

Abstract
The conserved lin-4 microRNA (miRNA) regulates the proper timing of stem cell fate decisions in C. elegans by regulating stemness genes such as lin-14 and lin-28.1-3 While lin-4 is upregulated toward the end of the first larval stage and functions as an essential developmental timing “switch”, little is known about how lin-4 expression is regulated.4 Here we show that in C. elegans hypodermal seam cells, transcription of lin-4 is positively regulated by lin-4 itself. In these cells, lin-4 activates its own transcription through a conserved lin-4-complementary element (LCE) in its promoter. We further show that lin-4 is required to recruit RNA polymerase II to its own promoter, and that lin-4 overexpression is sufficient for autoactivation. Finally, we show that a protein complex specifically binds the LCE in vitro, and that mutations that abolish this binding also reduce the in vivo expression of a plin-4:GFP reporter. Thus, we describe the first in vivo evidence of RNA activation (RNAa) by an endogenous miRNA, and provide new insights into an elegant autoregulatory mechanism that ensures the proper timing of stem cell fate decisions in development.
Keywords: lin-4, RNA activation, RNAa, heterochronic, miRNA, microRNA, seam cell, stem cell
Introduction
Small non-coding RNAs, such as small interfering RNAs (siRNAs) and miRNAs, are well known to silence gene expression through a variety of mechanisms, a phenomenon referred to as RNA interference (RNAi).5-7 Initially thought of as a post-transcriptional, cytoplasmic process, it has become apparent that nuclear RNAi pathways also exist, mediating the co-transcriptional and epigenetic silencing of many genomic loci.6,8-10 Interestingly, a number of promoter-targeting small RNAs have also been shown to activate the expression of the downstream gene.11-16 However, most RNAa studies have either only been shown in vitro or have been in the context of exogenous, synthetic RNAs.
In the nematode Caenorhabditis elegans, the timing of stem cell fate decisions is regulated by heterochronic genes.17-20 These genes, which include the miRNAs lin-4 and let-7 and the stemness factor lin-28, escort the blast cells of the developing nematode through 4 larval stages and into adulthood, ensuring that tissue and structures, like the hypodermis and vulva, form at the correct developmental stage. The lin-4 miRNA resides in the sense orientation in intron 9 of the gene F59G1.4. Detectable lin-4 expression begins in the late L1 larval stage and is the first known step in activating post-embryonic blast-cell fate determination by the heterochronic genes.4,21 Thus the timing of lin-4 expression is of critical importance; however, little is known about the regulation of lin-4 expression. Transcriptional control of lin-4 can be partially explained by the activity of the FLH-1 and FLH-2 transcription factors, which bind the lin-4 promoter and repress transcription until the late L1 stage of development,22 while lin-4 biogenesis is regulated post-transcriptionally by the RNA-binding protein RBM-28.4 To date, no positive trans-acting factors have been reported to affect transcription at the lin-4 locus. Here we provide evidence that lin-4 is itself a trans-acting factor necessary for its own expression.
Results
In order to elucidate cis-acting regulatory elements that are necessary for activating transcription from the lin-4 locus, we examined sequence homology between 3 different nematode species: C. elegans, C. briggsae, and C. remanei (Fig. 1A). Three kilobases (kb) flanking the lin-4 locus were analyzed, with very high sequence homology found immediately surrounding the lin-4 gene. This homology extends upstream from the lin-4 site about 700 nucleotides23 (Fig. 1A). We hypothesized that cis-acting elements important for regulating lin-4 transcription were most likely to be contained within the highly homologous sequence common to the 3 nematode species. In the zaIs1 (plin-4:GFP) line, GFP is driven by this region of the lin-4 promoter (Fig. 1B), and GFP expression in the stem cell-like hypodermal seam cells17 is detectable beginning in the early L2 stage24 (Fig. 1D), consistent with the timing of the appearance of mature lin-4, as detected by northern blot.24 Further, lin-4 mutants have an obvious, long and “floppy” phenotype, resulting in part from disruption of fate-determining divisions in the seam cell lineage,25,26 indicating a functional role for lin-4 in the seam cells. Therefore, GFP expression in the seam cells of the zaIs1 line recapitulates the timing and localization of lin-4 expression. As such, we chose to use GFP expression as a measure of lin-4 promoter activity in the seam cells, though GFP expression in the zaIs1 line is not limited to the seam cells (GFP in this line is seen in the pharynx, hypodermal cells, the distal tip cell, and the developing vulva).24 Protein-coding/non-coding genes are subject to different processing pathways during biogenesis. Thus, a protein (GFP) and an miRNA (lin-4), driven by an identical promoter and expressed at the same time and same place during development, suggests that the major element regulating expression in this instance is the common promoter. Transgenic lines carrying DNA constructs containing mutant lin-4-promoter derivatives driving GFP transcription were assayed for GFP expression in the seam cells (Fig. 1C). We found a small, 56-nucleotide (nt) DNA region (DT.1) located 50 bases upstream of a recently mapped transcriptional start site for the lin-4 gene (Fig. 1B)4 that was sensitive to deletion and truncation (Fig. 1C), both of which lead to a significant loss of GFP expression when compared with GFP expression driven by the wild-type lin-4 promoter (Fig. 1C and D). Deleting each half of the DT.1 sequence from the full-length lin-4-promoter:GFP construct (constructs DT.1a/b) showed that both halves are necessary for driving GFP expression (Fig. 1C). We tested if a cis-acting element might bridge the junction between both halves of the DT.1 sequence, and made a corresponding deletion from the wild-type promoter (DT.1c) (Fig. 1D). GFP expression from the DT.1c construct was affected similarly as compared with that of DT.1a/b constructs, showing that proper expression of GFP depended on a 34-nucleotide (nt) sequence shared in part by all these deletions (Fig. 1C).

