells (CPCs),1 a resident population of c-kit-positive stem cells within the heart that is responsible for sustaining cardiomyocyte turnover during the course of adult life.1-4 CPCs possess enormous potential for stem cell therapy to facilitate cardiac repair following myocardial infarction (MI). Multiple studies previously published by this laboratory alone have demonstrated the effectiveness of genetically enhanced CPCs in reducing scar size and improving cardiac output following an infarction.5,6 Phase 1 clinical trials have already begun to characterize the safety and efficacy of the use of CPCs for treatment in humans in the SCIPIO trial.7,8 However, there is much that remains to be learned about the biology of the stem cells themselves. While endogenous CPCs are capable of maintaining tissue homeostasis in the adult heart,4 they are inherently unable to sufficiently respond to myocardial challenge.9,10 Though CPCs participate in the response to MI, the heart remains permanently weakened and scarred following an infarction, because resident CPCs alone are unable to repair the massive tissue damage that takes place in the myocardium following MI.9-15 Furthermore, the risk of heart disease increases with age, and this can be attributed in part to the decline in regenerative potential of CPCs due to decreased telomere length and increased cellular senescence with time.16-19 Additional work is needed to understand and reverse the decline in CPC regenerative potential with age, as well as enhance the ability of resident CPCs to respond to myocardial challenge.
In an effort to identify potential factors that limit the regenerative potential of CPCs, this study explores the epigenetic mechanisms that influence CPC proliferation and commitment. Epigenetic regulation of the genome describes the cellular processes that are not encoded in the DNA, ranging from the expression or suppression of specific genes to the regulation of entire regions of a chromosome.20 One protein that plays a pivotal role is centromere-associated protein A (CENP-A), a histone protein homologous to histone H3 that forms structurally unique nucleosomes in the heterochromatic region of the chromosome that defines the centromere.21-23 While the sequence of DNA in this region is not conserved between species or even between members of a species, the centromere is distinguished from other regions of the chromosome through the way in which CENP-A is interspersed with histone H3 along the length of the heterochromatin.24,25 In eukaryotic cells the integration of CENP-A normally takes place during late M and early G1 phase of the cell cycle.25,26 In doing so, CENP-A becomes an epigenetic mark that serves to recruit the various components of the kinetochore to the heterochromatin during mitosis,27 without which the mitotic spindle cannot form, and proper chromosome segregation will not take place.25 This makes proper CENP-A deposition a critical step for the progression of the cell cycle. Depletion of CENP-A severely handicaps the cell cycle machinery and has been shown to cause apoptosis and increased senescence in numerous cell types.28-31
The age-associated decline in CPC regenerative potential in the heart, coupled with the compelling evidence that CENP-A decline is a precursor to senescence and cell death, supports the notion that CENP-A is essential for the cell cycle of CPCs. However, CENP-A has not been previously studied in the context of the heart. This study seeks to understand the role of CENP-A by investigating the levels of CENP-A expression within the heart in vivo, as well as the effect of acute CENP-A knockdown within CPCs in vitro, revealing that increased age correlates with CENP-A decline in the heart, and that premature decline of CENP-A via acute knockdown in cultured CPCs accelerates the age-associated increase in senescence.
Results
CENP-A expression declines in the adult mouse heart
The level of CENP-A in the pre-natal and adult heart was measured in a murine model to characterize CENP-A expression. CENP-A transcription drastically declines in the heart with age, decreasing by 80% within 3 mo as determined by qPCR analysis (Fig. 1A, P < 0.005). Reduced CENP-A expression in the adult mouse is confirmed by immunoblot, where CENP-A protein decreases by 90% (Fig. 1B, P < 0.0005). CENP-A localization was determined by immunohistochemistry of tissue sections. CENP-A is expressed throughout the myocardium in the pre-natal heart (Fig. 1C). However, CENP-A is dramatically reduced in sections of the 3-mo-old myocardium, consistent with assessment of protein expression by immunoblot (Fig. 1C). Given that CENP-A is necessary for cell proliferation, CENP-A expression in proliferating stem cells was measured in vivo.28,32 Pre-natal sections were co-stained for c-kit, phospho histone 3 (pH3), and CENP-A. CENP-A co-localizes with c-kit and pH3 as revealed by confocal analysis, confirming that CENP-A is present in stem cells undergoing mitosis within the heart (Fig. 1D). Furthermore, close inspection of c-kit-positive cells in the 3-mo-old heart indicates CPCs retain CENP-A during adulthood (Fig. 1E).

Figure 1. CENP-A expression declines in the adult heart. (A) qPCR analysis of CENP-A transcription (n = 3). (B) Immunoblot of CENP-A protein expression and quantification (n = 3). (C) Representative staining of CENP-A (green), tropomyosin or actin (red), and the nuclear stain topro (blue) in the pre-natal heart (left panel) and 3-mo-old heart (right panel). (D) Representative staining of c-kit (red) positive stem cells expressing CENP-A (green) and pH3 (white) in the pre-natal heart with respective magnified images. pH3 expression is detected in the mitotic stem cell shown in the far right panel but is absent from the non-mitotic stem cell in the center panel. The nuclear stain sytox is represented in blue. (E) Representative staining of c-kit (red) positive stem cells expressing CENP-A (green) together with actin (white) and nuclei (blue) in the 3-mo-old heart. **P < 0.005, ***P < 0.0005.
CENP-A expression decreases in old CPCs
CENP-A expression was analyzed in cultured CPCs to determine if their expression pattern is similar to what is seen in the heart. CENP-A concentrates around distinct foci in the nucleus, as shown by confocal microscopy, creating a speckled pattern consistent with CENP-A localization to centromeres (Fig. 2A).26,33 Co-staining with c-kit confirms that CENP-A is present in the CPCs (Fig. 2B). CENP-A expression also decreases in old CPCs by 90% relative to CPCs isolated from young mice, consistent with the age-associated decline observed in vivo as determined by immunoblot analysis (Fig. 2C, P < 0.05).

