

Abstract
The tumor protein (TP) p63/microRNAs functional network may play a key role in supporting the response of squamous cell carcinomas (SCC) to chemotherapy. We show that the cisplatin exposure of SCC-11 cells led to upregulation of miR-297, miR-92b-3p, and miR-485-5p through a phosphorylated ΔNp63α-dependent mechanism that subsequently modulated the expression of the protein targets implicated in DNA methylation (DNMT3A), histone deacetylation (HDAC9), and demethylation (KDM4C). Further studies showed that mimics for miR-297, miR-92b-3p, or miR-485-5p, along with siRNA against and inhibitors of DNMT3A, HDAC9, and KDM4C modulated the expression of DAPK1, SMARCA2, and MDM2 genes assessed by the quantitative PCR, promoter luciferase reporter, and chromatin immunoprecipitation assays. Finally, the above-mentioned treatments affecting epigenetic enzymes also modulated the response of SCC cells to chemotherapeutic drugs, rendering the resistant SCC cells more sensitive to cisplatin exposure, thereby providing the groundwork for novel chemotherapeutic venues in treating patients with SCC.
Keywords: TP63, microRNA, epigenetic regulation, DNA methylation, histone deacetylation, histone demethylation, chemoresistance, squamous cell carcinomas
Introduction
Multiple molecular mechanisms are implicated in regulation of gene expression in human cells in various physiologic and pathophysiologic conditions.1,2 They include heritable epigenetic alterations of DNA methylation, histone methylation/demethylation, histone acetylation/deacetylation, formation of multiple complexes between distinct chromatin components and transcription factors, RNA processing and translation, and post-translational modifications of nascent proteins.1,2 Finally, a modulation of gene expression by non-coding microRNAs is also implicated in epigenetic control of gene expression.3-6
microRNAs repress the expression of a variety of target genes involved in a plethora of distinct signaling pathways in development and disease.7,8 Primary microRNA transcripts are processed by the RNA-induced silencing complex to generate mature microRNAs; the latter form complexes with the specific sequences within mRNA targets based on complementarity.7-11 The microRNA/mRNA complexes then cause an inhibition of protein translation and/or degradation of the mRNAs. A single microRNA could modulate several mRNAs, and a few microRNAs might regulate the expression of the same mRNA target.10,11
Altered expression of microRNA genes has been found in a variety of tumor types, and specific microRNAs have shown the oncogenic, tumor-suppressive, or apoptotic potential.8,12-17 Certain microRNAs were shown to mediate epigenetic regulation of gene transcription and cell metabolism, the induction of cell death, cell cycle arrest, autophagy, and senescence.8,18-22 On one hand, microRNAs were shown to directly bind the gene promoter and gene terminus sequences, thereby modulating specific gene expression at the transcription level.23-26 On the other hand, transcriptional deregulation in cancer cells may lead to altered transcription of specific microRNA genes.27-29 For example, miR-34 was shown to be regulated by the tumor protein (TP)-p53 transcription factor, which regulates the cellular response to stress-induced DNA damage, cell cycle, apoptosis, autophagy, and metabolism.27
microRNAs may also have therapeutic applications, by which cancer-causing microRNAs could be modulated to restore the normal cellular function.30-33 The modified cholesterol-conjugated antisense RNA (“antagomirs”) were shown to effectively inhibit microRNA function in vivo.32 The competitive microRNA inhibitors (“microRNA sponges”) were reported to de-repress microRNA targets as strongly as chemically modified antisense oligonucleotides.33
We have previously shown that the SCC cells exposed to cisplatin treatment displayed a dramatic downregulation of ΔNp63α via an ATM-dependent phosphorylation mechanism.34 We have also shown that the phosphorylated (p)-ΔNp63α protein is critical for the transcriptional regulation of downstream mRNAs and microRNAs in SCC cells upon cisplatin exposure.35,36 Moreover, we have reported that p-ΔNp63α regulates microRNA expression in cisplatin-treated SCC cells through both transcriptional and post-transcriptional mechanisms.36 We have further showed that the specific microRNAs downregulated or upregulated in SCC cells in response to cisplatin treatment are involved in a broad plethora of cellular processes, including apoptosis, autophagy, and various metabolic and signaling pathways.36-39 P-ΔNp63α was also shown to transcriptionally activate or repress the specific microRNA promoters depending on the chromatin components bound to this transcriptional factor in SCC cells upon cisplatin exposure.28 In this report, we continue our quest to understand the role of the cisplatin-induced TP63-regulated microRNAs in epigenetic regulation and chemoresistance.
Results
P-ΔNp63α-dependent epi-microRNAs modulate the expression of epigenetic enzymes in SCC cells
We previously found that the SCC-11 cells exposed to cisplatin treatment expressed the ATM-dependent p-ΔNp63α, which appeared critical for the transcriptional regulation of downstream mRNAs and microRNAs in SCC-11 cells.35-39 Using knock-in technology, we generated SCC-11 cells, which have been shown to produce wild-type ΔNp63α, and SCC-11M cells that exclusively express ΔNp63α-S385G mutant protein, with an altered ability to be phosphorylated by ATM kinase.34
By global analysis of microRNA expression, we previously showed that cisplatin exposure led to a downregulation of 28 microRNAs (e.g., miR-519a-3p, miR-181a-5p, miR-374a-5p, miR-98-5p, miR-29c-3p, miR-22-3p, miR-34c-3p, miR-206, miR-429, miR-339-3p, miR-203a, miR-25-3p, miR-155-5p, and miR-148a-3p) by −5.18 to −19.27-fold, and upregulation of 15 microRNAs (e.g., miR-382-3p, miR-485-5p, miR-574-5p, miR-92b-3p, miR-297, miR-185-5p, miR-885-3p, miR-194-5p, and miR-630) by 3.95- to 7.46-fold in SCC-11 cells compared with SCC-11M cells upon cisplatin exposure.36-39 We further showed that cisplatin exposure altered microRNA expression in SCC-11 cells, resulting in downregulation of 7 microRNAs (e.g., miR-519-a-3p, miR-181a-5p, miR-374a-5p, miR-29c-3p, miR-98-5p, miR-22-3p, and miR-34c-3p, from −1.72 to −3.77-fold), and upregulation of 7 microRNAs (miR-382–3p, miR-485-5p, miR-574–5p, miR-297, miR-194-5p, miR-885-3p, and miR-630, from 2.08- to 4.98-fold), as shown in references 36–39.
To validate these data, we used quantitative (q)-PCR expression analysis and showed that miR-485-5p, miR-297, miR-382-3p, and miR-194-5p were upregulated by 5.2–6.3-fold, while miR-98-5p, miR-29c-3p, miR-101-3p, miR-22-3p, miR-34c-3p, miR-206, miR-429, miR-339-3p, miR-203a, miR-25-3p, miR-155-5p, and miR-148a-3p were downregulated (6.7–15.4-fold), as reported elsewhere.39
P-ΔNp63α was previously shown to regulate the expression of specific microRNAs in cisplatin-treated SCC-11 cells, subsequently leading to altering of tumor cell response to chemotherapy via mechanisms implicated in cell death and cell survival.36-39 We showed here that the p-ΔNp63α expressed in cisplatin-treated SCC-11 cells upregulated or downregulated a plethora of various “epi-microRNA” species,3,4 which are likely to affect the components of epigenetic regulatory machinery, defined by the web-based bioinformatics tools (Fig. 1A). These potential epigenetic-regulatory molecules include enzymes involved in DNA methylation (DNMT1, DNMT3A, DNMT3B, and MBD1), histone acetylation (KAT2B, KAT3B, and KAT6B), histone deacetylation (HDAC9), histone demethylation (KDM2A, KDM3A, KDM3B, KDM4C, and KDM5B), and members of the polycomb repressive complex (EZH2, BMI1, RNF2, EED, and RBBP4), as shown in Figure 1A. To investigate whether predicted microRNAs affect the expression of selected mRNAs, we employed the 3′-untranslated region (UTR)-mediated luciferase activity assay. We found that the microRNA mimics for miR-630, miR-34c-3p, miR-429, miR-485-5p, miR-297, miR-25-3p, miR-92b-3p, miR-519c-3p, miR-181a-5p, miR-720, miR-101a-3p, miR-27a-3p, miR-148a-5p, miR-185-5p, and miR-148a-3p inhibited the luciferase activity driven by the 3′-UTR of specific mRNAs by −1.4 to −2.2-fold compared with the scrambled microRNA (Fig. 1B; Fig. S1A–D). In addition, the direct effect of the specific microRNA mimics, as well as siRNAs, on the DNMT3A, HDAC9, KDNM4C, and DNMT1 protein levels was clearly seen in SCC-11 cells (Fig. S1E–D).

Figure 1. Cisplatin affects expression of microRNA targets in SCC-11 cells. (A) Schematic representation of the ATM-induced p-ΔNp63α-dependent protein targets involved in epigenetic regulation of gene transcription and their corresponding microRNAs in SCC-11 cells exposed to cisplatin. The microRNAs induced by cisplatin/p-ΔNp63α are indicated in black, while microRNAs repressed by cisplatin/p-ΔNp63α are indicated in gray. The corresponding microRNAs are shown next to the specific protein target. microRNAs listed on right next to ATM or ΔNp63α are shown to inhibit ATM or ΔNp63α expression, suggesting a feedback regulation through microRNA-dependent mechanism.28 (B) microRNA/3′-UTR luciferase reporter assays for indicated targets in SCC-11 cells. Cells were transfected with the 3-UTR luciferase plasmids along with the scrambled RNA (control) or microRNA mimics, as indicated below the graph. Target protein symbols are indicated above the graph. Data obtained from the control samples were presented in relative units (RU) and designated as 1. Data were expressed as means ± SD from 3 independent experiments in triplicate (P < 0.05).
Intriguingly, several epigenetic protein targets were affected by several microRNAs, while certain microRNAs could modulate several targets (Fig. 1; Fig. S1), as predicted elsewhere.40-42 Since it was difficult to predict a cumulative effect of cisplatin treatment on the protein targets that are likely to be modulated by p-ΔNp63α-dependent microRNAs, we tested the levels of certain epigenetic proteins in both cisplatin-sensitive SCC-11 cells and cisplatin-resistant SCC-11M cells, which were exposed to control medium (CON) or 10 μg/ml cisplatin (CIS). Target protein levels were monitored by immunoblotting, with the indicated antibodies followed by quantification imaging analysis. The obtained values were subsequently normalized to the β-actin levels (Fig. 2A and B). We observed that EZH2, RBBP4, DNMT3A, and KDM4C were downregulated, while RNF2, KDM2A, KDM3B, and KDM5B were upregulated in sensitive SCC-11 cells upon cisplatin exposure (Fig. 2A). BMI1, DNMT1, HDAC9, and KAT2B showed no significant changes under cisplatin exposure, probably due to opposing actions of cisplatin-/p-ΔNp63α-induced and -repressed microRNAs (Fig. 1A; Fig. S1). However, the resistant SCC-11M cells displayed a slightly distinct pattern of expression of tested protein targets (Fig. 2B), supporting the notion that some epigenetic biomarkers could be involved in the response of SCC-11 cells to cisplatin treatment.