Figure 1. A cis-acting element required for the transcription of lin-4. (A) Schematic illustration showing an alignment of the intronic sequence flanking lin-4 in the C. elegans genome (reference sequence) with the lin-4 locus of C. remanei and C. briggsae. The C. elegans graphic shows the position of the lin-4 sequence (gray box), as well as flanking exons of the host gene, F59G4.1 (blue boxes). C. remanei and C. briggsae schematics are shown using a heat bar to illustrate sequence homology at and around the lin-4 locus with the lin-4 locus in C. elegans. Sequence lengths are measured in kilobases (Kb). (B) Schematic illustration of the composition of the transcriptional fusion construct used to elucidate cis-acting regulatory elements in the putative lin-4 promoter.24 A short DNA sequence shown to rescue the lin-4-mutant phenotype is shown, within which is contained an FLH-1, -2 binding site (pink box), lin-4 (gray box), and 2 transcriptional start sites.4,22,23 The transcriptional start site represented by the dark blue arrow was independently verified in our laboratory. Homologous sequence upstream of the lin-4 mature sequence was fused to GFP, as previously described.24 Promoter length is measured in base pairs (bp). (C) On the left, an illustration of the promoter regions used to drive GFP expression in C. elegans. Truncations to, and deletions from, the original rescuing fragment promoter (RF) are indicated. On the right, transgenic lines developed with these constructs were assayed for expression of GFP in the seam cells. No GFP was seen in embryo and L1 animals, and the fraction of animals expressing GFP in the seam cells in stages L2 and L3 are indicated. Error bars represent the standard error of the proportion. Significance was measured using a z test, with calculated P values as indicated (*P < 0.005; **P < 0.0015; ***P < 0.00007); 23 < n < 73. (D) Images showing GFP expression in seam cells over 3 larval stages of development (L1, L2, L4). The DNA construct used to develop the transgenic lines is depicted at the left. Seam cells positive for GFP expression are indicated (yellow arrowheads). White lines indicate the regions where seam cells are found in GFP-negative animals.
We noticed a putative lin-4-complementary element (LCE) located precisely at the junction common to all 3 DT.1 deletions (a–c) (Fig. 2A). In C. elegans, this LCE is located ~400 bp upstream of the mature lin-4 miRNA. Intriguingly, the position of the LCE is precisely conserved in C. remanei and C. vulgaris. Furthermore, a similar LCE was found in C. briggsae ~1.4 kb upstream of the mature lin-4 sequence. The putative duplexes formed between lin-4 and these LCEs strikingly resemble previous experimentally validated duplexes formed when lin-4 binds to LCEs located in the mRNAs of lin-14 and lin-28 (Fig. 2B).23,27,28 A deletion of the 17-nt LCE from the plin-4:GFP construct resulted in a similar decrease in GFP expression as the earlier, larger deletions (Fig. 2C). Thus, our results suggest that a conserved LCE is necessary for promoting seam-cell GFP expression under the regulation of the lin-4 promoter.

Figure 2. A lin-4 complementary element is required for Plin-4:GFP expression and lin-4 rescue fragment activity. (A) DT.1 sequence shown, as well as the DT.1a/b/c regions. The putative LCE is shown in orange. (B) Top half: predicted lin-4/promoter LCE duplexes in C. elegans and other nematode species. The dashed blue box indicates a conserved sequence motif found in the bulged region across all duplexes. Bottom half: lin-4/LCE duplexes of experimentally validated 3′ UTR targets in C. elegans. (C) Quantification of the number of animals positive for seam cell GFP expression carrying wild-type (4 independent lines) vs. LCE-deleted Plin-4::GFP constructs (3 independent lines). (D) Comparison of wild-type (6 independent lines) and LCE-deleted lin-4 expression constructs (6 independent lines) in the ability to rescue the e912 vulvaless phenotype. In animals rescued by the LCE-deleted constructs, the proportion of vulvaless animals was significantly increased (P = 0.0006), indicating a lack of rescue. Similarly, the proportion of egg-laying animals was significantly decreased (P = 0.026), again indicating reduced rescuing activity. Significance was measured by a 2-tailed, Fischer exact test.
We tested whether the LCE was required for proper expression of lin-4 itself. Toward this end, we deleted the 17-nt LCE in a lin-4 rescue construct.23 We found that the LCE-deleted construct displayed a significantly reduced ability to rescue the lin-4(e912) phenotype (Fig. 2D). The e912 allele is a partially characterized deletion that removes the lin-4 locus from the genome.23 In unrescued e912 animals, 100% display a vulvaless phenotype and do not lay eggs (Fig. 2D). Upon injection with either the full-length or LCE-deleted lin-4 expression construct, a range of vulval phenotypes was observed. Transgenic animals bearing an LCE-deleted rescue construct showed a significantly higher fraction of a vulvaless phenotype (i.e., no rescue) and a significantly lower fraction of an egg-laying phenotype (i.e., full rescue) (Fig. 2D). The frequencies of bursting and protruding vulval phenotypes were similar from injections with either construct (Fig. 2D). Thus, the LCE appears to be a functional cis-regulatory element of the lin-4 promoter.