Figure 2. CENP-A declines in old CPCs. (A) Immunocytochemical analysis of CENP-A (green), the actin stain ph633 (red), and nuclear stain sytox (blue) in CPCs cultured in vitro. (B) Immunocytochemical analysis confirming CENP-A staining (green) co-localizes with c-kit (red). (C) Immunoblot demonstrating that CENP-A is significantly diminished in CPCs derived from old mice. Quantification shown in right panel (n = 3). *P < 0.05.
Inhibition of CENP-A results in reduced CPC proliferation
CENP-A was knocked down via targeted silencing with shRNA (CENP-A KD) and compared with scrambled shRNA transduced controls (ScrCon) to determine whether CENP-A is necessary for normal progression of CPCs through the cell cycle. CENP-A expression is reduced by greater than 50% upon knockdown (Fig. 3A, P < 0.005). Reduced CENP-A expression following shRNA treatment is also evident by immunofluorescence microscopy (Fig. S1). The proliferation rate of CENP-A KD CPCs significantly declines as shown by CyQUANT analysis, where after 5 d of culture the number of cells drops by 25% relative to ScrCon CPCs (Fig. 3B, P < 0.05). The number of CENP-A KD CPCs in the G2/M phase of the cell cycle also goes down by 50% relative to ScrCon CPCs (Fig. 3C, P < 0.05). This reduction in the G2/M population is accompanied by a 10% increase of cells in G1, although this augmentation is not statistically significant relative to the vast majority of CPCs that are normally present in G1. Together, these results indicate that CENP-A silencing is causing fewer CPCs to enter G2/M phase.
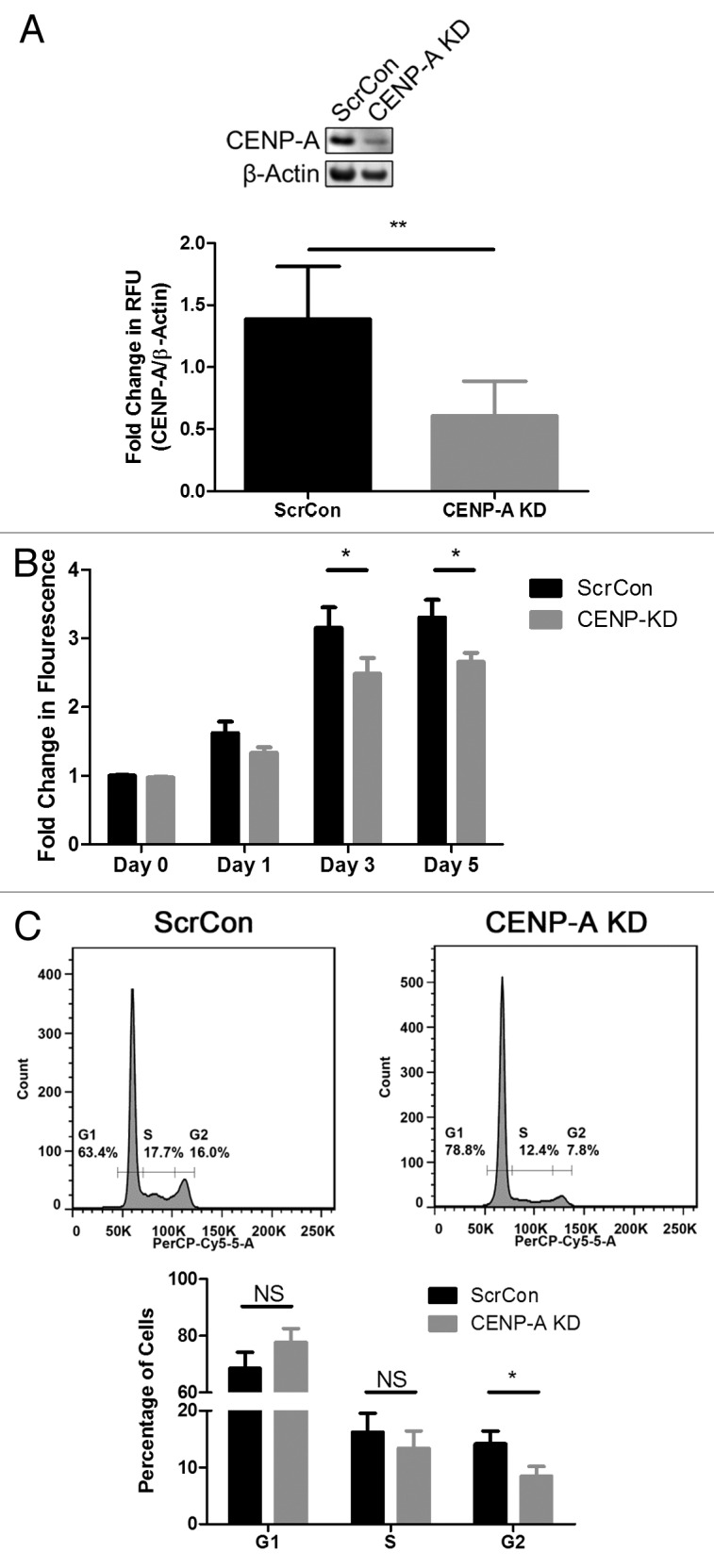
Figure 3. CENP-A silencing slows CPC proliferation. (A) Representative immunoblot showing effective CENP-A knockdown via shRNA (CENP-A KD). Quantification shown in lower panel (n = 3). (B) CyQUANT assay showing slowed growth rate in CENP-A KD CPCs (n = 3). (C) Cell cycle analysis of CENP-A KD CPCs reveals the number of cells in G2 is significantly reduced. Representative histograms shown in top panel, quantification given in lower panel (n = 3). *P < 0.05; **P < 0.005; NS, not significant.