Figure 2. Expression of epigenetic protein targets in SCC-11 cells and SCC-11M cells upon cisplatin exposure. Immunoblot analysis with indicated antibodies. SCC-11 cells (A) and SCC-11M cells (B) were exposed to control medium (CON) or 10 μg/ml cisplatin (CIS) for 16 h. Each lysate was divided into 2 aliquots: (1) to detect the levels of indicated proteins, and (2) to detect the β-actin level. Lines between images indicate the separate gel runs and blots with various antibodies. Aliquots for β-actin were run on one gel and blotted altogether. Blots were scanned and quantified in triplicate by the Image Quant software version 3.3. Values indicated above the blots were normalized by β-actin levels and expressed as a fold change to a control sample defined as 1. (C–E). Immunoprecipitation (IP) of ΔNp63α with DNMT3A (C). HDAC9 (D) or KDM4C (E) in SCC-11 and SCC-11M cells upon cisplatin exposure.
ΔNp63α is forming protein complexes with epigenetic enzymes in SCC cells
Previous protein–protein interaction studies showed that TP63, and specifically ΔNp63α, is capable of binding to numerous proteins implicated in epigenetic regulation of gene expression.43 We, therefore, examined whether both sensitive SCC-11 cells and resistant SCC-11M cells exposed to cisplatin treatment displayed the formation of protein complexes between ΔNp63α and tested epigenetic enzymes. We showed the increased ΔNp63α binding to DNMT3A, HDAC9, and KDM4C in SCC-11M cells compared with SCC-11 cells (Fig. 2C), suggesting that these complexes, which preferentially occurred in cisplatin-treated SCC-11M cells, could recruit the epigenetic enzymes to the target gene promoters. To support this hypothesis, we examined whether ΔNp63α binds to the DAPK1, SMARCA2, and MDM2 gene promoters (Figs. S2–4) in larynx-derived sensitive SCC-11/resistant SCC-11M cells and tongue-derived sensitive SCC-25/resistant SCC-25CP cells upon cisplatin exposure.39,44 Using the chromatin immunoprecipitation (ChIP) assay, we found that under cisplatin pressure ΔNp63α bound more efficiently to the DAPK1 (Fig. S5), SMARCA2 (Fig. S6), and MDM2 (Fig. S7) promoters in SCC-11M cells/SCC-25CP cells than in SCC-11 cells/SCC-25 cells (Fig. S5–7). Since, sensitive SCC-11 and SCC-25 cells exclusively express or have the higher p-ΔNp63α/non-p-ΔNp63α ratio, one could notice the binding of p-ΔNp63α in these cells, which is a part of the total ΔNp63α binding (Fig. S5–7). Taken together, we propose that ΔNp63α contributes to recruiting the epigenetic enzymes to the certain gene promoters in order to regulate their transcription by DNA methylation, histone deacetylation, and demethylation, as shown for many transcription factors, including TP63.45-47
Modulation of DNA methylation affects the DAPK1 expression in SCC cells upon cisplatin exposure
Accumulating evidence shows that promoter DNA hypermethylation of various genes involved in cell cycle arrest or apoptosis leads to their epigenetic repression and subsequently to chemoresistance of tumor cells to anticancer drugs.48-51 Several DNA methyltransferases, DNMT1, DNMT3A, and DNMT3B, are involved in the addition of methyl groups to the 5′-cytosine at the CpG islands within the specific promoter DNA sequences, subsequently repressing the transcription of these genes. DNMT1 preserves the methylation DNA patterns throughout each cell division, while DNMT3A and 3B transfer a methyl group to unmethylated DNA sequences.52-56 Although DNMT3A and 3B are believed to play a role of de novo DNA methyltransferases in development, recent studies showed that both DNMT3A and DNMT3B could also serve as maintenance enzymes that are responsible for copying DNA methylation patterns to the daughter strands during DNA replication.52-56 Therefore, DNA methylation mediated by a combined action of DNMT1, DNMT3A, and DNMT3B is essential for understanding the epigenetic mechanisms underlying cellular transformation.52-56
Our initial studies that employed the high-throughput DNA methylation chip arrays showed that many sequences were exclusively hypermethylated in SCC-11M cells upon cisplatin exposure, compared with SCC-11 cells treated with cisplatin (data not shown). Among these sequences, the DAPK1 promoter area was found starting at −1768 bp (Fig. S2). For example, the hypermethylation of the death-associated protein kinase-1 (DAPK1) promoter was reported to contribute to chemoresistance of cancer cells to several therapeutic agents.50,51 Intriguingly, the putative TP63 binding sequences in the specific DNMT3A promoter area (−1763 to −1344 bp; Fig. S2) are shown to overlap with the potential DNMT3A consensus sequence.45
We examined whether the expression of DAPK1 was affected in SCC-11 cells and SCC-11M cells exposed to control media and 10 μg/ml cisplatin for 16 h (Fig. 3). Since, miR-297 is upregulated in SCC-11 cells compared with SCC-11M cells upon cisplatin exposure,39 and was shown to target DNMT3A expression (Fig. 1; Fig. S1A and E), we suggested that the p-ΔNp63α-upregulated miR-297 might be implicated in epigenetic regulation of the DAPK1 expression. Using the qPCR, luciferase reporter, and ChIP assays, we tested whether miR-297, siRNA to DNMT3A, and DNA methylation inhibitor, 5′-azacytidine, (5′-AzaC) would affect the DAPK1 expression in SCC-11 cells and SCC-11M cells treated with control media (Fig. 3). We showed that the cisplatin exposure of SCC-11 cells induced DAPK1 mRNA expression by 2.45-fold, while miR-297, siRNA to DNMT3A, and 5′-AzaC increased the DAPK1 mRNA expression in SCC-11 cells by 3.34-, 3.41-, and 2.06-fold, respectively (Fig. 3A). Although SCC-11M cells exposed to cisplatin displayed no change in the DAPK1 mRNA expression compared with control treatment, miR-297, siRNA to DNMT3A, and 5′-AzaC increased the DAPK1 mRNA expression in SCC-11M cells by 3.11-, 3.21-, and 2.01-fold, respectively (Fig. 3A). We further showed that the DAPK1 promoter-driven luciferase activity was increased in SCC-11 cells upon cisplatin exposure, and under influence of miR-297, siRNA to DNMT3A, and 5′-AzaC by 5.98-, 7.88-, 8.18-, and 5.65-fold, respectively (Fig. 3B). At the same time, SCC-11M cells exposed to cisplatin showed no change in the DAPK1 promoter function compared with control treatment, while miR-297, siRNA to DNMT3A, and 5′-AzaC increased the DAPK1 promoter–reporter activity in SCC-11M cells by 7.62-, 7.72-, and 3.45-fold, respectively (Fig. 3B). We next showed that the DNMT3A binding to the DAPK1 promoter (Fig. S2) was markedly decreased in SCC-11 cells upon cisplatin exposure and after treatment of SCC-11 cells with miR-297 and siRNA to DNMT3A (Fig. 3C). However, the DNMT3A binding to the DAPK1 promoter (Fig. S2) in SCC-11M cells was practically unchanged after cisplatin, but was decreased in SCC-11M cells treated with miR-297 and siRNA to DNMT3A (Fig. 3C). Inactivation of DNMT3A activity with 5′-AzaC had only a slight effect on the DNMT3A binding to the DAPK1 promoter (compared with scrambled control) in both SCC-11 and SCC-11M cells (Fig. 3C). Similar ChIP assay results were shown using the tongue-derived cisplatin-sensitive SCC-25/cisplatin-resistant SCC-25CP pair of cells (Fig. S8A).
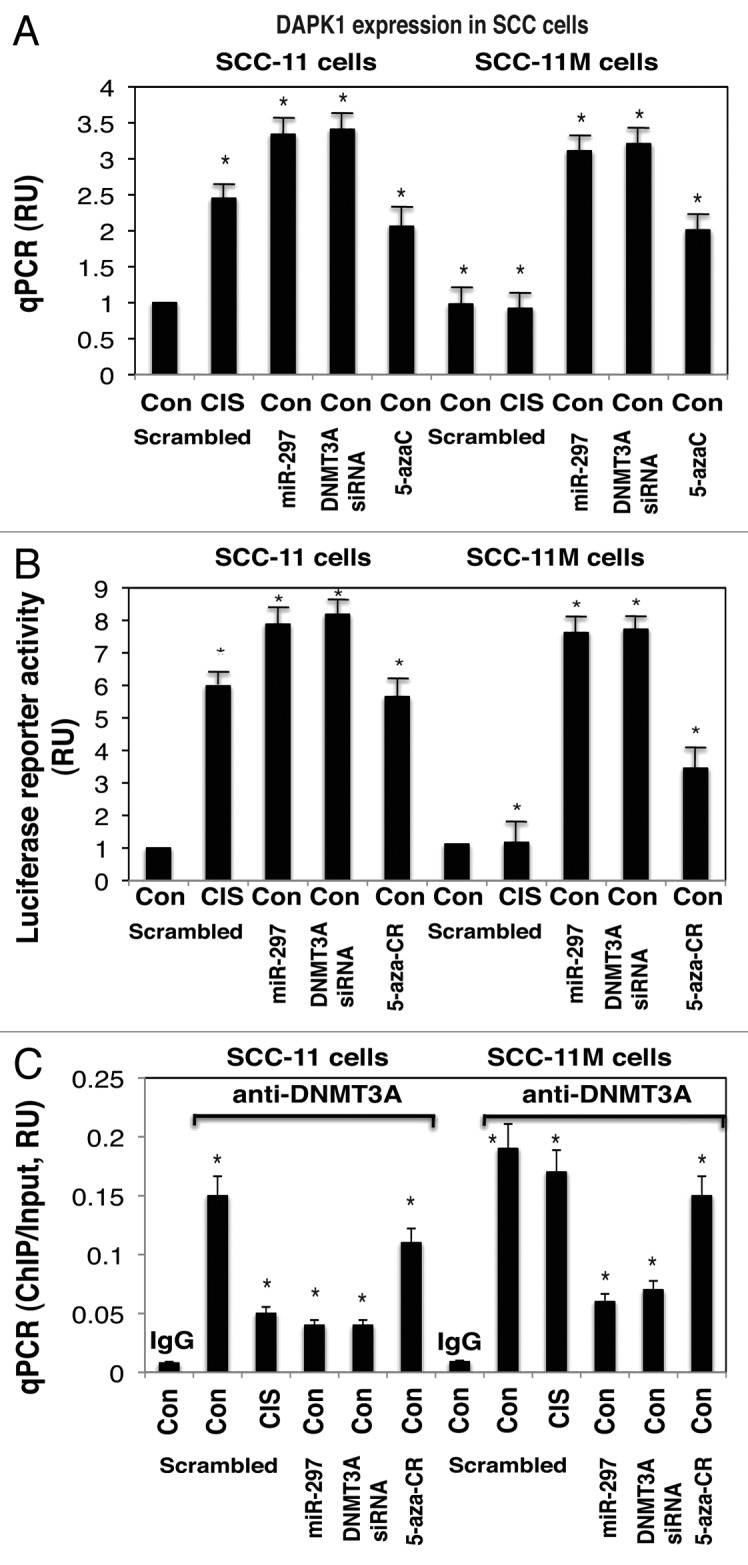
Figure 3. Expression of DAPK1 is modulated by DNA methylation in SCC-11 cells upon cisplatin exposure. SCC-11 cells and SCC-11M cells were transfected with the scrambled (Scr) miRNA for 32 h, and then exposed to control medium (Con) or 10 μg/ml cisplatin (CIS) for an additional 16 h. Cells were also transfected with the miR-297 mimic, or DNMT3A siRNA and exposed to control media for 48 h. Cells were also treated with the 1.5 μM 5′-AzaC for 16 h. (A) QPCR assay for the DAPK1 expression was performed from 3 independent experiments in triplicate (P < 0.05). (B) SCC-11 cells and SCC-11M cells were additionally transfected with 100 ng of the LightSwitch_Pro reporter plasmid for the DAPK1 promoter for 24 h. Renilla luciferase reporter activity assay was conducted from 3 independent experiments in triplicate (P < 0.05). (C) ChIP-qPCR assay of the DNMT3A binding to the specific region of the DAPK1 promoter. QPCR assay was performed using 3 independent experiments in triplicate (P < 0.05). The amount of ChIP-enriched DNA (ChIP/input) represented as a signal relative to the total amount of chromatin DNA (Input) using the same primers.