Having demonstrated the consequence of loss of the LCE in positive plin-4 activity, we hypothesized that lin-4 itself may positively regulate its own expression. To test this, we crossed the integrated plin-4:GFP fusion line (zaIs1) into the lin-4(e912) mutant strain. In a wild-type background, basal levels of GFP expression from the plin-4:GFP transgene are first detectable in the early L2 stage in the seam cells of the worm (Fig. 3A). Interestingly, 2 distinct bursts of augmented GFP expression are observed, initiated in the early L2 and early L4 stages of development (Fig. 3A). Comparatively, we detected significantly fewer plin-4:GFP-positive seam cells in both the L2 and L4 in the lin-4 mutants, where no endogenous lin-4 is produced (Fig. 3A; Fig. S2). Quantitative PCR (qPCR) analysis of total GFP mRNA from mixed-stage wild-type and mutant animals confirmed that this observation in the seam cells is also true as a general trend for GFP expression when assayed from throughout the animal: 3 times more GFP mRNA is transcribed from the reporter construct in the wild-type background than in the lin-4 mutant background (Fig. 3B). Thus, lin-4 is required to promote activity from its own promoter.

Figure 3.lin-4 is necessary and sufficient for transcriptional autoactivation. (A) GFP expression data in animals carrying the lin-4:GFP integrated transgene in the wild-type (zaIs1) and the lin-4 (e912) deletion background. Seam cells positive for GFP were counted at each larval stage of development and in adulthood. Error bars represent the standard error of the mean, and significance was measured using a 2-sample t test; P values are as indicated (*P < 1 × 10−6); n = 50, zaIs1; n = 150, zaIs1;e912. (B) Whole-body GFP mRNA levels collected from mixed-stage animals were measured by qPCR and represented as the mean ± SEM. Expression levels were normalized to pmp-3 mRNA. Significance was measured using a 2-tailed t test (*P < 0.0001). (C) GFP+ seam cell counts in synchronized, early L3 (eL3) animals, represented as the mean ± SEM. GFP+ seam cell counts for the lin-4 overexpressor lines (O/E) are an average from 4 independent transgenic lines. Significance was measured using a 2-tailed t test (*P < 1 × 10−10). (D) RNAP II chromatin immunoprecipitation from zaIs1 and zaIs1;e912 mixed-stage populations grown in liquid culture (see methods). Left panel: semi-quantitative PCR detecting a ~500 bp fragment of the lin-4 promoter upstream of GFP, from 2 technical ChIP replicates. Right panel: ImageJ quantification of band intensities. The data shown is an average of the 2 replicates from the gel in the left panel and an additional biological replicate (not shown in gel). Significance was measured using a 2-tailed t test (*P = 0.002). Error bars represent standard deviation of the mean.
To test whether lin-4 was sufficient for autoactivation, we overexpressed lin-4 in the zaIs1 line. We scored synchronized early L3 animals, a stage where we found that zaIs1 animals express relatively low basal levels of GFP. Importantly, transgenic lines bearing a lin-4 expression construct showed a ~3-fold increase in the number of GFP-expressing seam cells (Fig. 3C) compared with control animals. Together, our results suggest that the lin-4 miRNA is both necessary and sufficient for the activation of its own promoter.
To determine whether lin-4 was required to recruit RNA polymerase II (RNAP II) to its own promoter, we performed chromatin immunoprecipitation followed by semi-quantitative PCR. Since the e912 strain carries a deletion that also removes the endogenous lin-4 promoter, we compared the amount of RNAP II binding in zaIs1 and zaIs1; e912 animals, only measuring RNAP II binding to the lin-4 promoter upstream of GFP (Fig. 3D). Interestingly, we observed an 8-fold decrease in promoter-associated RNAP II in lin-4 mutants (Fig. 3D), suggesting that lin-4 autoactivation indeed involves the recruitment of RNAP II.
We next tested by electromobility shift assays (EMSAs) whether the LCE may be bound by an RNA/protein complex. The LCE-containing DT.1c sequence forms a proteinase-K and RNase-sensitive complex in whole-worm extract (protein plus nucleic acid), but only when the LCE was in single-stranded form (Fig. S1). In the RNase-containing reactions, we observed the formation of a secondary complex, larger in molecular weight than the complex formed under normal reaction conditions. While we are uncertain about the composition of this complex, its size roughly correlates with the size of the RNase. Whether this be the case or not, the complex formed under RNase-positive conditions differs from that formed under conditions without RNAase, and these data confirm that the LCE is a binding site for a ribonuclear protein complex in vitro.
To investigate the specificity of the interaction between the LCE and the LCE-binding complex, we performed competition EMSAs. Non-radiolabeled strands of DNA were added to the EMSA reactions to test their ability to compete with the radiolabeled probe for protein binding. We found that the unlabeled LCE was able to compete for protein binding with much greater efficiency than an unlabeled sequence found downstream and on the opposing strand of the LCE (Fig. 4A), suggesting that the LCE complex formation was sequence-specific. Furthermore, we introduced mutations into the LCE probe and tested the ability of the mutant probe to compete with the wild-type probe for protein binding (Fig. 4B). While each mutation affected the ability of the probe to compete for binding, mutation 2 had the most pronounced effect (Fig. 4B). An LCE with all 3 mutations likewise failed to compete with the probe for protein association (Fig. 4B). Consistent with these observations, when we radiolabeled each of the mutant LCE probes, mutant 2 displayed the most dramatic reduction in shifting activity. This was somewhat surprising, as the 2 nucleotide changes in the mutant-2 probe are situated within the central bulge of the putative lin-4/LCE duplex. Our data suggests that a protein of interest binds the LCE directly, potentially at the sequence within the bulge.