Silencing of CENP-A results in cellular senescence arising prematurely in young CPCs
Given that CENP-A diminution correlates with increased senescence,28,29 young CPCs with CENP-A KD were stained for senescence-associated β-galactosidase (β-gal) to determine if suppression of CENP-A causes an accelerated senescent phenotype. The number of senescent cells significantly increases by 30% in CENP-A KD CPCs as shown by the analysis of bright field microscopy images of β-gal-positive cells (Fig. 4A, P < 0.05). To verify that cell cycle arrest caused by CENP-A KD is not a consequence of cellular commitment, α smooth muscle actin (αSMA), cardiac troponin T (cTnT), SM22, and Nkx2.5 expression were screened. CPCs treated with dexamethasone (Dex), a nonspecific inducer of CPC differentiation, were used as a positive control. No significant changes were evident in αSMA expression between ScrCon and CENP-A KD CPCs as shown by immunoblot (Fig. 4B). Likewise, CENP-A KD CPCs exhibit no significant increase in transcription of cTnT, SM22, or Nkx2.5 relative to ScrCon CPCs as shown by qPCR analysis (Fig. S2). Therefore, cell cycle arrest caused by CENP-A KD is a consequence of CPCs acquiring a senescent phenotype, not a result of CPC differentiation.

Figure 4. CENP-A knockdown results in premature senescence in CPCs. (A) Representative bright field images from 2 independent experiments show increased number of β-gal-positive CPCs following CENP-A knockdown (top right panels). Quantification of cell counts reveals 3-fold increase in the percentage of senescent CPCs (n = 4). (B) Immunoblot showing no increase in expression of αSMA in CENP-A KD CPCs relative to ScrCon CPCs. CPCs treated with Dex for 7 d to induce cellular commitment serves as a positive control. Two different exposures of the immunoblot are shown (n = 3). *P < 0.05; NS, not significant. Scale bars = 100 μm.
Silencing of CENP-A increases cell death in committed CPCs
The level of apoptosis in CENP-KD CPCs was analyzed to determine if increased cell death contributes to the diminished proliferation observed in these cells. FACS analysis of CPCs stained for the apoptotic marker Annexin V indicates no significant differences between ScrCon and CENP-A KD CPCs (Fig. 5A). However, previous studies that investigated the effects of silencing CENP-A show that committed cell types are inherently more vulnerable to CENP-A depletion.28,30 Therefore, the effect of CENP-A silencing on the level of cell death in committed CPCs was investigated. The number of apoptotic cells increases 2-fold in CENP-A KD CPCs subjected to Dex-induced differentiation as measured by flow cytometry (Fig. 5B, P < 0.05). CENP-A expression declines upon treatment with Dex, as shown by immunoblot, but the effect is more severe in the CENP-A KD group, where CENP-A expression has been dramatically attenuated (Fig. 5C). Thus, silencing CENP-A clearly impairs CPC survival when challenged with a differentiation stimulus.

Figure 5. Complete ablation of CENP-A in Dex-treated CPCs increases apoptosis. (A) FACS analysis of Annexin V staining shows no increase in basal apoptosis in CENP-A KD CPCs (n = 3). (B) Annexin V staining significantly increases in CENP-A KD CPCs following Dex induced differentiation. Representative scatter plots shown in left panels, quantifications in right panels (n = 3). (C) Immunoblot analysis indicating that CENP-A expression declines in CPCs treated with Dex, and is completely absent in CENP-A KD CPCs treated with Dex. Two different exposures of the immunoblot are shown (n = 3). *P < 0.05, NS, not significant.
Discussion
The literature shows that deregulation of CENP-A disrupts the normal functions of eukaryotic cells.12,28-31,34 However, few published works have looked at the role of CENP-A in stem cells, and none have studied CENP-A in the cardiac context. This study reveals CENP-A to be a novel component of stem cells within the heart and demonstrates that proper regulation of CENP-A is essential for the normal growth of CPCs and their survival following lineage commitment. Similar to the pattern of CENP-A expression in the pancreas,31 CENP-A shows marked decline in the heart early in life and is almost entirely absent by adulthood (Fig. 1). Diminished CENP-A expression is also observed in the stem cells themselves when old CPCs in vitro are compared with their young counterparts (Fig. 2). CPCs silenced for CENP-A show a significant reduction in proliferation accompanied by a 3-fold increase in the number of senescent CPCs (Figs. 3 and 4). The amount of senescence is comparable to the phenotype that arises in old CPCs naturally (unpublished observation), demonstrating that CENP-A diminution is expediting the acquisition of the senescent phenotype in young CPCs. Similar observations have been reported in human primary and dermal fibroblasts and correlates with decreased proliferation and increased levels of senescence in these cell types.28,29
Furthermore, CENP-A knockdown results in a significantly decreased proportion of cells that can be found in the G2/M phase of the cell cycle (Fig. 3), consistent with previous studies that characterize the cyclical pattern of CENP-A loading in mammalian cells. Past research identifies G1 as the stage of the cell cycle in which the chaperone protein HJURP loads new CENP-A nucleosomes onto the centromere of the chromosomes.25,26,34 If CENP-A loading takes place just before DNA replication in CPCs as well, then removing CENP-A should prevent CPCs from completing G1. However, delay in G1 is not true for all cell types. Cell cycle arrest can take place in G1 or G2/M, and the specific stage of the cell cycle in which it occurs varies between cell types and the stimulus used to induce it.35-38 For example, studies characterizing CENP-A knockdown in BJ fibroblasts have reported delay in G2/M and increased apoptosis to prevent defective mitosis. Alternatively, human primary fibroblasts initiate p53-mediated senescence to prevent chromosome missegregation from taking place.28 Human pluripotent stem cells (hPSCs), in contrast, exhibit no cell cycle delay or reduction in proliferation.30