Modulation of histone deacetylation affects the SMARCA2 expression in SCC cells
Histone deacetylation is a well-known molecular mechanism underlying the transcription repression, and certain histone deacetylases (HDAC) have been shown to repress the transcription of specific cancer-related genes.47,57-62 SMARCA2 (BRM) is not mutated in tumor cells; however, it is epigenetically silenced by various HDACs confirmed by the use of HDAC inhibitors, shown to reverse SMARCA2 silencing and subsequently to inhibit cancer cell growth.61,62 Using the knockdown approach, the high-throughput screening of HDAC showed that the class II HDACs, HDAC4 and HDAC9, regulate SMARCA2 expression.61-64
We examined whether SMARCA2 expression was affected in SCC-11 cells and SCC-11M cells exposed to control media and 10 μg/ml cisplatin for 16 h (Fig. 4). Since, miR-92b-3p is upregulated in SCC-11 cells compared with SCC-11M cells upon cisplatin exposure,39 and was shown to modulate HDAC9 expression in vitro (Fig. 1B; Fig. S1B and F), we suggested that the p-ΔNp63α-upregulated miR-92b-3p might be implicated in epigenetic regulation of the SMARCA2 expression. Using the qPCR, luciferase reporter, and ChIP assays, we tested whether miR-92b-3p, siRNA to HDAC9, and class IIb histone deacetylase inhibitor, MC1568 (refs. 62–64) would affect the SMARCA2 expression in SCC-11 cells and SCC-11M cells treated with control media (Fig. 4).
Figure 4.
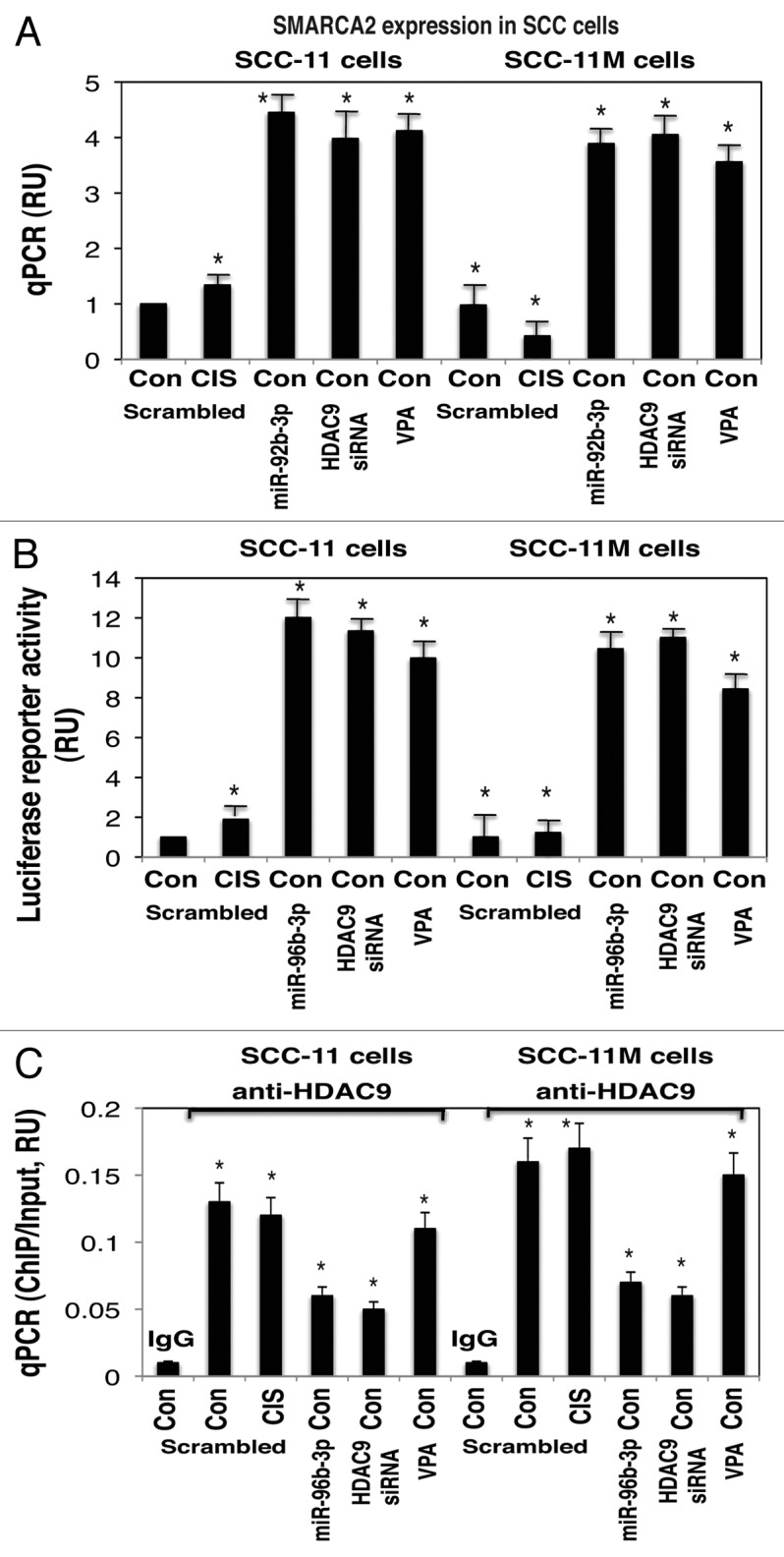
Expression of SMARCA2 is modulated by histone deacetylation in SCC-11 cells upon cisplatin exposure. SCC-11 cells and SCC-11M cells were transfected with the scrambled (Scr) miRNA for 32 h, and then exposed to control medium (Con) or 10 μg/ml cisplatin (CIS) for an additional 16 h. Cells were also transfected with the miR-92b-3p mimic, or HDAC9 siRNA and exposed to control media for 48 h. Cells were also treated with the 5 μM MC1568 for 16 h. (A) QPCR assay for the SMARCA2 expression was performed from 3 independent experiments in triplicate (P < 0.05). (B) SCC-11 cells and SCC-11M cells were additionally transfected with 100 ng of the LightSwitch_Pro reporter plasmid for the SMARCA2 promoter for 24 h. Renilla luciferase reporter activity assay was conducted from 3 independent experiments in triplicate (P < 0.05). (C) ChIP-qPCR assay of the HDAC9 binding to the specific region of the SMARCA2 promoter. QPCR assays were performed using 3 independent experiments in triplicate (P < 0.05). The amount of ChIP-enriched DNA (ChIP/Input) represented as a signal relative to the total amount of chromatin DNA (input) using the same primers.
We showed that the cisplatin exposure of SCC-11 cells induced SMARCA2 mRNA expression by 1.63-fold, while miR-92b-3p, siRNA to HDAC9, and MC1568 increased the SMARCA2 mRNA expression in SCC-11 cells by 4.53-, 3.94-, and 4.16-fold, respectively (Fig. 4A). Although SCC-11M cells exposed to cisplatin displayed the −1.87-fold decrease in the SMARCA2 mRNA expression compared with control treatment, miR-92b-3p, siRNA to HDAC9, and MC1568 increased the SMARCA2 mRNA expression in SCC-11M cells by 4.02-, 4.15-, and 3.58-fold, respectively (Fig. 4A). We further showed that the SMARCA2 promoter-driven luciferase activity was increased in SCC-11 cells upon cisplatin exposure by 2.03-fold, while miR-92b-3p, siRNA to HDAC9, and MC1568 increased the SMARCA2 luciferase activity by 12.03-, 11.19-, and 9.94-fold, respectively (Fig. 4B). At the same time, SCC-11M cells exposed to cisplatin showed no change in the SMARCA2 promoter function compared with control treatment, while miR-92b-3p, siRNA to HDAC9, and MC1568 activated the SMARCA2 mRNA expression in SCC-11M cells by 10.52-, 11.25-, and 8.64-fold, respectively (Fig. 4B). We next showed that HDAC9 binding to the SMARCA2 promoter (Fig. S3) was unchanged in both SCC-11 and SCC-11M cells upon cisplatin exposure; however, this was markedly decreased after treatment of SCC-11 cells and SCC-11M cells with miR-92b-3p, and siRNA to HDAC9 (Fig. 4C). Inhibition of HDAC9 activity with MC1568 had only a slight effect on the HDAC9 binding to the SMARCA2 promoter (compared with scrambled control) in both SCC-11 and SCC-11M cells (Fig. 4C). Similar ChIP results were observed using the tongue-derived cisplatin-sensitive SCC-25/cisplatin-resistant SCC-25CP pair of cells (Fig. S8B).