Figure 4A and B. Sequence-specific complex formation with the LCE from whole-animal lysate. All probes are labeled (DT.1c); arrows indicate expected LCE/protein complexes. (A) In the top left panel, a competition EMSA performed using cold competitors (concentration ranges depicted in colored triangles; 50–200× probe concentration). Orange represents the DT.1c sequence (shown below the left panel, within an orange box; the LCE is highlighted in orange lettering), while blue represents the DT.1b complimentary sequence (also depicted below the left panel). The right panel shows quantification of the percent of probe bound by protein in the presence of cold competitor. (B) Two EMSA experiments using mutated versions of the DT.1c sequence to compete with wild-type DT.1c sequence for protein binding (left EMSA) or to assay the ability of a mutant sequence to be bound in a complex (right EMSA). Gel lanes are labeled so as to correspond with shift quantification shown above each EMSA. Competitors were present at a concentration 200× over that of the probe. At the top right, a schematic depicts the full DT.1c sequence, with the LCE highlighted in orange. Also shown are 4 mutant DT.1c sequences, numbered 1–4. The nature of each mutation is indicated, with mutant 4 including all mutations 1–3.
To test these results in vivo, we generated a series of transgenic lines carrying mutated derivatives of the plin-4:GFP construct, corresponding to the LCE point mutations studied in vitro (Fig. 4C). Compared with the full-length construct, each individual point mutant pair resulted in a small decrease in GFP-expressing seam cells. Although the decrease caused by mutation 2 is statistically significant, it is not as dramatic as the reduction in shifting activity observed in the EMSAs. However, in transgenic lines carrying the plin-4:GFP construct containing all 3 LCE mutations, seam-cell GFP expression was dramatically reduced (Fig. 4C). This “triple” LCE mutant would not be expected to bind the lin-4 miRNA at all; consistent with this, the degree of reduction in seam cell GFP is almost identical to that observed when lin-4 expression is lost in zaIs1 animals (Fig. 3A; Fig. S2).
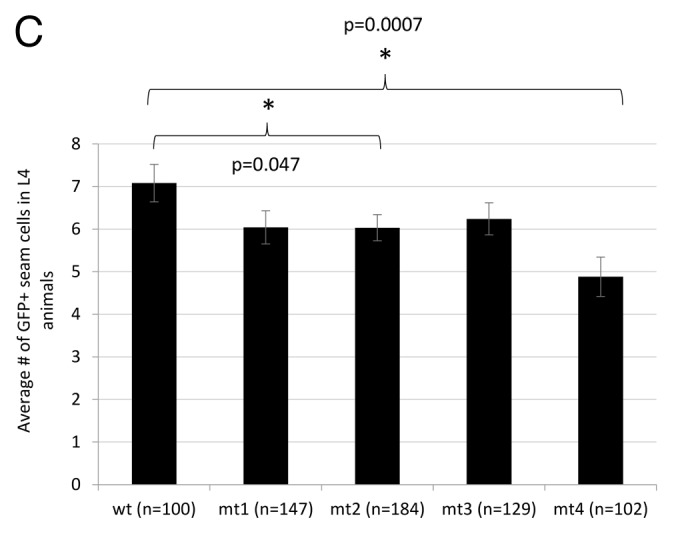
Figure 4C. (C) In vivo effects of mutations 1–3 on seam cell GFP expression; 3, 4, 3, 5, and 6 independent transgenic lines were generated for the wild-type, mt1, mt2, mt3, and mt4 constructs, respectively. All scoring was blinded; error bars represent the standard error of the mean. Significance was measured using a 2-tailed t test.
Discussion
Taken together, our data support the hypothesis that lin-4 functions to activate its own expression, providing the first in vivo example of miRNA-mediated RNAa. We found a lin-4 complementary element (LCE) in the lin-4 promoter, whose position—though not the exact sequence—was conserved in 3 of 4 nematode species examined. We showed that deletion of this LCE in C. elegans significantly reduced lin-4 promoter activity. We further demonstrated that the LCE is bound by a yet-to-be-identified complex in an RNA-, protein-, and structure-specific manner, and that this binding event was sequence-specific. We showed that a series of point mutations in the LCE abrogated binding of the complex in vitro, while the same mutations in the plin-4:GFP construct reduced GFP expression in vivo. Moreover, we showed that lin-4 itself is both necessary and sufficient to upregulate plin-4:GFP expression, at least in part by recruiting RNAP II to its promoter.
In C. elegans, lin-4 resides within the ninth intron of a host gene (F59G1.4) of unknown function. During development, lin-4 is specifically upregulated (i.e., independently of host gene transcription) toward the end of the first larval stage and is required for the proper timing of subsequent developmental events.21 We and others have shown that this upregulation is at the level of transcription and is followed by efficient processing of the primary transcript into the mature lin-4 miRNA.24,29 Prior to late L1, the FLYWCH family of transcription factors FLH-1 and FLH-2 are known to bind the lin-4 promoter and repress lin-4 expression.22 However, even in these early stages of development, RNA sequences containing lin-4 have been shown to be present.4 The levels of these transcripts correlate exactly with levels of the host gene mRNA, suggesting that they are exclusively products of host gene transcription.
However, beginning in late L1, the number of lin-4 transcripts have been shown to increase over 4-fold relative to the number of host gene transcripts,4 suggesting the activation of 1 or more independent promoters. We propose that the autoregulation of lin-4 expression shown in our study provides a self-timing mechanism for the initiation of lin-4-mediated developmental events (Fig. 5). In this model, throughout early and mid L1, low levels of lin-4-containing host gene transcripts may provide the primary RNA substrates for Drosha and Dicer processing, leading to the accumulation of a small pool of mature lin-4 (top panel). By the late L1 stage, the levels of lin-4 may pass a threshold level required for autoactivation, initiating the positive feedback loop through direct interaction with sequences in or from the lin-4 promoter region and resulting in the burst of transcription observed in early L2.