Thus, different cell types possess varying responses to CENP-A diminution. More importantly, however, stem cells appear to possess an inherent resistance to the challenge of CENP-A silencing,30 and this also appears to be true of CPCs. As the results of this study indicate, the decline of CPCs in G2 is significant but small, and there is no significant increase in the G1 population due to the large majority of CPCs that normally are present at this phase of the cell cycle (Fig. 3). While there is a significant decline in CPC growth rate following CENP-A silencing, the overall proliferation of CPCs remains strong (Fig. 3). Moreover, CENP-A KD CPCs show no significant change in the level of basal apoptosis (Fig. 5). Unlike committed cell types such as fibroblasts, the majority of CPCs are clearly able to compensate for diminished levels of CENP-A expression, which is in apparent contradiction to the fact that proper regulation of CENP-A expression is critical in maintaining a functional epigenetic mark for the centromere.27 One possible explanation proposed by Ambartsumyan et al.30 is that hPSCs possess an open chromatin configuration, in which the amount of heterochromatin at the centromere is relatively small compared with committed cell types.39-41 Therefore, the threshold of CENP-A depletion below which cells cannot survive is much lower in stem cells, because the amount of CENP-A deposition needed is much less. Furthermore, this model accounts for the fact that the level of apoptosis drastically rises when hPSCs depleted of CENP-A are forced to commit.30 A similar increase in cell death is exhibited by CPCs, where the level of apoptosis increases roughly 2-fold following Dex-induced differentiation (Fig. 5). Consistent findings in vivo show that complete ablation of CENP-A results in early embryonic lethality 6.5 d post-coitum,42 whereas subtle perturbations in CENP-A structure have detrimental effects on mouse embryonic development, resulting in increased apoptosis of cells undergoing the earliest stages of differentiation in structures like the neural tube and somites.43 Increased apoptosis coincides with abnormal morphology and lethality delayed until 10.5 d post-coitum,43 suggesting that small changes in CENP-A structure and function do not provoke lethality until the early stages of cell commitment.
The response of CPCs and hPSCs to forced cellular commitment highlights the fact that the amount of CENP-A protein needed to maintain a functional centromere is determined by the extent of cell differentiation, and, in turn, the epigenetic configuration of the genome. CPCs appear to adopt an intermediate phenotype, possibly because they are more committed than hPSCs but not completely differentiated into the cell types of the heart. While many CPCs continue to survive following CENP-A depletion, they begin to exit the cell cycle and acquire a senescent phenotype.
CPCs form the foundation on which tissue homeostasis in the heart depends. However, the regenerative capacity of the endogenous stem cell pool remains extremely limited. Given these limitations, it is vital to identify the molecular characteristics that contribute to CPC senescence and cell death. The finding that CENP-A declines in the heart and CPCs with age provides new insight into what could be an important contributor to the reduced regenerative capacity of CPCs, and lays further groundwork that will be critical for engineering a stem cell population that is more robust in the face of pathological load. Pim-1 kinase has already been identified as a promising candidate, shown to mitigate the senescent phenotype of aged human CPCs.44 However, the capacity of Pim-1 to rescue and restore the growth reserve of cells depleted of CENP-A is yet to be determined, and remains a topic of future investigation.
Materials and Methods
Preparation of heart tissue samples
Heart tissue lysates were prepared from samples harvested from FVB mice at 11 d post-coitum and 3 mo after birth. Samples were suspended in isolation buffer at a concentration of 100 mg/mL and homogenized with a Qiagen Retsch TissueLyser (QIAGEN). Isolation buffer was prepared from a recipe of sucrose (70 mM), mannitol (190 mM), HEPES solution (20 mM), and EDTA solution (0.2 mM) in de-ionized water. Samples were then used for immunoblot and qPCR analysis as needed.
Immunohistochemistry of paraffin sections
Immunohistochemistry of mouse sections has been previously described.45 In brief, retroperfused hearts were fixed in formalin for 24 h at room temperature and then embedded in paraffin. Sections were cut and deparaffinized using standard techniques. Slides were then blocked in TNB for 1 h, and primary antibodies were applied overnight at 4 °C. Slides were washed in TN buffer and secondary antibodies were then applied for 2 h at room temperature. Amplification with tyramide was performed as needed for select antibodies. Primary antibodies used include those against CENP-A (Cell Signaling Technology, C5187), c-kit (R&D Systems, AF1356), pH3 (Millipore, 05-806), tropomyosin (Sigma, T2780), and actin (Sigma, A2172). CENP-A required tyramide amplification. TO-PRO-3 Iodide and Sytox Blue were used to stain for nuclei. Slides were imaged using confocal microscopy.