Modulation of histone demethylation affects the MDM2 expression in SCC cells
Histone methylation/demethylation can either activate or repress gene transcription. While methylation of histone 3 (H3) at lysine (K)-4 and K36 is linked to actively transcribed genes, the methylation at H3K9 and H3K27 is associated with transcriptional repression.65-67 Histone lysine methylation was regulated by a large number of histone methyltransferases containing SET domain and demethylases (e.g., LSD1 and JMJC-domain containing proteins), as reviewed in references 65-67. The histone demethylase JMJD2C (KMD4C, GASC1) can demethylate trimethylated H3K9 (H3K9me3) and H3K36 (H3K36me3).68,69 Previously known as GASC1, this histone demethylase found amplified in esophageal SCC.70,71 Moreover, knockdown of KDM4C caused decreased proliferation of the tumor cells.71 The transcription of MDM2 (known to reduce the TP53 protein levels in tumor cells) was induced by histone demethylase KDM4C through the changes of histone H3 methylation on the MDM2 promoter.72,73
We examined whether the MDM2 expression was affected in SCC-11 cells and SCC-11M cells exposed to control media and 10 μg/ml cisplatin for 16 h (Fig. 5). Since, miR-485-5p was upregulated in SCC-11 cells compared with SCC-11M cells upon cisplatin exposure,39 and is shown to target KDM4C expression (Fig. 1; Fig. S1C and G), we suggested that the p-ΔNp63α-upregulated miR-485-5p might be implicated in epigenetic regulation of the KDM4C expression. Using the qPCR, luciferase reporter, and ChIP assays, we tested whether miR-485-5p, siRNA to KDM4C, and histone demethylase inhibitor, IOX1 would affect the MDM2 expression in SCC-11 cells and SCC-11M cells treated with control media (Fig. 5). We showed that the cisplatin exposure of SCC-11 cells reduced the MDM2 mRNA expression by −1.4-fold, while miR-485-5p, siRNA to KDM4C and IOX1 reduced the MDM2 mRNA expression in SCC-11 cells by −1.85-, −2.38-, and −2.04-fold, respectively (Fig. 5A).
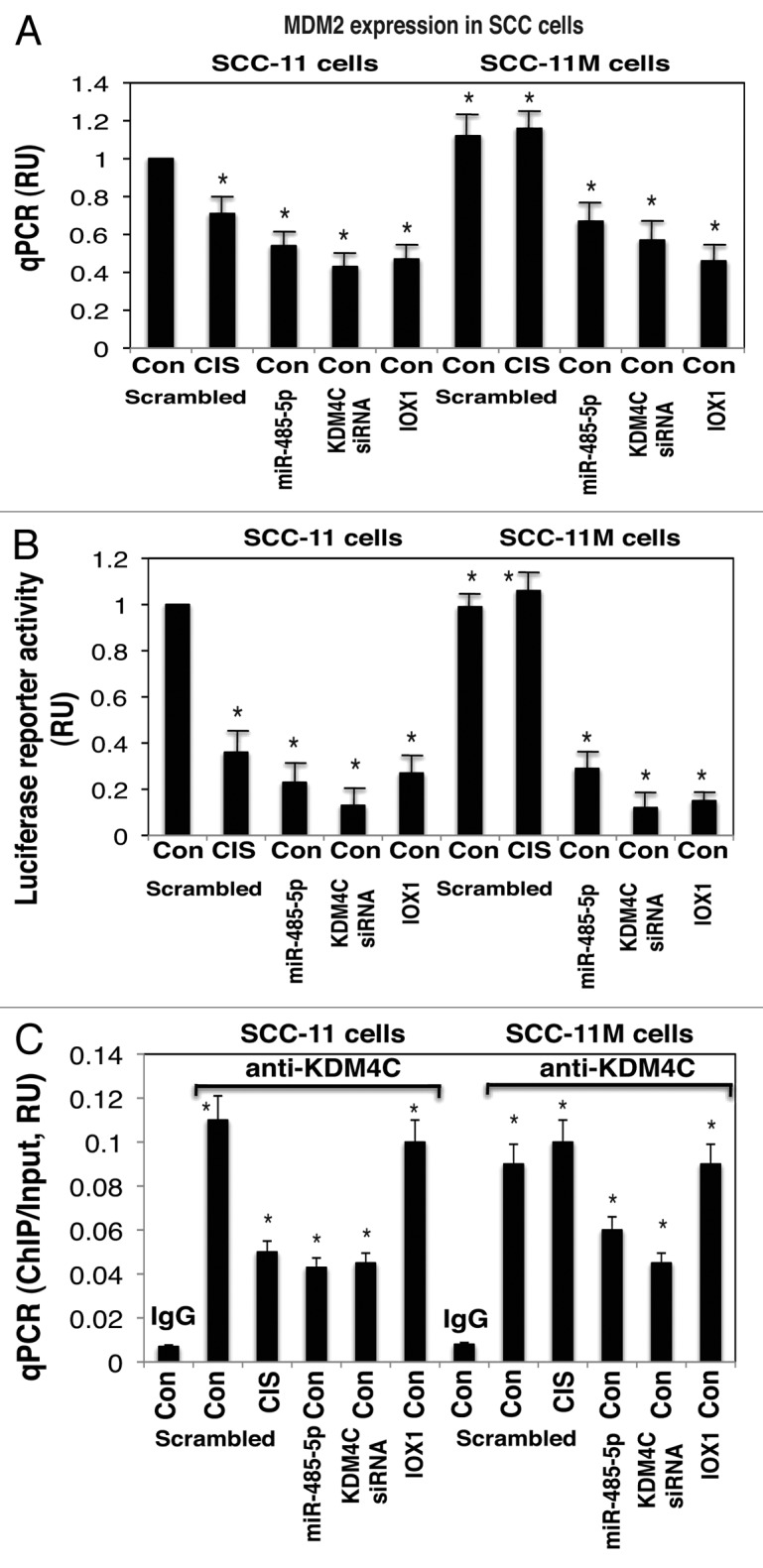
Figure 5. Expression of MDM2 is modulated by histone demethylation in SCC-11 cells upon cisplatin exposure. SCC-11 cells and SCC-11M cells were transfected with the scrambled (Scr) miRNA for 32 h, and then exposed to control medium (Con) or 10 μg/ml cisplatin (CIS) for an additional 16 h. Cells were also transfected with the miR-485-5p mimic, or KDM4C siRNA, and exposed to control media for 48 h. Cells were also treated with 1 mM IOX1 for 16 h. (A) QPCR assay for the MDM2 expression was performed from 3 independent experiments in triplicate (P < 0.05). (B) SCC-11 cells and SCC-11M cells were additionally transfected with 100 ng of the LightSwitch_Pro reporter plasmid for the MDM2 promoter for 24 h. Renilla luciferase reporter activity assay was conducted from 3 independent experiments in triplicate (P < 0.05). (C) ChIP-qPCR assay of the KDM4C binding to the specific region of the MDM2 promoter. QPCR assay was performed using 3 independent experiments in triplicate (P < 0.05). The amount of ChIP-enriched DNA (ChIP/Input) represented as a signal relative to the total amount of chromatin DNA (Input) using the same primers.
Although SCC-11M cells exposed to cisplatin displayed only a slight change in the MDM2 mRNA expression compared with control treatment, miR-485-5p, siRNA to KDM4C, and IOX1 activated the MDM2 mRNA expression in SCC-11M cells by −1.61-, −1.87-, and −2.22-fold, respectively (Fig. 5A). We further showed that the MDM2 promoter-driven luciferase activity was decreased in SCC-11 cells upon cisplatin exposure by −2.63-fold, while miR-485-5p, siRNA to KDM4C, and IOX1 decreased this activity by −3.45-, −5.88-, and −3.23-fold, respectively (Fig. 5B).
At the same time, SCC-11M cells exposed to cisplatin showed no significant change in the MDM2 promoter function compared with control treatment, while miR-485-5p, siRNA to KDM4C, and IOX1 activated the MDM2 mRNA expression in SCC-11M cells by −2.78-, −6.67-, and −5.06-fold, respectively (Fig. 5B). We next showed that the KDM4C binding to the MDM2 promoter (Fig. S4) was unchanged in both SCC-11 and SCC-11M cell lines, however, was greatly decreased after treatment of SCC-11 cells with cisplatin, miR-485-5p, and siRNA to KDM4C (Fig. 5C). However, in SCC-11M cells treated with cisplatin the KDM4C binding to the MDM2 promoter (Fig. S4) showed no significant changes compared with control treatment, while miR-485-5p and siRNA to KDM4C markedly decreased that binding (Fig. 5C). Inhibition of KDM4C activity with IOX1 had only a slight effect on the KDM4C binding to the MDM2 promoter (compared with scrambled control) in both SCC-11 and SCC-11M cells (Fig. 5C). Similar ChIP results were detected using the tongue-derived cisplatin-sensitive SCC-25/cisplatin-resistant SCC-25CP pair of cells (Fig. S8C).
Modulation of SCC cell chemoresistance to cisplatin by epi-microRNAs
While the SCC-11 cells (expressing the wild-type ΔNp63α capable to undergo phosphorylation by ATM kinase) were shown to display the sensitivity to cisplatin exposure, the SCC-11M cells (expressing the mutated ΔNp63α-S385G with an altered ability to undergo the ATM-dependent phosphorylation) were found to be more cisplatin-resistant than SCC-11 cells.35-39
To increase the chemosensitivity of SCC-11M cells, we finally examined the potential effect of selected epi-microRNA mimics on the viability of SCC-11M cells upon cisplatin exposure. SCC-11 cells and SCC-11M cells were transfected with the scrambled microRNA for 32 h and then exposed to 10 μg/ml cisplatin (CIS) for 1–6 d (Fig. 6). SCC-11M cells transfected with the scrambled RNA for 32 h and were also exposed to control medium (Con) for 1–6 d (Fig. 6). SCC-11M cells were also transfected with indicated epi-microRNA mimics (miR-297, Fig. 6A; miR-92b-3p, Fig. 6B; and miR-485-5p, Fig. 6C) for 32 h, and then were exposed to 10 μg/ml cisplatin (CIS) for 1–6 d (Fig. 6A–C). We showed that the cisplatin treatment led to a dramatic decline in survival of SCC-11 cells (3.52–4.75-fold, Fig. 6A–C), while its effect on SCC-11M cells appeared to be less dramatic (1.71–1.89-fold, Fig. 6A–C), suggesting that the altered ability to phosphorylate ΔNp63α by ATM kinase in SCC-11M cells rendered them more resistant to cisplatin treatment than SCC-11 cells. Intriguingly, the introduction of miR-297 mimic, DNMT3A siRNA, and 5′-Aza5C into SCC-11M cells rendered them more sensitive to cisplatin exposure (Fig. 6A). Similarly, the treatment of SCC-11M cells with miR-92b-3p mimic, HDAC9 siRNA, and MC1568, or with miR-485-5p mimic, KDM4C siRNA and IOX1 decreased the cell viability of SCC-11M cells upon cisplatin treatment (Fig. 6B and C, respectively). Finally, we showed that while individual microRNA mimics decreased the viability of SCC-11M cells by −22.2% (for miR-297), −18.9% (for miR-92b-3p), and −21.5% (for miR-485-5p), their combined treatment decreased the cell viability by −60.5% compared with control SCC-11M cells with the scrambled RNA treated with cisplatin showing additivity rather than synergy (Fig. S9).