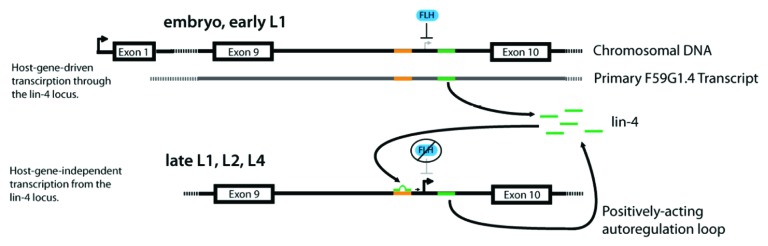
Figure 5. Model for lin-4 autoregulation by RNA activation. The full intron in which lin-4 is located is depicted from its genomic locus within host gene F59G1.4. Exons are depicted as boxes, introns as lines. The LCE in the lin-4 promoter is shown in orange, while lin-4 is shown in green. FLH proteins, orthologs of D. melanogaster FLYWCH transcription factors, are represented by a blue oval. Transcriptional start sites are shown as active (bold and black arrows) or inactive (gray arrows). The model illustrates both initiation of lin-4 expression by host-gene transcription at early larval stages, as well as activation of lin-4 transcription via an intronic, host-gene independent lin-4 promoter at later developmental stages. Beginning in the embryo and continuing into the L1 stage, transcription of F59G1.4 progresses through the lin-4 locus, while FLH proteins inhibit transcription from the intronic lin-4 promoter. By the late L1, early L2 stages, FLH proteins release the lin-4 locus, and lin-4 generated by host-gene-mediated transcription binds to the LCE in the lin-4 promoter to activate transcription. lin-4 generated by activated transcription from the lin-4 locus then becomes available to further activate lin-4 transcription during later larval stages and into adulthood, forming a positive autoregulatory loop.
The let-7 miRNA has also recently been shown to positively regulate its own expression through a complementary element in its primary transcript.30 While our ChIP experiments suggest that lin-4 autoactivation may be at the transcriptional level, similar autoregulatory modules are likely not unique to these 2 well-characterized miRNAs. The conservation of the LCE in other nematode species suggests that lin-4 autoactivation may be a common strategy to ensure the proper expression of lin-4¸ and thus the proper progression of larval development. Moreover, our in vivo findings add to a small but growing body of reports describing gene-activating functions for small non-coding RNAs.
RNA activation (RNAa) is a poorly understood phenomenon whereby small RNA duplexes targeting promoter regions have been shown to mediate the specific activation of the downstream gene.11,12,15,16,31-33 To our knowledge, RNAa by an endogenous miRNA has never been directly shown in vivo; furthermore, the activated gene has always been protein-coding, and never the non-coding RNA itself. Our finding that the lin-4 miRNA activates its own transcription in vivo suggests, perhaps unsurprisingly, that RNA-mediated gene activation is not limited to typical protein-coding genes, and may apply to the thousands of non-coding RNAs throughout the genome. lin-4 has played a major role as a model for studying the general principles and mechanisms of miRNA pathways, and we suspect that our work will prime further research into other non-canonical functions of miRNAs and other small RNAs.
Experimental Procedures
Lines and crosses
N2 is the wild-type strain; zaIs1 carries an integrated transgene containing the lin-4 promoter driving GFP followed by the unc-54 3′UTR (plin-4:GFP:UNC-54).24
Genetic crosses were performed as follows: N2 males were allowed to mate with zaIs1 hermaphrodites. GFP-expressing males from this cross were then used to mate with the lin-4(e912)/mnC1 hermaphrodites. Two hundred GFP-expressing hermaphrodites from this cross were individually plated and allowed to self-fertilize. All plates with less than 100% GFP expression in the progeny (indicating GFP heterozygosity) were discarded. Those that remained were screened for the lin-4 homozygous phenotype (long, floppy, uncoordinated animals; no egg laying). Animals homozygous for both GFP and lin-4 were assayed for GFP expression in the seam cells.
plin-4:GFP transgenic lines were developed through microinjection.
DNA for injection was prepared as follows: PCR amplicons made from cloned plin-4:GFP:UNC-54 constructs were purified in 0.8% agarose gels. Excised gel fragments were loaded into 0.45 micron Spin-X Centrifuge Tube Filters (Costar) and centrifuged at 16 000 × g for 10 min. Eluted DNA was phenol extracted and ethanol precipitated. Injection mixes were comprised of PCR-amplified and gel-purified promoter:GFP fusion DNA (25 ng/ul), injection marker myo-2:dsRED (5 ng/ul), EcoRI-digested 1 kb ladder (NEB) (25 ng/ul).
A lin-4 expression construct was microinjected at 16 ng/ul into the zaIs1 line to generate lin-4 overexpressor lines. For assaying rescue of the e912 phenotype, the MT3316 (lin-4[e912]/mnC1; dpy-10[e128] unc-52[e444] II) balanced strain was also microinjected with 16 ng/ul of either wild-type or LCE-deleted lin-4 expression constructs. Only independent transgenic lines determined to be e912/e912 were scored, allowing for an accurate measure of lin-4 production solely from the transgenic arrays. As above, injection mixes consisted of PCR amplified expression constructs, with 5 ng/ul of the myo-2:dsRED injection marker and 25 ng/ul EcoRI-digested 1-kb ladder (NEB).