CPC isolation, cell culture, and transduction
CPCs were isolated as previously described and culture-expanded in cardiac stem cell growth media.6 Young and old CPCs were isolated from 3-mo- and 13-mo-old FVB mice. For CENP-A knockdown experiments, CPCs were genetically modified with lentiviral vectors to deliver shRNA directed against CENP-A (CENP-A KD) or scrambled control (ScrCon) targets. The construct used for CENP-A silencing was a Thermo Scientific TRC mouse shRNA clone (Clone ID: TRCN0000093123) directed against CENP-A mRNA (NCBI Accession Number: NM_007681) with an antibiotic resistance gene for puromycin selection of the infected population. Lentivirus was generated as previously described.46 CPCs were plated in 10-cm dishes at a density of 100 000 cells per dish and transduced with lentivirus for 48 h. CPCs were then treated with puromycin for 24 h and then re-plated as needed for analysis. To induce cellular commitment, CPCs were plated in 6-well plates at a density of 10 000 cells/well and treated with differentiation media for 7 d. Differentiation media was prepared from α-MEM (SIGMA, M0644) supplemented with 10% FBS, 1%PSG, and dexamethasone (SIGMA, D2915-100 mg) at a final concentration of 10−8 M.
Immunocytochemistry of fixed cells
CPCs were plated on 2-well permanox slides at a density of 3000 cells/chamber and cultured for 48 h. Cell were fixed in 4% paraformaldehyde for 30 min and then washed in PBS. Cells were then permeabalized with 0.2% TritonX in PBS for 10 min, blocked with 10% horse serum in PBS for 1 h, and then incubated with primary antibodies overnight at 4 °C. Slides were washed with PBS the next day and secondary antibodies were applied for 1 h. Primary antibodies and nuclear staining reagents were used as described above for paraffin-embedded sections, and slides were imaged using confocal microscopy.
Immunoblot analysis
Heart tissue and CPC protein samples were harvested in sample buffer containing 150 mM Tris (pH 6.8), 150 mg/ml sucrose, 2 mM EDTA (pH 7.5–8), 480 mg/ml urea, 8 mg/ml dithiothreitol, 0.2% sodium dodecyl sulfate, and 0.2 mg/ml bromophenol blue at a final pH of 6.8. Samples were then separated on a 4–12% Bis-Tris gel (Invitrogen) for 1 h and 10 min and transferred to an Immobilon-PSQ polyvinylidene diflouride membrane (Millipore) for 1.5 h. Membranes were blocked for 1 h with 0.2% i-BLOCK (Applied Biosystems, T2015) in TBST. Primary antibodies were then applied for 24 h. Membranes were then washed with TBST and incubated with secondary antibodies for 1 h. Membranes were once again washed in TBST and imaged on a Typhoon TRIO scanner. Primary antibodies used include those against CENP-A (Cell Signaling Technology, C5187), α-SMA (Sigma, A2547), and β-actin (Santa Cruz, sc-81178).
RNA isolation and qPCR analysis
Total RNA was isolated from heart tissue samples with a Quick-RNA MiniPrep kit (Zymo Research, R1055) and reverse transcribed into cDNA with an iScript cDNA Synthesis kit (Bio-Rad, 170-8891). Real-time quantitative PCR was then performed on all samples using an iQ SYBR Green PCR kit (Bio-Rad, 170-8885). Samples were amplified using an Opticon thermocycler (MJ Research). Primer sequences used to amplify genes of interest are as follows: mouse CENP-A forward primer TTGGCCCTTC AGGAGGCAGC A, mouse CENP-A reverse primer AAGCGTGACC CGACCAGCAT; mouse 18s forward primer CGAGCCGCCT GGATACC, mouse 18s reverse primer CATGGCCTCA GTTCCGAAAA; mouse SM22 forward primer GACTGCACTT CTCGGCTCAT, mouse SM22 reverse primer CCGAAGCTAC TCTCCTTCCA; mouse cTnT forward primer ACCCTCAGGC TCAGGTTCA, mouse cTnT reverse primer GTGTGCAGTC CCTGTTCAGA; mouse Nkx2.5 forward primer TCAATGCCTA TGGCTACAAC GCCT, mouse Nkx2.5 reverse primer GACGCCAAG TTCACGAAGT TGCT. CENP-A transcription was normalized to 18s for analysis.
CyQUANT analysis
To measure CPC growth rate samples were subjected to a CyQUANT NF Cell Proliferation Assay (Invitrogen, C35011). The assay serves as an indirect measurement of cell number that is based on the measurement of cellular DNA content though the binding of a fluorescent dye. For this assay CPCs were plated in a transparent 96-well flat bottom plate at a cell density of 500 cell/well in a total volume of 100 µL/well. CPCs were cultured in growth media and subjected to a time-course analysis, in which samples were stained and 1, 3, and 5 d after plating. At the given time points the CyQUANT staining solution was added to the growth media of each well at a ratio of 1:1. At the end of 5 d, the fluorescence intensity of the samples in the 96-well was measured using a Tecan SpectraFluor plate reader at excitation and emission wavelengths of 485 nm and 530 nm, respectively.
Cell cycle analysis
The cell cycle distribution of CPCs was measured by staining cellular DNA content with propidium iodide (PI). CPCs were plated in 10-cm dishes at a density of 100 000 cells/dish and cultured for 3 d. Cells were then fixed in 70% ethanol and stored at −80 °C for later analysis. Samples were thawed, washed with PBS, and then incubated with PI/RNase staining buffer (BD Biosciences, 550825) for 15 min at room temperature. Samples were then subjected to FACS analysis using a BD Canto flow cytometer (BD Biosciences). The distribution of cells in phases G1, S, and G2 of the cell cycle were finally quantified using FlowJo software.
Senescence-associated β-galactosidase staining
CPCs were plated on 2-well permanox slides at a density of 3000 cells/chamber and cultured for 48 h. Cells were then stained using a Senescence Detection Kit (Abcam, ab65351) designed to histochemically detect SA-β-Gal (β-gal) activity known to be a characteristic of senescence.47 Protocols were followed in accordance with the manufacturer’s instructions. In brief, cells were fixed with the provided fixative solution for 15 min at room temperature. The staining solution was then applied overnight at 37 °C. The following day samples were imaged using bright field microscopy, and senescent cells were counted. The development of a blue color indicates senescence-associated β-gal activity.