Figure 6. Modulation of epigenetic regulatory components sensitize SCC-11M cells to cisplatin exposure. Cell viability assay. (A–C). SCC-11M cells were transfected with the scrambled RNA for 32 h, and then exposed to control medium (Con) or 10 μg/ml cisplatin (CIS) for indicated time. Cells were also transfected with indicated microRNA mimics (A, miR-297; B, miR-92b-3p; C, miR-485-5p), or siRNAs against DNMT3A (A), HDAC9 (B), and KDM4C (C) for 32 h, and then exposed to 10 μg/ml cisplatin (CIS) for indicated time. Cells were also exposed to chemical inhibitors for DNMT3A (A, 1.5 μM 5′-AzaC), HDAC9 (B, class II HDAC inhibitor, 5 μM MC1568), or KDM4C (C, 1 mM, IOX1) along with 10 μg/ml cisplatin (CIS) for indicated time periods. Cell viability (MTT assay) was monitored in triplicate in 3 independent experiments.
Discussion
Cancer initiation and progression is triggered by a combined program of epigenetic and genetic alterations resulting in deregulated gene expression and, subsequently, function.1,2,6,56,67,73,74 DNA hypermethylation represses the gene transcription, whereas DNA demethylation induces the transcription of genes, thereby controlling the expression and function of genes involved in cell differentiation, proliferation, survival, and apoptosis, which are often deregulated in cancer cells, leading to malignant phenotypes.2,56,74 Finally, the intricate network of epigenetic regulation of gene expression has been further enriched by the non-coding microRNAs affecting gene expression via binding to the mRNA sequences and by modulation of the epigenetic machinery.6,12-17,74-78
Transcriptional regulation of gene expression, ultimately leading to activation or repression of target genes, involves many layers of control including activating mechanisms, such as demethylation of promoter DNA sequences, acetylation, or demethylation of histones, subsequently affecting chromatin remodeling and repression mechanisms, such as methylation of promoter DNA sequences and methylation or deacetylation of histones forming nucleosome structures around promoter sequences, and microRNA.2,25,56,58,65,67,79-84 Our current studies shed a light on the potential role for p-ΔNp63α/microRNA network in these epigenetic regulatory molecular layers, essentially leading to modulation of tumor cell response to chemotherapeutic drugs through cell cycle arrest and apoptosis.39,78
We found that the p-ΔNp63α-dependent epi-microRNAs modulate the protein targets involved in DNA methylation (DNMT1 and DNMT3A), histone acetylation (KAT2B), histone deacetylation (HDAC9), histone demethylation (KDM2A, KDM3B, KDM4C, and KDM5B), polycomb repressive complex (EZH2, BMI1, RNF2, and RBBP4). We showed that the levels of EZH2, RBBP4, DNMT3A, and KDM4C proteins were downregulated, while levels for RNF2, KDM2A, KDM3B, and KDM5B proteins were upregulated in the larynx-derived SCC-11 cells compared with SCC-11M cells upon cisplatin exposure. We next found that DNMT3A, HDAC9, and KDM4C were forming protein–protein complexes with ΔNp63α, noting that this ability increased in SCC-11M cells, therefore supporting the idea that non-p-ΔNp63α is likely to recruit these epigenetic enzymes to certain gene promoters (DAPK1, SMARCA2, and MDM2) through TP63 binding sequence (Figs. S2–4). We suggested that ΔNp63α along with DNMT3A, HDAC9, and KDM4C could transcriptionally regulate the expression of tested genes, thereby contributing to SCC cell response to platinum chemotherapeutic compounds. We showed that the expression of DAPK1, SMARCA2, and MDM2 was affected through a modulation of DNMT3A (for DAPK1), HDAC9 (for SMARCA2), and KDM4C (for MDM2), respectively, by the specific epi-microRNA (miR-297, miR-92b-3p, and miR-485–5p), siRNAs, and chemical inhibitors against DNMT3A, HDAC9, and KDM4C. By qPCR and promoter luciferase reporter assays, we showed that the inactivation of DNMT3A and HDAC9 led to activation of DAPK1 and SMARCA2 expression, while inactivation of KDM4C resulted in repression of MDM2 expression in the larynx-derived SCC-11 cells. Additionally, we found that the binding of DNMT3A, HDAC9, and KDM4C to the DAPK1, SMARCA2, and MDM2 promoters was affected by the tested epi-microRNAs, siRNAs, and chemical inhibitors against DNMT3A, HDAC9, and KDM4C in SCC of larynx (SCC-11/11M) and tongue (SCC-25/25CP) origin. Finally, we observed that SCC-11M cells were markedly more resistant to cisplatin treatment than SCC-11 cells; however, the former could be sensitized to cisplatin treatment by inactivation of DNMT3A, HDAC9, and KDM4C using the tested microRNA, siRNAs, and chemical inhibitors described in this study.
Although tp53 and tp63 were shown to transcriptionally control microRNA expression, the ability of microRNAs to regulate the components of the epigenetic machinery, targeting molecules involved in the DNA methylation, histone acetylation, and modulation of transcription factors (e.g., TP53 and TP63) has also started to emerge, creating a controlled feedback mechanism.19,28,76-78,85
The miR-29 family was shown to directly target DNMT3A and DNMT3B and indirectly target DNMT1 through regulation of the transactivator SP1 or RBL2,86-88 while miR-148 and miR-140 were shown to target DNMT1 and DNMT3B.89-91 miR-101 was shown to regulate the expression of EZH2, catalytic subunit of the polycomb repressive complex 2, which mediates epigenetic gene silencing by trimethylating histone H3 lysine 27.92,93 miR-200a was shown to target HDAC4, while miR-449a was found to modulate HDAC1 and subsequently induce cell cycle arrest, apoptosis, and a senescent phenotype in prostate and hepatocellular cancers and myeloid leukemia cells.94-96 Introduction of miR-148a and miR-34b/c in cancer cells was shown to inhibit their cell motility, reduce tumor growth, and impair metastasis formation in xenograft models, and led to a downregulation of microRNA-dependent protein targets, such as c-MYB, c-MYC, E2F3, CDK6, HDAC, and TGIF2.97
Once it was widely demonstrated that an aberrant microRNA-ome is a hallmark in cancer, accumulating evidence showed that the microRNA expression is affected by the same epigenetic mechanisms as mRNA transcription.29,74,83,85 microRNA expression can be regulated by several epigenetic mechanisms, including transcriptional modulation of microRNA genes by transcription factors, promoter methylation, or histone acetylation, and/or altered microRNA maturation.83,85 The ability of microRNAs to regulate the components of the epigenetic machinery, targeting molecules involved in the DNA methylation, histone acetylation, and modulation of transcription factors is also started to emerge creating a controlled feedback mechanism.3,4,21,22,78,85 Furthermore, accumulating evidence supports a strong potential role for microRNA-dependent regulation in the tumor response to anti-cancer chemotherapeutic treatments, thereby increasing the significance of microRNA-based approaches in personalized therapies of human cancers.30,31,98,99
Materials and Methods
Cells, reagents, and antibodies
Squamous cell carcinoma (SCC)-11 cells (wt-TP53 is expressed, wt-TP63 is amplified, and ΔNp63α is overexpressed, express both α and β isoforms of TAp73) were derived from the primary larynx SCC and authenticated.34-39,100 Stable SCC cell lines expressing wild-type ΔNp63α (SCC-11) or ΔNp63α-S385G (SCC-11M) were generated using Flp-In technology.34 We also used SCC-25 cells (expressing mutated TP53 [R209] and CDKN2A, expressing ΔNp63α) and SCC-25CP cells (with a spontaneously acquired cisplatin resistance) derived from the primary tongue SCC, as previously reviewed.39,44,101 Cells were maintained in a 1:1 mixture of Dulbecco modified Eagle medium and Ham F12 medium containing 1.2 g/L sodium bicarbonate, 2.5 mM L-glutamine, 15 mM HEPES, and 0.5 mM sodium pyruvate and supplemented with 400 ng/ml hydrocortisone and 10% fetal bovine serum. Cells were incubated with control medium or 10 μg/ml cis-diammine-dichloro-platinum-dichloride (cisplatin [CIS], P4394) along with 0.5–1.5 μM of 5′-azacytidine (5′-AzaC, A2385), 1–5 μM of MC1568 (M1824), or 0.5–1 mM of 8- hydroxy-5-quinolinecarboxylic acid (IOX1, SML0067), all from Sigma-Aldrich. Total lysates were used for immunoblotting and immunoprecipitation35 with the following antibodies against β-actin (Sigma), EZH2 (07–689), ΔNp63 (PC373), and BMI1 (05–1322) both from Millipore/EMD, DNMT1 (GTX30364), DNMT3A (GTX30365) from GenTex, HDAC9 (PA5–11246, Thermo Fisher Scientific), KDM2A (A301–475A), KDM4C (A300–885A), RNF2 (A302–869A) from Bethyl Laboratories, RBBP4 (LS-C53331), and KDM3B (LS-C71162), KDM5B (LS-C71115), KAT6B (LS-C125982) from LifeSpan Biosciences. The custom rabbit polyclonal antibody against phosphorylated peptide encompassing the ΔNp63α protein sequence (ATM motif, NKLPSV-pS-QLINPQQ, residues 379–392) was also used.34,35
Transfection with microRNA mimics
The following individual human mirVana® microRNA mimics (hsa-miR-297, hsa-miR-92b-3p, and hsa-miR-485-5p) were purchased from Ambion/Life Technologies. Cells in a 6-well plate were transfected with 100 pmol of the mimic or scrambled RNA in 500 μl serum-free media with 5 μl of Lipofectamine-2000 reagent (Invitrogen) for 32 h. Each experiment was performed independently 3 times and in triplicate. Cells were also transiently transfected with the scrambled siRNA and the following siRNAs: DNMT3A (sc-37757), HDAC9 (sc-35550), or KDM4C (sc-92765), all from Santa Cruz Biotechnology. Transfection of cells with 20 nM of siRNA was carried-out using Lipofectamine SiRNAMAX (Invitrogen) for 32 h.39 Resulting cells were treated with control medium, 10 μg/ml cisplatin, or other chemical reagents for an additional 16 h.
Quantitative (q)-PCR
We performed a qPCR using the High-Capacity RNA-to-cDNA Kit (#4387406), and TaqMan® PreAmp Master Mix Kit with the Gene Expression Master Mix) (#4384267). The DAPK1 mRNA, SMARCA2 mRNA, and MDM2 mRNA were amplified using the TaqMan PCR kits (105 bp, Hs00234489_m1, 67 bp, Hs01030846_m1, and 149 bp, Hs00242813_m1), respectively. The reaction (20 μl) was carried out at 50 °C for 2 min, 95 °C for 10 min, 40 cycles of 95 °C for 15 s and 60 °C for 1 min. All reagents were obtained from Ambion/Life Technologies). Three independent biological experiments were performed. Each RNA sample was amplified in triplicate. Expression was normalized to the 18S RNA TaqMan probe (187 bp, #445332), and expression levels were determined as the average Ct of this control, subsequently used to normalize the sample’s Ct. The average mRNA expression was determined using the Mann–Whitney U test. Data presented as relative values (RU) to data obtained from the control samples (SCC-11 cells transfected with the scrambled RNA and exposed to control media) designated as 1.