Constructs
Generation of the lin-4:GFP fusion construct cloned into vector pPD95.70 was previously described, here called RF. Truncated promoter derivatives of the RF construct, p450 and p450T.1, were created by PCR using primer pairs 19/17 and 138/17, respectively (Table S1). Promoter derivatives containing deletions, pDT.1, pDT.1a/b/c, were created using fusion PCR. In short, each deletion construct was generated using PCR to fuse together the 5′ and 3′ DNA sequence that flanked the region of deletion. 5′ and 3′ sequences were generated by PCR as follows: the DT.1:GFP construct was made using primer pairs, 32/143 for the 5′ sequence, and 138/17 for the 3′ sequence (Table S1). Likewise, 5′ sequences for constructs DT.1a/b/c were generated using primer pairs 32/142, 32, 166, 32/164, and 3′ sequences were generated with primer pairs 141/17, 138/17, 165,17, respectively. The above fusion PCR products were cloned into vector PCR2.1 (Invitrogen) and sequenced. Specific LCE deletions in the Plin-4:GFP and lin-4 expression constructs were made by the 3-fragment Gateway Cloning system.
GFP analysis
Transgenic (non-integrated) lines were screened for GFP expression in the seam cells, as noted in Figure 1C, using an upright Zeiss Axioplan microscope. Because of mosaic effects often seen in transgenic animals, nematodes showing any GFP expression in the seam cells were scored as positive, even if GFP was seen in only 1/16 seam cells. For each transgenic construct, 3–4 lines were established and then examined for GFP expression in the seam cells over the first 3 larval stages of development. One representative line from each transgenic construct was chosen (the line that represented the average GFP expression levels seen when comparing all generated lines), and GFP expression in the seam cells was compared with that of the zaIs1-integrated strain (Fig. 1C). Statistical significance was measured by performing a z test on the standard error of the proportion (shown by error bars) of animals expressing GFP in the seam cells of each line, as noted in the figure caption.
For homozygous mutant strains (lin-4[e912];zaIs1), GFP in the seam cells was assayed by counting the number of GFP-expressing seam cells on the near side of the animal (the side closest to the microscope lens). We noticed differences in GFP expression penetrance, though GFP was integrated into the genome, and thus counted a “positive” seam cell as one that had discernable GFP expression above noise level (zaIs1 nematodes express GFP throughout the nematode at low levels), as determined visually. We counted the number of seam cells “positive” for GFP as a total (Fig. 2) and as a fraction of total seam cells (Fig. S2A). For the mutant strains, the data from 3 independently derived mutant lines were pooled together into 1 sample. For both the wild-type and mutant lines, the standard error of the mean was calculated using the total number of seam cells counted as “n”, which ranged from 490 to 1500 cells. Significance was calculated as described in the figure captions. Data was shown in 2 formats due to complications with measuring fractions of GFP-positive seam cells in the L2 stage. In wild-type nematodes, the V seam cells undergo a double division during the L2 stage, increasing the total number of seam cells from 10 to 16. Thus, early L2 nematodes have 10 seam cells, and late L2 have 16 seam cells. lin-4 mutants only have 10 seam cells throughout development. All L2 nematodes were pooled together in these analyses. In Figure S2A, proportions shown were calculated on the premise that all wild-type L2 nematodes had 10 seam cells. As some of these L2s contained 16 seam cells, the actual proportion of GFP-expressing seam cells is lower than indicated here.
RNA extraction and quantitative PCR
RNA from mixed-stage animals was extracted following a standard Trizol (Invitrogen) protocol. cDNA synthesis was performed using Superscript III (Invitrogen) and random hexamers. Quantitative real-time PCR was performed using SYBR Green (Applied Biosystems) on a Roche LightCycler 480. GFP expression levels were normalized to pmp-3 mRNA.
Chromatin immunoprecipitation
Mixed-stage animals were grown in 500 mL liquid culture as described.34 Samples were cross-linked in 37% formaldehyde for 30 min, and flash-frozen in liquid nitrogen. Extract prep and immunoprecipitation was performed as previously described,34 with the following modifications. After sonification, 10 μL of 8WG16 mouse monoclonal IgG (Santa Cruz Biotech) was added to ~2 mg of protein without the addition of sarkosyl solution. Immunocomplexes were collected with 25 μL of protein G-conjugated sepharose beads (GE healthcare). Eluted samples were treated with 3 μL of 6 mg/mL proteinase K, and incubated at 65 °C for ~16 h to reverse crosslinks. DNA was purified with the Qiaquick PCR Purification Kit (Qiagen) and interrogated by semi-quantitative PCR, with primers 32 and 255 to detect the lin-4 promoter. PCR products were run on a 1.5% agarose gel and quantified by ImageJ.
Electro-mobility shift assay (EMSA)
Probes for EMSA experiments (Table S1) were generated as follows: single-stranded oligonucleotides were incubated at 95 °C for 5 min, and then placed on ice. Labeling reactions were performed using T4 PNK (NEB) and gamma-ATP (NEN), according to the manufacturer’s protocols. Labeled oligonucleotides were purified through G-25 spin columns (Roche), according to recommended protocol. All EMSA experiments, unless otherwise indicated, were performed under the following conditions: 2.5 fmol radio-labeled probe, 15–30 μg total protein from whole-animal extract, 50 ng/ul poly dI-dC (Sigma), 1× Buffer D (20 mM HEPES-KOH, ph7.9; 20% glycerol; 0.2 mM EDTA; 20 mM KCl; 1 mM DTT, Complete Protease Inhibitor [Roche]), 1 μM DTT (Sigma), 50 ng/ul BSA (NEB). All reagents, except whole-animal extract, were mixed on ice. Extract was then added to a final reaction volume of 10 μl. Reactions were incubated on ice for 20 min and then loaded onto pre-run 5% native polyacrylamide gels and run at 4 °C. Gels were dried and analyzed using phospho-image screens. Quantification of radioactive bands was performed using Imagequant 5.1 software.