Apoptosis assay
CPCs entering into programmed cell death were measured using an APC Annexin V Apoptosis Detection Kit (BD PharMingen, 559763). CPCs were plated in 6-well plates at a density of 10 000 cells/well and subjected to dexamethasone induced differentiation as described above. Cells were then harvested with 0.25% trypsin, washed once with PBS, and resuspended in 100 µL Annexin V binding buffer. Samples were incubated with 5 µL of Annexin V conjugated to allophycocyanin (APC) for 15 min at room temperature, after which 400 µL of binding buffer were added to each sample. Samples were then subjected to FACS analysis using a BD Canto flow cytometer (BD Biosciences). FlowJo software was used to quantify the percentage of cells positive for the APC fluorescent probe.
Image analysis
All bright field images were taken with an Olympus IX70 light microscope. All immunohistochemical images were taken with a Leica DMRE confocal microscope. Photoshop was used for the analysis of β-gal-positive cells in bright field images and the generation of overlaid pseudocolored images of single channel scans taken on the confocal microscope. Immunoblots imaged on the Typhoon Trio scanner were quantified with ImageQuant 5.0 software.
Statistical analysis
Data are represented as Mean +/− SEM. Statistical analysis was performed using GraphPad Prism 5. A 2-tailed paired Student t test was applied to compare 2 groups. P values less than or equal to 0.05 were considered statistically significant.
Supplementary Material
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank all members of the Sussman laboratory for their feedback and support. The authors thank the FACS core and genomics core at SDSU for help with flow cytometry analyses and for animal maintenance.
This work was supported by the National Heart, Lung, and Blood Institute Grants 1R37HL091102, 1R01HL105759, 5R01HL067245, 1R01HL113656, 1R01HL117163, 1R01HL113647 (to M.A.S.), American Heart Association Post-Doctoral Fellowship 12POST12060191 (to N.H.).
Author Contributions
M.M. designed and performed experiments, analyzed data and wrote the manuscript; N.H. designed and performed experiments and edited the manuscript; A.Y.J. performed experiments; R.L.M. provided technical and experimental advice; M.A.S. supervised all procedures, edited and approved the final version of the manuscript to be published.
Glossary
Abbreviations:
- CENP-A
centromere-associated protein A
- CENP-A KD
CENP-A knockdown
- CPC
cardiac progenitor cell
- Dex
dexamethasone
- FACS
fluorescence activated cell sorting
- G1 phase
gap 1 phase of cell cycle
- G2 phase
gap 2 phase of cell cycle
- HJURP
Holliday junction recognition protein
- hPSC
human pluripotent stem cell
- M phase
mitosis phase of cell cycle
- MI
myocardial infarction
- pH3
phospho histone 3
- PI
propidium iodide
- S phase
synthesis phase of cell cycle
- αSMA
alpha smooth muscle actin
- β-gal
senescence-associated beta galactosidase
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/27549
References
- 1.Beltrami AP, Barlucchi L, Torella D, Baker M, Limana F, Chimenti S, Kasahara H, Rota M, Musso E, Urbanek K, et al. Adult cardiac stem cells are multipotent and support myocardial regeneration. Cell. 2003;114:763–76. doi: 10.1016/S0092-8674(03)00687-1. [DOI] [PubMed] [Google Scholar]
- 2.Frati C, Savi M, Graiani G, Lagrasta C, Cavalli S, Prezioso L, Rossetti P, Mangiaracina C, Ferraro F, Madeddu D, et al. Resident cardiac stem cells. Curr Pharm Des. 2011;17:3252–7. doi: 10.2174/138161211797904181. [DOI] [PubMed] [Google Scholar]
- 3.Leri A, Kajstura J, Anversa P. Cardiac stem cells and mechanisms of myocardial regeneration. Physiol Rev. 2005;85:1373–416. doi: 10.1152/physrev.00013.2005. [DOI] [PubMed] [Google Scholar]
- 4.Anversa P, Kajstura J, Rota M, Leri A. Regenerating new heart with stem cells. J Clin Invest. 2013;123:62–70. doi: 10.1172/JCI63068. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 5.Mohsin S, Khan M, Toko H, Bailey B, Cottage CT, Wallach K, Nag D, Lee A, Siddiqi S, Lan F, et al. Human cardiac progenitor cells engineered with Pim-I kinase enhance myocardial repair. J Am Coll Cardiol. 2012;60:1278–87. doi: 10.1016/j.jacc.2012.04.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fischer KM, Cottage CT, Wu W, Din S, Gude NA, Avitabile D, Quijada P, Collins BL, Fransioli J, Sussman MA. Enhancement of myocardial regeneration through genetic engineering of cardiac progenitor cells expressing Pim-1 kinase. Circulation. 2009;120:2077–87. doi: 10.1161/CIRCULATIONAHA.109.884403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chugh AR, Beache GM, Loughran JH, Mewton N, Elmore JB, Kajstura J, Pappas P, Tatooles A, Stoddard MF, Lima JA, et al. Administration of cardiac stem cells in patients with ischemic cardiomyopathy: the SCIPIO trial: surgical aspects and interim analysis of myocardial function and viability by magnetic resonance. Circulation. 2012;126(Suppl 1):S54–64. doi: 10.1161/CIRCULATIONAHA.112.092627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bolli R, Chugh AR, D’Amario D, Loughran JH, Stoddard MF, Ikram S, Beache GM, Wagner SG, Leri A, Hosoda T, et al. Cardiac stem cells in patients with ischaemic cardiomyopathy (SCIPIO): initial results of a randomised phase 1 trial. Lancet. 