Chromatin immunoprecipitation (ChIP)
5 × 106 cell equivalents of chromatin (2–2.5 kbp in size) were immunoprecipitated with 5 μg of the ChIP-grade antibodies against DNMT3A (ab2850, Abcam), anti-HDAC9 (ab59718, Abcam), or anti-KDM4C (NB110–38884, Novus Biologicals), as previously described.28,38 The ChIP-grade normal rabbit immunoglobulin (IgG, ab37415, Abcam) was used as a negative control. After reversal of formaldehyde cross-linking, RNA-ase A, and proteinase K treatments, IP-enriched DNAs were used for qPCR assays. To amplify the specific regions, we used the following primers: sense, (−1804) 5′-GATAGCGCAA ATAAACTCTG CG-3′, and antisense, 5′-GCCTATGGTC GGCCTCCGAC AG-3′ (−900), yielding the 905-bp PCR fragment for the DAPK1 promoter; sense, (−860) 5′-TTATAAGGCG TTCAGCCTCT-3′, and antisense, 5′-TCATCAATGA AGTCATATTC AT-3′ (−23), yielding the 837-bp PCR fragment for the SMARCA2 promoter; and sense, (−997) 5′-AAACGTTTTT GCCACATCTC-3′, and antisense, 5′-CAGCCCGCCG CGCCCGC (−157), yielding the 841-bp PCR fragment for the MDM2 promoter. QPCR consisted of 40 cycles of 94 °C for 30 s, 60 °C for 30 s, and 72 °C for 30 s using Taq DNA polymerase (Invitrogen). The ChIP-qPCR values were obtained from the ChIP and Input samples and then normalized for GAPDH qPCR values. Values obtained from the Input samples were designated as 1. ChIP/Input ratio was plotted using the Microsoft Excel software. Experiments were performed in triplicate.
Luciferase reporter assay
We used the LightSwitch_Pro reporter plasmids for the DAPK1 (S719576), SMARCA2 (S706761), and MDM2 (S704939) promoters all obtained from SwitchGear Genomics. 5 × 104 cells/well in a 24-well plate were transfected with the control (empty) pLightSwitch_Prom vector (#S707592) using Fugene HD reagent (Roche) for 32 h, as previously described.39 Resulting cells were then treated with control media or with 10 μg/ml cisplatin for an additional 16 h. Renilla luciferase activity was measured at 480 nm using a luminometer. Data presented as relative values (RU) to data obtained from the control samples (SCC-11 cells transfected with the scrambled RNA and exposed to control media) designated as 1.
For microRNA/3′-UTR luciferase reporter assays, we used the 3′-UTR luciferase reporter plasmids for EZH2 (S811982), BMI1 (S810388), EED (S806207), RNF2 (S811266), RBBP4 (S808163), DNMT1 (S802002), DNMT3A (S808608), DNMT3B (S809202), MBD1 (S807532), HDAC9 (S811202), KDM2A (S811640), KDM3A (S804904), KDM3B (S808966), KDM4C (S806873), KDM5B (S810136), KAT2B (S810567), KAT3B (S808354), and KAT6B (S810388), all from SwitchGear Genomics. 5 × 104 cells/well in a 24-well plate were transfected with the control (empty) pLightSwitch_3UTR vector (S890005), respectively, using Fugene HD reagent (Roche) as previously described.36-38 Cells were also transfected with the selected 3′-UTR plasmids along with 100 ng of the tested microRNA mimics for 48 h. Data obtained from the control samples were presented in relative units (RU) and designated as 1. Data were expressed as means ± SD from 3 independent experiments in triplicate.
Cell viability assay
104 cells/well in 96-well plates were incubated in serum-free medium with 5 μg/ml of the 3-(4,5-dimethyl thiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT assay, American Tissue Culture Collection) in the dark for 4 h at 37 °C. Cells were lysed and incubated for 2 h at 37 °C, and the measurements (A570 nm to A650 nm) were obtained on a Spectra Max 250 plate reader (Molecular Devices). Each assay was repeated 3 times in triplicate.39
Statistical analysis and bioinformatics
Differences in variables between experimental and control groups were assessed by using the Student t test. For prediction of the microRNA “seed” sequences in the 3′-UTRs, we used miRDB-microRNA Target Prediction and Functional Study Database, v3.0 (http://www.mirdb.org).
Supplementary Material
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This study was supported in part by the Flight Attendant Research Institutions grant (#082469).
Glossary
Abbreviations:
- ATM
ataxia telangiectasia
- ChIP
chromatin immunoprecipitation
- CIS
cisplatin
- CON
control
- DAPK1
death-associated protein kinase
- DNMT
DNA methyltransferase
- HDAC
histone deacetylase
- KDM
K (lysine) histone demethylase
- MDM2
MDM2 oncogene, E3 ubiquitin protein ligase
- miR
microRNA
- p
phosphorylated
- qPCR
quantitative PCR
- RU
relative unit
- RLU
relative luciferase unit
- SCC
squamous cell carcinoma
- siRNA
small interfering RNA
- SMARCA2 (BRM)
SWI/SNF-related, matrix-associated, actin-dependent regulator of chromatin, subfamily a, member 2
- TP
tumor protein
- UTR
untranslated region
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/27676
References
- 1.Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007;128:683–92. doi: 10.1016/j.cell.2007.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tsai HC, Baylin SB. Cancer epigenetics: linking basic biology to clinical medicine. Cell Res. 2011;21:502–17. doi: 10.1038/cr.2011.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Iorio MV, Piovan C, Croce CM. Interplay between microRNAs and the epigenetic machinery: an intricate network. Biochim Biophys Acta. 2010;1799:694–701. doi: 10.1016/j.bbagrm.2010.05.005. [DOI] [PubMed] [Google Scholar]
- 4.Wiklund ED, Kjems J, Clark SJ. Epigenetic architecture and miRNA: reciprocal regulators. Epigenomics. 2010;2:823–40. doi: 10.2217/epi.10.51. [DOI] [PubMed] [Google Scholar]
- 5.Esteller M. Non-coding RNAs in human disease. Nat Rev Genet. 2011;12:861–74. doi: 10.1038/nrg3074. [DOI] [PubMed] [Google Scholar]
- 6.Lovat F, Valeri N, Croce CM. microRNAs in the pathogenesis of cancer. Semin Oncol. 2011;38:724–33. doi: 10.1053/j.seminoncol.2011.08.006. [DOI] [PubMed] [Google Scholar]
- 7.Pratt AJ, MacRae IJ. The RNA-induced silencing complex: a versatile gene-silencing machine. J Biol Chem. 2009;284:17897–901. doi: 10.1074/jbc.R900012200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van Kouwenhove M, Kedde M, Agami R. microRNA regulation by RNA-binding proteins and its implications for cancer. Nat Rev Cancer. 2011;11:644–56. doi: 10.1038/nrc3107. [DOI] [PubMed] [Google Scholar]
- 9.Sethupathy P, Megraw M, Hatzigeorgiou AG. A guide through present computational approaches for the identification of mammalian microRNA targets. Nat Methods. 2006;3:881–6. doi: 10.1038/nmeth954. [DOI] [PubMed] [Google Scholar]
- 10.Griffiths-Jones S, Grocock RJ, van Dongen S, Bateman A, Enright AJ. miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res. 2006;34:D140–4. doi: 10.1093/nar/gkj112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pasquinelli AE. microRNAs and their targets: recognition, regulation and an emerging reciprocal relationship. Nat Rev Genet. 2012;13:271–82. doi: 10.1038/nrg3162. [DOI] [PubMed] [Google Scholar]
- 12.Calin GA, Croce CM. microRNA signatures in human cancers. Nat Rev Cancer. 2006;6:857–66. doi: 10.1038/nrc1997. [DOI] [PubMed] [Google Scholar]
- 13.Volinia S, Calin GA, Liu CG, Ambs S, Cimmino A, Petrocca F, Visone R, Iorio M, Roldo C, Ferracin M, et al. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc Natl Acad Sci U S A. 2006;103:2257–61. doi: 10.1073/pnas.0510565103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thomson JM, Newman M, Parker JS, Morin-Kensicki EM, Wright T, Hammond SM. Extensive post-transcriptional regulation of microRNAs and its implications for cancer. Genes Dev. 2006;20:2202–7. doi: 10.1101/gad.1444406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gaur A, Jewell DA, Liang Y, Ridzon D, Moore JH, Chen C, Ambros VR, Israel MA. Characterization of microRNA expression levels and their biological correlates in human cancer cell lines. Cancer Res. 2007;67:2456–68. doi: 10.1158/0008-5472.CAN-06-2698. [DOI] [PubMed] [Google Scholar]
- 16.Krutovskikh VA, Herceg Z. Oncogenic microRNAs (OncomiRs) as a new class of cancer biomarkers. Bioessays. 2010;32:894–904. doi: 10.1002/bies.201000040. [DOI] [PubMed] [Google Scholar]
- 17.Kumar MS, Lu J, Mercer KL, Golub TR, Jacks T. Impaired microRNA processing enhances cellular transformation and tumorigenesis. Nat Genet. 2007;39:673–7. doi: 10.1038/ng2003. [DOI] [PubMed] [Google Scholar]
- 18.Saito Y, Liang G, Egger G, Friedman JM, Chuang JC, Coetzee GA, Jones PA. Specific activation of microRNA-127 with downregulation of the proto-oncogene BCL6 by chromatin-modifying drugs in human cancer cells. Cancer Cell. 2006;9:435–43. doi: 10.1016/j.ccr.2006.04.020. [DOI] [PubMed] [Google Scholar]
- 19.Chang TC, Wentzel EA, Kent OA, Ramachandran K, Mullendore M, Lee KH, Feldmann G, Yamakuchi M, Ferlito M, Lowenstein CJ, et al. Transactivation of miR-34a by p53 broadly influences gene expression and promotes apoptosis. Mol Cell. 2007;26:745–52. doi: 10.1016/j.molcel.2007.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gonzalez S, Pisano DG, Serrano M. Mechanistic principles of chromatin remodeling guided by siRNAs and miRNAs. Cell Cycle. 2008;7:2601–8. doi: 10.4161/cc.7.16.6541. [DOI] [PubMed] [Google Scholar]
- 21.Juan AH, Sartorelli V. microRNA-214 and polycomb group proteins: a regulatory circuit controlling differentiation and cell fate decisions. Cell Cycle. 2010;9:1445–6. doi: 10.4161/cc.9.8.11472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim T, Veronese A, Pichiorri F, Lee TJ, Jeon YJ, Volinia S, Pineau P, Marchio A, Palatini J, Suh SS, et al. p53 regulates epithelial-mesenchymal transition through microRNAs targeting ZEB1 and ZEB2. J Exp Med. 2011;208:875–83. doi: 10.1084/jem.20110235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim DH, Saetrom P, Snøve O, Jr., Rossi JJ. microRNA-directed transcriptional gene silencing in mammalian cells. Proc Natl Acad Sci U S A. 2008;105:16230–5. doi: 10.1073/pnas.0808830105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Place RF, Li LC, Pookot D, Noonan EJ, Dahiya R. microRNA-373 induces expression of genes with complementary promoter sequences. Proc Natl Acad Sci U S A. 2008;105:1608–13. doi: 10.1073/pnas.0707594105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Suzuki K, Kelleher AD. Transcriptional regulation by promoter targeted RNAs. Curr Top Med Chem. 2009;9:1079–87. doi: 10.2174/156802609789630875. [DOI] [PubMed] [Google Scholar]