EMSA reactions performed using RNase or protease were performed as above, except whole-animal extracts were incubated with enzymes for 20 min on ice before adding them to the EMSA reaction. Competition assays were also prepared as described above, except cold competitors (at indicated concentrations) were mixed with other EMSA reagents before addition of whole-animal extract to the reactions.
Extract prep and purification
Animals were grown to the desired developmental stage using liquid culture, as described34 and flash frozen in liquid nitrogen. To produce protein extracts, animals were thawed in lysis buffer using a buffer:animal volume ratio of 2:3. Resulting slurries were placed over 1.0 mm Zirconia Beads (Biospec Products) and subjected to 4 rounds of homogenization (full-speed for 20 s) and chilling (ice for 1 min). Homogenization was performed using a Fastprep-24 (MP Biomedicals). Resulting extracts were centrifuged at 16 000 × g at 4 °C. Supernatant was taken and centrifuged at 100 K × g at 4 °C for 20 min. Supernatants were then flash frozen in liquid nitrogen and stored at −80 °C.
Supplementary Material
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We would like to thank Drs Alexandre de-Lencastre, Zachary Pincus and Giovanni Stefani for critical reading of this manuscript. M.J.T. was funded by an NIH Training Grant in CMB to Yale University. A.L.J. was funded by the Gruber Foundation and the Dwight N. and Noyes D. Clark Scholarship Fund. This work was supported by NIH grants (GM06401 and AG033921).
Some strains were provided by the CGC, which is funded by NIH Office of Research Infrastructure Programs (P40 OD010440).
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/27679
References
- 1.Moss EG, Lee RC, Ambros V. The cold shock domain protein LIN-28 controls developmental timing in C. elegans and is regulated by the lin-4 RNA. Cell. 1997;88:637–46. doi: 10.1016/S0092-8674(00)81906-6. [DOI] [PubMed] [Google Scholar]
- 2.Arasu P, Wightman B, Ruvkun G. Temporal regulation of lin-14 by the antagonistic action of two other heterochronic genes, lin-4 and lin-28. Genes Dev. 1991;5:1825–33. doi: 10.1101/gad.5.10.1825. [DOI] [PubMed] [Google Scholar]
- 3.Slack F, Ruvkun G. Temporal pattern formation by heterochronic genes. Annu Rev Genet. 1997;31:611–34. doi: 10.1146/annurev.genet.31.1.611. [DOI] [PubMed] [Google Scholar]
- 4.Bracht JR, Van Wynsberghe PM, Mondol V, Pasquinelli AE. Regulation of lin-4 miRNA expression, organismal growth and development by a conserved RNA binding protein in C. elegans. Dev Biol. 2010;348:210–21. doi: 10.1016/j.ydbio.2010.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Siomi H, Siomi MC. On the road to reading the RNA-interference code. Nature. 2009;457:396–404. doi: 10.1038/nature07754. [DOI] [PubMed] [Google Scholar]
- 6.Castel SE, Martienssen RA. RNA interference in the nucleus: roles for small RNAs in transcription, epigenetics and beyond. Nat Rev Genet. 2013;14:100–12. doi: 10.1038/nrg3355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Filipowicz W, Bhattacharyya SN, Sonenberg N. Mechanisms of post-transcriptional regulation by microRNAs: are the answers in sight? Nat Rev Genet. 2008;9:102–14. doi: 10.1038/nrg2290. [DOI] [PubMed] [Google Scholar]
- 8.Robb GB, Brown KM, Khurana J, Rana TM. Specific and potent RNAi in the nucleus of human cells. Nat Struct Mol Biol. 2005;12:133–7. doi: 10.1038/nsmb886. [DOI] [PubMed] [Google Scholar]
- 9.Morris KV, Chan SW, Jacobsen SE, Looney DJ. Small interfering RNA-induced transcriptional gene silencing in human cells. Science. 2004;305:1289–92. doi: 10.1126/science.1101372. [DOI] [PubMed] [Google Scholar]
- 10.Guang S, Bochner AF, Burkhart KB, Burton N, Pavelec DM, Kennedy S. Small regulatory RNAs inhibit RNA polymerase II during the elongation phase of transcription. Nature. 2010;465:1097–101. doi: 10.1038/nature09095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li L-C, Okino ST, Zhao H, Pookot D, Place RF, Urakami S, Enokida H, Dahiya R. Small dsRNAs induce transcriptional activation in human cells. Proc Natl Acad Sci U S A. 2006;103:17337–42. doi: 10.1073/pnas.0607015103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Janowski BA, Younger ST, Hardy DB, Ram R, Huffman KE, Corey DR. Activating gene expression in mammalian cells with promoter-targeted duplex RNAs. Nat Chem Biol. 2007;3:166–73. doi: 10.1038/nchembio860. [DOI] [PubMed] [Google Scholar]