2011;378:1847–57. doi: 10.1016/S0140-6736(11)61590-0. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 9.Jesty SA, Steffey MA, Lee FK, Breitbach M, Hesse M, Reining S, Lee JC, Doran RM, Nikitin AY, Fleischmann BK, et al. c-kit+ precursors support postinfarction myogenesis in the neonatal, but not adult, heart. Proc Natl Acad Sci U S A. 2012;109:13380–5. doi: 10.1073/pnas.1208114109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hou J, Wang L, Jiang J, Zhou C, Guo T, Zheng S, Wang T. Cardiac stem cells and their roles in myocardial infarction. Stem Cell Rev. 2013;9:326–38. doi: 10.1007/s12015-012-9421-4. [DOI] [PubMed] [Google Scholar]
- 11.Krzemiński TF, Nozyński JK, Grzyb J, Porc M. Wide-spread myocardial remodeling after acute myocardial infarction in rat. Features for heart failure progression. Vascul Pharmacol. 2008;48:100–8. doi: 10.1016/j.vph.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 12.Fransioli J, Bailey B, Gude NA, Cottage CT, Muraski JA, Emmanuel G, Wu W, Alvarez R, Rubio M, Ottolenghi S, et al. Evolution of the c-kit-positive cell response to pathological challenge in the myocardium. Stem Cells. 2008;26:1315–24. doi: 10.1634/stemcells.2007-0751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ferreira-Martins J, Ogórek B, Cappetta D, Matsuda A, Signore S, D’Amario D, Kostyla J, Steadman E, Ide-Iwata N, Sanada F, et al. Cardiomyogenesis in the developing heart is regulated by c-kit-positive cardiac stem cells. Circ Res. 2012;110:701–15. doi: 10.1161/CIRCRESAHA.111.259507. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 14.Kajstura J, Rota M, Cappetta D, Ogórek B, Arranto C, Bai Y, Ferreira-Martins J, Signore S, Sanada F, Matsuda A, et al. Cardiomyogenesis in the aging and failing human heart. Circulation. 2012;126:1869–81. doi: 10.1161/CIRCULATIONAHA.112.118380. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 15.Leri A, Kajstura J, Anversa P. Role of cardiac stem cells in cardiac pathophysiology: a paradigm shift in human myocardial biology. Circ Res. 2011;109:941–61. doi: 10.1161/CIRCRESAHA.111.243154. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 16.Cesselli D, Beltrami AP, D’Aurizio F, Marcon P, Bergamin N, Toffoletto B, Pandolfi M, Puppato E, Marino L, Signore S, et al. Effects of age and heart failure on human cardiac stem cell function. Am J Pathol. 2011;179:349–66. doi: 10.1016/j.ajpath.2011.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sussman MA, Anversa P. Myocardial aging and senescence: where have the stem cells gone? Annu Rev Physiol. 2004;66:29–48. doi: 10.1146/annurev.physiol.66.032102.140723. [DOI] [PubMed] [Google Scholar]
- 18.Torella D, Rota M, Nurzynska D, Musso E, Monsen A, Shiraishi I, Zias E, Walsh K, Rosenzweig A, Sussman MA, et al. Cardiac stem cell and myocyte aging, heart failure, and insulin-like growth factor-1 overexpression. Circ Res. 2004;94:514–24. doi: 10.1161/01.RES.0000117306.10142.50. [DOI] [PubMed] [Google Scholar]
- 19.Cottage CT, Neidig L, Sundararaman B, Din S, Joyo AY, Bailey B, Gude N, Hariharan N, Sussman MA. Increased mitotic rate coincident with transient telomere lengthening resulting from pim-1 overexpression in cardiac progenitor cells. Stem Cells. 2012;30:2512–22. doi: 10.1002/stem.1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bird A. Perceptions of epigenetics. Nature. 2007;447:396–8. doi: 10.1038/nature05913. [DOI] [PubMed] [Google Scholar]
- 21.Palmer DK, O’Day K, Trong HL, Charbonneau H, Margolis RL. Purification of the centromere-specific protein CENP-A and demonstration that it is a distinctive histone. Proc Natl Acad Sci U S A. 1991;88:3734–8. doi: 10.1073/pnas.88.9.3734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bui M, Dimitriadis EK, Hoischen C, An E, Quénet D, Giebe S, Nita-Lazar A, Diekmann S, Dalal Y. Cell-cycle-dependent structural transitions in the human CENP-A nucleosome in vivo. Cell. 2012;150:317–26. doi: 10.1016/j.cell.2012.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bui M, Walkiewicz MP, Dimitriadis EK, Dalal Y. The CENP-A nucleosome: a battle between Dr Jekyll and Mr Hyde. Nucleus. 2013;4:37–42. doi: 10.4161/nucl.23588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.González-Barrios R, Soto-Reyes E, Herrera LA. Assembling pieces of the centromere epigenetics puzzle. Epigenetics. 2012;7:3–13. doi: 10.4161/epi.7.1.18504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Valente LP, Silva MC, Jansen LE. Temporal control of epigenetic centromere specification. Chromosome Res. 2012;20:481–92. doi: 10.1007/s10577-012-9291-2. [DOI] [PubMed] [Google Scholar]