- 26.Younger ST, Corey DR. Transcriptional regulation by miRNA mimics that target sequences downstream of gene termini. Mol Biosyst. 2011;7:2383–8. doi: 10.1039/c1mb05090g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.He L, He X, Lim LP, de Stanchina E, Xuan Z, Liang Y, Xue W, Zender L, Magnus J, Ridzon D, et al. A microRNA component of the p53 tumour suppressor network. Nature. 2007;447:1130–4. doi: 10.1038/nature05939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huang Y, Kesselman D, Kizub D, Guerrero-Preston R, Ratovitski EA. Phospho-ΔNp63α/microRNA feedback regulation in squamous carcinoma cells upon cisplatin exposure. Cell Cycle. 2013;12:684–97. doi: 10.4161/cc.23598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Baer C, Claus R, Plass C. Genome-wide epigenetic regulation of miRNAs in cancer. Cancer Res. 2013;73:473–7. doi: 10.1158/0008-5472.CAN-12-3731. [DOI] [PubMed] [Google Scholar]
- 30.Iguchi H, Kosaka N, Ochiya T. Versatile applications of microRNA in anti-cancer drug discovery: from therapeutics to biomarkers. Curr Drug Discov Technol. 2010;7:95–105. doi: 10.2174/157016310793180648. [DOI] [PubMed] [Google Scholar]
- 31.Galasso M, Sana ME, Volinia S. Non-coding RNAs: a key to future personalized molecular therapy? Genome Med. 2010;2:12. doi: 10.1186/gm133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Krützfeldt J, Rajewsky N, Braich R, Rajeev KG, Tuschl T, Manoharan M, Stoffel M. Silencing of microRNAs in vivo with ‘antagomirs’. Nature. 2005;438:685–9. doi: 10.1038/nature04303. [DOI] [PubMed] [Google Scholar]
- 33.Ebert MS, Neilson JR, Sharp PA. microRNA sponges: competitive inhibitors of small RNAs in mammalian cells. Nat Methods. 2007;4:721–6. doi: 10.1038/nmeth1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huang Y, Sen T, Nagpal J, Upadhyay S, Trink B, Ratovitski E, Sidransky D. ATM kinase is a master switch for the Δ Np63 α phosphorylation/degradation in human head and neck squamous cell carcinoma cells upon DNA damage. Cell Cycle. 2008;7:2846–55. doi: 10.4161/cc.7.18.6627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang Y, Chuang AY, Romano RA, Liégeois NJ, Sinha S, Trink B, Ratovitski E, Sidransky D. Phospho-DeltaNp63α/NF-Y protein complex transcriptionally regulates DDIT3 expression in squamous cell carcinoma cells upon cisplatin exposure. Cell Cycle. 2010;9:328–38. doi: 10.4161/cc.9.2.10432. [DOI] [PubMed] [Google Scholar]
- 36.Huang Y, Chuang A, Hao H, Talbot C, Sen T, Trink B, Sidransky D, Ratovitski E. Phospho-ΔNp63α is a key regulator of the cisplatin-induced microRNAome in cancer cells. Cell Death Differ. 2011;18:1220–30. doi: 10.1038/cdd.2010.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huang Y, Chuang AY, Ratovitski EA. Phospho-ΔNp63α/miR-885-3p axis in tumor cell life and cell death upon cisplatin exposure. Cell Cycle. 2011;10:3938–47. doi: 10.4161/cc.10.22.18107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huang Y, Guerrero-Preston R, Ratovitski EA. Phospho-ΔNp63α-dependent regulation of autophagic signaling through transcription and micro-RNA modulation. Cell Cycle. 2012;11:1247–59. doi: 10.4161/cc.11.6.19670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ratovitski EA. Phospho-ΔNp63α-dependent microRNAs modulate chemoresistance of squamous cell carcinoma cells to cisplatin: at the crossroads of cell life and death. FEBS Lett. 2013;587:2536–41. doi: 10.1016/j.febslet.2013.06.020. [DOI] [PubMed] [Google Scholar]
- 40.Lim LP, Lau NC, Garrett-Engele P, Grimson A, Schelter JM, Castle J, Bartel DP, Linsley PS, Johnson JM. Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature. 2005;433:769–73. doi: 10.1038/nature03315. [DOI] [PubMed] [Google Scholar]
- 41.Bartel DP. microRNAs: target recognition and regulatory functions. Cell. 2009;136:215–33. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Guo H, Ingolia NT, Weissman JS, Bartel DP. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature. 2010;466:835–40. doi: 10.1038/nature09267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huang Y, Jeong JS, Okamura J, Sook-Kim M, Zhu H, Guerrero-Preston R, Ratovitski EA. Global tumor protein p53/p63 interactome: making a case for cisplatin chemoresistance. Cell Cycle. 2012;11:2367–79. doi: 10.4161/cc.20863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Huang Y, Ratovitski EA. Phospho-ΔNp63α/Rpn13-dependent regulation of LKB1 degradation modulates autophagy in cancer cells. Aging (Albany NY) 2010;2:959–68. doi: 10.18632/aging.100249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hervouet E, Vallette FM, Cartron PF. Dnmt3/transcription factor interactions as crucial players in targeted DNA methylation. Epigenetics. 2009;4:487–99. doi: 10.4161/epi.4.7.9883. [DOI] [PubMed] [Google Scholar]
- 46.Hervouet E, Vallette FM, Cartron PF. Dnmt1/Transcription factor interactions: an alternative mechanism of DNA methylation inheritance. Genes Cancer. 2010;1:434–43. doi: 10.1177/1947601910373794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ramsey MR, He L, Forster N, Ory B, Ellisen LW. Physical association of HDAC1 and HDAC2 with p63 mediates transcriptional repression and tumor maintenance in squamous cell carcinoma. Cancer Res. 2011;71:4373–9. doi: 10.1158/0008-5472.CAN-11-0046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ho AS, Turcan S, Chan TA. Epigenetic therapy: use of agents targeting deacetylation and methylation in cancer management. Onco Targets Ther. 2013;6:223–32. doi: 10.2147/OTT.S34680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Karaca B, Atmaca H, Bozkurt E, Kisim A, Uzunoglu S, Karabulut B, Sezgin C, Sanli UA, Uslu R. Combination of AT-101/cisplatin overcomes chemoresistance by inducing apoptosis and modulating epigenetics in human ovarian cancer cells. Mol Biol Rep. 2013;40:3925–33. doi: 10.1007/s11033-012-2469-z. [DOI] [PubMed] [Google Scholar]
- 50.Sugita H, Iida S, Inokuchi M, Kato K, Ishiguro M, Ishikawa T, Takagi Y, Enjoji M, Yamada H, Uetake H, et al. Methylation of BNIP3 and DAPK indicates lower response to chemotherapy and poor prognosis in gastric cancer. Oncol Rep. 2011;25:513–8. doi: 10.3892/or.2010.1085. [DOI] [PubMed] [Google Scholar]
- 51.Ogawa T, Liggett TE, Melnikov AA, Monitto CL, Kusuke D, Shiga K, Kobayashi T, Horii A, Chatterjee A, Levenson VV, et al. Methylation of death-associated protein kinase is associated with cetuximab and erlotinib resistance. Cell Cycle. 2012;11:1656–63. doi: 10.4161/cc.20120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kim GD, Ni J, Kelesoglu N, Roberts RJ, Pradhan S. Co-operation and communication between the human maintenance and de novo DNA (cytosine-5) methyltransferases. EMBO J. 2002;21:4183–95. doi: 10.1093/emboj/cdf401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yokochi T, Robertson KD. Preferential methylation of unmethylated DNA by Mammalian de novo DNA methyltransferase Dnmt3a. J Biol Chem. 2002;277:11735–45. doi: 10.1074/jbc.M106590200. [DOI] [PubMed] [Google Scholar]
- 54.Oka M, Meacham AM, Hamazaki T, Rodić N, Chang LJ, Terada N. De novo DNA methyltransferases Dnmt3a and Dnmt3b primarily mediate the cytotoxic effect of 5-aza-2′-deoxycytidine. Oncogene. 2005;24:3091–9. doi: 10.1038/sj.onc.1208540. [DOI] [PubMed] [Google Scholar]
- 55.Jeong S, Liang G, Sharma S, Lin JC, Choi SH, Han H, Yoo CB, Egger G, Yang AS, Jones PA. Selective anchoring of DNA methyltransferases 3A and 3B to nucleosomes containing methylated DNA. Mol Cell Biol. 2009;29:5366–76. doi: 10.1128/MCB.00484-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jin B, Ernst J, Tiedemann RL, Xu H, Sureshchandra S, Kellis M, Dalton S, Liu C, Choi JH, Robertson KD. Linking DNA methyltransferases to epigenetic marks and nucleosome structure genome-wide in human tumor cells. Cell Rep. 2012;2:1411–24. doi: 10.1016/j.celrep.2012.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ling Y, Sankpal UT, Robertson AK, McNally JG, Karpova T, Robertson KD. Modification of de novo DNA methyltransferase 3a (Dnmt3a) by SUMO-1 modulates its interaction with histone deacetylases (HDACs) and its capacity to repress transcription. Nucleic Acids Res. 2004;32:598–610. doi: 10.1093/nar/gkh195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Joshi P, Greco TM, Guise AJ, Luo Y, Yu F, Nesvizhskii AI, Cristea IM. The functional interactome landscape of the human histone deacetylase family. Mol Syst Biol. 2013;9:672. doi: 10.1038/msb.2013.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Clements EG, Mohammad HP, Leadem BR, Easwaran H, Cai Y, Van Neste L, Baylin SB. DNMT1 modulates gene expression without its catalytic activity partially through its interactions with histone-modifying enzymes. Nucleic Acids Res. 2012;40:4334–46. doi: 10.1093/nar/gks031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bourachot B, Yaniv M, Muchardt C. Growth inhibition by the mammalian SWI-SNF subunit Brm is regulated by acetylation. EMBO J. 2003;22:6505–15. doi: 10.1093/emboj/cdg621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Glaros S, Cirrincione GM, Muchardt C, Kleer CG, Michael CW, Reisman D. The reversible epigenetic silencing of BRM: implications for clinical targeted therapy. Oncogene. 2007;26:7058–66. doi: 10.1038/sj.onc.1210514. [DOI] [PubMed] [Google Scholar]