- 13.Matsui M, Chu Y, Zhang H, Gagnon KT, Shaikh S, Kuchimanchi S, Manoharan M, Corey DR. Janowski B a. Promoter RNA links transcriptional regulation of inflammatory pathway genes. Nucleic Acids Res. 2013;41:10086–109. doi: 10.1093/nar/gkt777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Matsui M, Sakurai F, Elbashir S, Foster DJ, Manoharan M, Corey DR. Activation of LDL receptor expression by small RNAs complementary to a noncoding transcript that overlaps the LDLR promoter. Chem Biol. 2010;17:1344–55. doi: 10.1016/j.chembiol.2010.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang V, Place RF, Portnoy V, Wang J, Qi Z, Jia Z, Yu A, Shuman M, Yu J, Li L-C. Upregulation of Cyclin B1 by miRNA and its implications in cancer. Nucleic Acids Res. 2012;40:1695–707. doi: 10.1093/nar/gkr934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schwartz JC, Younger ST, Nguyen N-B, Hardy DB, Monia BP, Corey DR, Janowski BA. Antisense transcripts are targets for activating small RNAs. Nat Struct Mol Biol. 2008;15:842–8. doi: 10.1038/nsmb.1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nimmo RA, Slack FJ. An elegant miRror: microRNAs in stem cells, developmental timing and cancer. Chromosoma. 2009;118:405–18. doi: 10.1007/s00412-009-0210-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Slack F, Ruvkun G. Heterochronic genes in development and evolution. Biol Bull. 1998;195:375–6. doi: 10.2307/1543152. [DOI] [PubMed] [Google Scholar]
- 19.Ambros V. Control of developmental timing in Caenorhabditis elegans. Curr Opin Genet Dev. 2000;10:428–33. doi: 10.1016/S0959-437X(00)00108-8. [DOI] [PubMed] [Google Scholar]
- 20.Ambros V, Moss EG. Heterochronic genes and the temporal control of C. elegans development. Trends Genet. 1994;10:123–7. doi: 10.1016/0168-9525(94)90213-5. [DOI] [PubMed] [Google Scholar]
- 21.Feinbaum R, Ambros V. The timing of lin-4 RNA accumulation controls the timing of postembryonic developmental events in Caenorhabditis elegans. Dev Biol. 1999;210:87–95. doi: 10.1006/dbio.1999.9272. [DOI] [PubMed] [Google Scholar]
- 22.Ow MC, Martinez NJ, Olsen PH, Silverman HS, Barrasa MI, Conradt B, Walhout AJM, Ambros V. The FLYWCH transcription factors FLH-1, FLH-2, and FLH-3 repress embryonic expression of microRNA genes in C. elegans. Genes Dev. 2008;22:2520–34. doi: 10.1101/gad.1678808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993;75:843–54. doi: 10.1016/0092-8674(93)90529-Y. [DOI] [PubMed] [Google Scholar]
- 24.Esquela-Kerscher A, Johnson SM, Bai L, Saito K, Partridge J, Reinert KL, Slack FJ. Post-embryonic expression of C. elegans microRNAs belonging to the lin-4 and let-7 families in the hypodermis and the reproductive system. Dev Dyn. 2005;234:868–77. doi: 10.1002/dvdy.20572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chalfie M, Horvitz HR, Sulston JE. Mutations that lead to reiterations in the cell lineages of C. elegans. Cell. 1981;24:59–69. doi: 10.1016/0092-8674(81)90501-8. [DOI] [PubMed] [Google Scholar]
- 26.Sulston JE, Horvitz HR. Abnormal cell lineages in mutants of the nematode Caenorhabditis elegans. Dev Biol. 1981;82:41–55. doi: 10.1016/0012-1606(81)90427-9. [DOI] [PubMed] [Google Scholar]
- 27.Wightman B, Ha I, Ruvkun G. Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans. Cell. 1993;75:855–62. doi: 10.1016/0092-8674(93)90530-4. [DOI] [PubMed] [Google Scholar]
- 28.Ha I, Wightman B, Ruvkun G. A bulged lin-4/lin-14 RNA duplex is sufficient for Caenorhabditis elegans lin-14 temporal gradient formation. Genes Dev. 1996;10:3041–50. doi: 10.1101/gad.10.23.3041. [DOI] [PubMed] [Google Scholar]
- 29.Martinez NJ, Ow MC, Reece-Hoyes JS, Barrasa MI, Ambros VR, Walhout AJM. Genome-scale spatiotemporal analysis of Caenorhabditis elegans microRNA promoter activity. Genome Res. 2008;18:2005–15. doi: 10.1101/gr.083055.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zisoulis DG, Kai ZS, Chang RK, Pasquinelli AE. Autoregulation of microRNA biogenesis by let-7 and Argonaute. Nature. 2012;486:541–4. doi: 10.1038/nature11134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huang V, Qin Y, Wang J, Wang X, Place RF, Lin G, Lue TF, Li L-C. RNAa is conserved in mammalian cells. PLoS One. 2010;5:e8848. doi: 10.1371/journal.pone.0008848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Johnsson P, Ackley A, Vidarsdottir L, Lui W-O, Corcoran M, Grandér D, Morris KV. A pseudogene long-noncoding-RNA network regulates PTEN transcription and translation in human cells. Nat Struct Mol Biol. 2013;20:440–6. doi: 10.1038/nsmb.2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Morris KV, Santoso S, Turner A-M, Pastori C, Hawkins PG. Bidirectional transcription directs both transcriptional gene activation and suppression in human cells. PLoS Genet. 2008;4:e1000258. doi: 10.1371/journal.pgen.1000258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhong M, Niu W, Lu ZJ, Sarov M, Murray JI, Janette J, Raha D, Sheaffer KL, Lam HYK, Preston E, et al. Genome-wide identification of binding sites defines distinct functions for Caenorhabditis elegans PHA-4/FOXA in development and environmental response. PLoS Genet. 2010;6:e1000848. doi: 10.1371/journal.pgen.1000848. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.