- 26.Nechemia-Arbely Y, Fachinetti D, Cleveland DW. Replicating centromeric chromatin: spatial and temporal control of CENP-A assembly. Exp Cell Res. 2012;318:1353–60. doi: 10.1016/j.yexcr.2012.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.De Rop V, Padeganeh A, Maddox PS. CENP-A: the key player behind centromere identity, propagation, and kinetochore assembly. Chromosoma. 2012;121:527–38. doi: 10.1007/s00412-012-0386-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Maehara K, Takahashi K, Saitoh S. CENP-A reduction induces a p53-dependent cellular senescence response to protect cells from executing defective mitoses. Mol Cell Biol. 2010;30:2090–104. doi: 10.1128/MCB.01318-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Heo JI, Cho JH, Kim JR. HJURP regulates cellular senescence in human fibroblasts and endothelial cells via a p53-dependent pathway. J Gerontol A Biol Sci Med Sci. 2013;68:914–25. doi: 10.1093/gerona/gls257. [DOI] [PubMed] [Google Scholar]
- 30.Ambartsumyan G, Gill RK, Perez SD, Conway D, Vincent J, Dalal Y, Clark AT. Centromere protein A dynamics in human pluripotent stem cell self-renewal, differentiation and DNA damage. Hum Mol Genet. 2010;19:3970–82. doi: 10.1093/hmg/ddq312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee SH, Itkin-Ansari P, Levine F. CENP-A, a protein required for chromosome segregation in mitosis, declines with age in islet but not exocrine cells. Aging (Albany NY) 2010;2:785–90. doi: 10.18632/aging.100220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li Y, Zhu Z, Zhang S, Yu D, Yu H, Liu L, Cao X, Wang L, Gao H, Zhu M. ShRNA-targeted centromere protein A inhibits hepatocellular carcinoma growth. PLoS One. 2011;6:e17794. doi: 10.1371/journal.pone.0017794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lando D, Endesfelder U, Berger H, Subramanian L, Dunne PD, McColl J, Klenerman D, Carr AM, Sauer M, Allshire RC, et al. Quantitative single-molecule microscopy reveals that CENP-A(Cnp1) deposition occurs during G2 in fission yeast. Open Biol. 2012;2:120078. doi: 10.1098/rsob.120078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Foltz DR, Jansen LE, Bailey AO, Yates JR, 3rd, Bassett EA, Wood S, Black BE, Cleveland DW. Centromere-specific assembly of CENP-a nucleosomes is mediated by HJURP. Cell. 2009;137:472–84. doi: 10.1016/j.cell.2009.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rayess H, Wang MB, Srivatsan ES. Cellular senescence and tumor suppressor gene p16. Int J Cancer. 2012;130:1715–25. doi: 10.1002/ijc.27316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Olivares-Illana V, Fåhraeus R. p53 isoforms gain functions. Oncogene. 2010;29:5113–9. doi: 10.1038/onc.2010.266. [DOI] [PubMed] [Google Scholar]
- 37.Houtgraaf JH, Versmissen J, van der Giessen WJ. A concise review of DNA damage checkpoints and repair in mammalian cells. Cardiovasc Revasc Med. 2006;7:165–72. doi: 10.1016/j.carrev.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 38.Cmielová J, Rezáčová M. p21Cip1/Waf1 protein and its function based on a subcellular localization [corrected] J Cell Biochem. 2011;112:3502–6. doi: 10.1002/jcb.23296. [corrected] [DOI] [PubMed] [Google Scholar]
- 39.Meshorer E, Yellajoshula D, George E, Scambler PJ, Brown DT, Misteli T. Hyperdynamic plasticity of chromatin proteins in pluripotent embryonic stem cells. Dev Cell. 2006;10:105–16. doi: 10.1016/j.devcel.2005.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gaspar-Maia A, Alajem A, Polesso F, Sridharan R, Mason MJ, Heidersbach A, Ramalho-Santos J, McManus MT, Plath K, Meshorer E, et al. Chd1 regulates open chromatin and pluripotency of embryonic stem cells. Nature. 2009;460:863–8. doi: 10.1038/nature08212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Melcer S, Meshorer E. Chromatin plasticity in pluripotent cells. Essays Biochem. 2010;48:245–62. doi: 10.1042/bse0480245. [DOI] [PubMed] [Google Scholar]
- 42.Howman EV, Fowler KJ, Newson AJ, Redward S, MacDonald AC, Kalitsis P, Choo KH. Early disruption of centromeric chromatin organization in centromere protein A (Cenpa) null mice. Proc Natl Acad Sci U S A. 2000;97:1148–53. doi: 10.1073/pnas.97.3.1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kalitsis P, Fowler KJ, Earle E, Griffiths B, Howman E, Newson AJ, Choo KH. Partially functional Cenpa-GFP fusion protein causes increased chromosome missegregation and apoptosis during mouse embryogenesis. Chromosome Res. 2003;11:345–57. doi: 10.1023/A:1024044008009. [DOI] [PubMed] [Google Scholar]
- 44.Mohsin S, Khan M, Nguyen J, Alkatib M, Siddiqi S, Hariharan N, Wallach K, Monsanto M, Gude N, Dembitsky W, et al. Rejuvenation of human cardiac progenitor cells with Pim-1 kinase. Circ Res. 2013;113:1169–79. doi: 10.1161/CIRCRESAHA.113.302302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Muraski JA, Rota M, Misao Y, Fransioli J, Cottage C, Gude N, Esposito G, Delucchi F, Arcarese M, Alvarez R, et al. Pim-1 regulates cardiomyocyte survival downstream of Akt. Nat Med. 2007;13:1467–75. doi: 10.1038/nm1671. [DOI] [PubMed] [Google Scholar]
- 46.Swan CH, Bühler B, Steinberger P, Tschan MP, Barbas CF, 3rd, Torbett BE. T-cell protection and enrichment through lentiviral CCR5 intrabody gene delivery. Gene Ther. 2006;13:1480–92. doi: 10.1038/sj.gt.3302801. [DOI] [PubMed] [Google Scholar]
- 47.Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, Medrano EE, Linskens M, Rubelj I, Pereira-Smith O, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci U S A. 1995;92:9363–7. doi: 10.1073/pnas.92.20.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.