- 62.Kahali B, Gramling SJ, Marquez SB, Thompson K, Lu L, Reisman D. Identifying targets for the restoration and reactivation of BRM. Oncogene. 2014;33:653–64. doi: 10.1038/onc.2012.613. [DOI] [PubMed] [Google Scholar]
- 63.Duong V, Bret C, Altucci L, Mai A, Duraffourd C, Loubersac J, Harmand PO, Bonnet S, Valente S, Maudelonde T, et al. Specific activity of class II histone deacetylases in human breast cancer cells. Mol Cancer Res. 2008;6:1908–19. doi: 10.1158/1541-7786.MCR-08-0299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Giannini G, Cabri W, Fattorusso C, Rodriquez M. Histone deacetylase inhibitors in the treatment of cancer: overview and perspectives. Future Med Chem. 2012;4:1439–60. doi: 10.4155/fmc.12.80. [DOI] [PubMed] [Google Scholar]
- 65.Cloos PA, Christensen J, Agger K, Helin K. Erasing the methyl mark: histone demethylases at the center of cellular differentiation and disease. Genes Dev. 2008;22:1115–40. doi: 10.1101/gad.1652908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pedersen MT, Helin K. Histone demethylases in development and disease. Trends Cell Biol. 2010;20:662–71. doi: 10.1016/j.tcb.2010.08.011. [DOI] [PubMed] [Google Scholar]
- 67.Varier RA, Timmers HT. Histone lysine methylation and demethylation pathways in cancer. Biochim Biophys Acta. 2011;1815:75–89. doi: 10.1016/j.bbcan.2010.10.002. [DOI] [PubMed] [Google Scholar]
- 68.Luo W, Chang R, Zhong J, Pandey A, Semenza GL. Histone demethylase JMJD2C is a coactivator for hypoxia-inducible factor 1 that is required for breast cancer progression. Proc Natl Acad Sci U S A. 2012;109:E3367–76. doi: 10.1073/pnas.1217394109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Berry WL, Janknecht R. KDM4/JMJD2 histone demethylases: epigenetic regulators in cancer cells. Cancer Res. 2013;73:2936–42. doi: 10.1158/0008-5472.CAN-12-4300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yang ZQ, Imoto I, Fukuda Y, Pimkhaokham A, Shimada Y, Imamura M, Sugano S, Nakamura Y, Inazawa J. Identification of a novel gene, GASC1, within an amplicon at 9p23-24 frequently detected in esophageal cancer cell lines. Cancer Res. 2000;60:4735–9. [PubMed] [Google Scholar]
- 71.Cloos PA, Christensen J, Agger K, Maiolica A, Rappsilber J, Antal T, Hansen KH, Helin K. The putative oncogene GASC1 demethylates tri- and dimethylated lysine 9 on histone H3. Nature. 2006;442:307–11. doi: 10.1038/nature04837. [DOI] [PubMed] [Google Scholar]
- 72.Ishimura A, Terashima M, Kimura H, Akagi K, Suzuki Y, Sugano S, Suzuki T. Jmjd2c histone demethylase enhances the expression of Mdm2 oncogene. Biochem Biophys Res Commun. 2009;389:366–71. doi: 10.1016/j.bbrc.2009.08.155. [DOI] [PubMed] [Google Scholar]
- 73.Suzuki T, Terashima M, Tange S, Ishimura A. Roles of histone methyl-modifying enzymes in development and progression of cancer. Cancer Sci. 2013;104:795–800. doi: 10.1111/cas.12169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 75.Drakaki A, Iliopoulos D. microRNA Gene Networks in Oncogenesis. Curr Genomics. 2009;10:35–41. doi: 10.2174/138920209787581299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sotiropoulou G, Pampalakis G, Lianidou E, Mourelatos Z. Emerging roles of microRNAs as molecular switches in the integrated circuit of the cancer cell. RNA. 2009;15:1443–61. doi: 10.1261/rna.1534709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Fabbri M, Calin GA. Epigenetics and miRNAs in human cancer. Adv Genet. 2010;70:87–99. doi: 10.1016/B978-0-12-380866-0.60004-6. [DOI] [PubMed] [Google Scholar]
- 78.Ratovitski EA. Tumor Protein p63/microRNA network in epithelial cancer cells. Curr Genomics. 2013;14:441–52. doi: 10.2174/13892029113146660011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lee TI, Young RA. Transcriptional regulation and its misregulation in disease. Cell. 2013;152:1237–51. doi: 10.1016/j.cell.2013.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Shu XS, Li L, Tao Q. Chromatin regulators with tumor suppressor properties and their alterations in human cancers. Epigenomics. 2012;4:537–49. doi: 10.2217/epi.12.50. [DOI] [PubMed] [Google Scholar]
- 81.Kulaeva OI, Nizovtseva EV, Polikanov YS, Ulianov SV, Studitsky VM. Distant activation of transcription: mechanisms of enhancer action. Mol Cell Biol. 2012;32:4892–7. doi: 10.1128/MCB.01127-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zentner GE, Henikoff S. Regulation of nucleosome dynamics by histone modifications. Nat Struct Mol Biol. 2013;20:259–66. doi: 10.1038/nsmb.2470. [DOI] [PubMed] [Google Scholar]
- 83.Sethupathy P. Illuminating microRNA Transcription from the Epigenome. Curr Genomics. 2013;14:68–77. doi: 10.2174/138920213804999183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Shalgi R, Brosh R, Oren M, Pilpel Y, Rotter V. Coupling transcriptional and post-transcriptional miRNA regulation in the control of cell fate. Aging (Albany NY) 2009;1:762–70. doi: 10.18632/aging.100085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Samantarrai D, Dash S, Chhetri B, Mallick B. Genomic and epigenomic cross-talks in the regulatory landscape of miRNAs in breast cancer. Mol Cancer Res. 2013;11:315–28. doi: 10.1158/1541-7786.MCR-12-0649. [DOI] [PubMed] [Google Scholar]
- 86.Fabbri M, Garzon R, Cimmino A, Liu Z, Zanesi N, Callegari E, Liu S, Alder H, Costinean S, Fernandez-Cymering C, et al. microRNA-29 family reverts aberrant methylation in lung cancer by targeting DNA methyltransferases 3A and 3B. Proc Natl Acad Sci U S A. 2007;104:15805–10. doi: 10.1073/pnas.0707628104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Garzon R, Liu S, Fabbri M, Liu Z, Heaphy CE, Callegari E, Schwind S, Pang J, Yu J, Muthusamy N, et al. microRNA-29b induces global DNA hypomethylation and tumor suppressor gene reexpression in acute myeloid leukemia by targeting directly DNMT3A and 3B and indirectly DNMT1. Blood. 2009;113:6411–8. doi: 10.1182/blood-2008-07-170589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Benetti R, Gonzalo S, Jaco I, Muñoz P, Gonzalez S, Schoeftner S, Murchison E, Andl T, Chen T, Klatt P, et al. A mammalian microRNA cluster controls DNA methylation and telomere recombination via Rbl2-dependent regulation of DNA methyltransferases. Nat Struct Mol Biol. 2008;15:268–79. doi: 10.1038/nsmb.1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Duursma AM, Kedde M, Schrier M, le Sage C, Agami R. miR-148 targets human DNMT3b protein coding region. RNA. 2008;14:872–7. doi: 10.1261/rna.972008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Xu Q, Jiang Y, Yin Y, Li Q, He J, Jing Y, Qi YT, Xu Q, Li W, Lu B, et al. A regulatory circuit of miR-148a/152 and DNMT1 in modulating cell transformation and tumor angiogenesis through IGF-IR and IRS1. J Mol Cell Biol. 2013;5:3–13. doi: 10.1093/jmcb/mjs049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Takata A, Otsuka M, Yoshikawa T, Kishikawa T, Hikiba Y, Obi S, Goto T, Kang YJ, Maeda S, Yoshida H, et al. microRNA-140 acts as a liver tumor suppressor by controlling NF-κB activity by directly targeting DNA methyltransferase 1 (Dnmt1) expression. Hepatology. 2013;57:162–70. doi: 10.1002/hep.26011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Varambally S, Cao Q, Mani RS, Shankar S, Wang X, Ateeq B, Laxman B, Cao X, Jing X, Ramnarayanan K, et al. Genomic loss of microRNA-101 leads to overexpression of histone methyltransferase EZH2 in cancer. Science. 2008;322:1695–9. doi: 10.1126/science.1165395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Zhang JG, Guo JF, Liu DL, Liu Q, Wang JJ. microRNA-101 exerts tumor-suppressive functions in non-small cell lung cancer through directly targeting enhancer of zeste homolog 2. J Thorac Oncol. 2011;6:671–8. doi: 10.1097/JTO.0b013e318208eb35. [DOI] [PubMed] [Google Scholar]
- 94.Noonan EJ, Place RF, Pookot D, Basak S, Whitson JM, Hirata H, Giardina C, Dahiya R. miR-449a targets HDAC-1 and induces growth arrest in prostate cancer. Oncogene. 2009;28:1714–24. doi: 10.1038/onc.2009.19. [DOI] [PubMed] [Google Scholar]
- 95.Buurman R, Gürlevik E, Schäffer V, Eilers M, Sandbothe M, Kreipe H, Wilkens L, Schlegelberger B, Kühnel F, Skawran B. Histone deacetylases activate hepatocyte growth factor signaling by repressing microRNA-449 in hepatocellular carcinoma cells. Gastroenterology. 2012;143:811–20, e1-15. doi: 10.1053/j.gastro.2012.05.033. [DOI] [PubMed] [Google Scholar]
- 96.Yuan JH, Yang F, Chen BF, Lu Z, Huo XS, Zhou WP, Wang F, Sun SH. The histone deacetylase 4/SP1/microrna-200a regulatory network contributes to aberrant histone acetylation in hepatocellular carcinoma. Hepatology. 2011;54:2025–35. doi: 10.1002/hep.24606. [DOI] [PubMed] [Google Scholar]
- 97.Lujambio A, Calin GA, Villanueva A, Ropero S, Sánchez-Céspedes M, Blanco D, Montuenga LM, Rossi S, Nicoloso MS, Faller WJ, et al. A microRNA DNA methylation signature for human cancer metastasis. Proc Natl Acad Sci U S A. 2008;105:13556–61. doi: 10.1073/pnas.0803055105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kutanzi KR, Yurchenko OV, Beland FA, Checkhun VF, Pogribny IP. microRNA-mediated drug resistance in breast cancer. Clin Epigenetics. 2011;2:171–85. doi: 10.1007/s13148-011-0040-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Haenisch S, Cascorbi I. miRNAs as mediators of drug resistance. Epigenomics. 2012;4:369–81. doi: 10.2217/epi.12.39. [DOI] [PubMed] [Google Scholar]
- 100.DeYoung MP, Johannessen CM, Leong CO, Faquin W, Rocco JW, Ellisen LW. Tumor-specific p73 up-regulation mediates p63 dependence in squamous cell carcinoma. Cancer Res. 2006;66:9362–8. doi: 10.1158/0008-5472.CAN-06-1619. [DOI] [PubMed] [Google Scholar]
- 101.Rheinwald JG, Beckett MA. Tumorigenic keratinocyte lines requiring anchorage and fibroblast support cultured from human squamous cell carcinomas. Cancer Res. 1981;41:1657–63. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.