

Abstract
The Arf tumor suppressor gene product, p19Arf, regulates cell proliferation in incipient cancer cells and during embryo development. Beyond its commonly accepted p53-dependent actions, p19Arf also acts independently of p53 in both contexts. One such p53-independent effect with in vivo relevance includes its repression of Pdgfrβ, a process that is essential for vision in the mouse. We have utilized cell culture-based and mouse models to define a new role for miR-34a in this process. Ectopic expression of Arf in cultured cells enhanced the expression of several microRNAs predicted to target Pdgfrß synthesis, including the miR-34 family. Because miR-34a has been implicated as a p53-dependent effector, we investigated whether it also contributed to p53-independent effects of p19Arf. Indeed, in mouse embryo fibroblasts (MEFs) lacking p53, Arf-driven repression of Pdgfrβ and its blockade of Pdgf-B stimulated DNA synthesis were both completely interrupted by anti-microRNA against miR-34a. Ectopic miR-34a directly targeted Pdgfrβ and a plasmid reporter containing wild-type Pdgfrβ 3′UTR sequence, but not one in which the miR-34a target sequence was mutated. Although miR-34a expression has been linked to p53—a well-known effector of p19Arf—Arf expression and its knockdown correlated with miR-34a level in MEFs lacking p53. Finally, analysis of the mouse embryonic eye demonstrated that Arf controlled expression of miR-34a, and the related miR-34b and c, in vivo during normal mouse development. Our findings indicate that miR-34a provides an essential link between p19Arf and its p53-independent capacity to block cell proliferation driven by Pdgfrβ. This has ramifications for developmental and tumor suppressor roles of Arf.
Keywords: Pdgfrβ, cell cycle, miR-34a, p19Arf, p53, tumor suppression, vascular remodeling
Introduction
Cell proliferation is governed by 2 major pathways: the p16Ink4a/RB pathway and the p19Arf/p53 pathway.1,2 The p19Arf tumor suppressor was originally discovered as a protein encoded at the mouse (and human) Cdkn2a locus in an alternate reading frame (Arf) when compared with p16Ink4a, the first tumor suppressor identified at that locus.3,4 The simplest paradigm describing p19Arf biology posits that its expression is induced by oncogenic stimuli, at which point it sequesters and inactivates Mdm2, a negative regulator of p53.5-7 The resulting p53 elevation fosters numerous anti-cancer events, such as cell cycle arrest and the induction of pro-apoptotic or DNA damage repair pathways.8
Soon after its initial discovery, it became clear that the seemingly linear pathway stemming from Arf induction to p53-dependent tumor suppression was incomplete. In the first compelling challenge to that prevailing model, members of the Zambetti laboratory showed that ectopic expression of p19Arf could still induce cell cycle arrest in mouse embryo fibroblasts (MEFs) lacking Arf, Mdm2, and p53 (TKO MEFs).9 Although the arrest in TKO MEFs was slower than when Arf was expressed in Arf−/− MEFs retaining Mdm2 and p53, the findings argued for p53-independent activities of p19Arf. Over the ensuing years, the portfolio of p53-independent biochemical effects of p19Arf grew to now include: (1) inhibition of rRNA processing;10 (2) physical interactions with E2F111 and c-Myc12,13 to repress their trans-activating potential; (3) blockade of the RelA subunit of NFκB;14 (4) blunting of Pdgfrβ expression;15 (5) promotion of p53-independent sumoylation of Mdm2, nucleophosmin, and other proteins;16,17 and recently, post-transcriptional repression of Drosha, a microRNA-processing enzyme.18
The relevance of the p53-independent effects of p19Arf for cancer suppression is supported by several observations in mouse models. For example, mice that lack p53, Arf, or both develop tumors spontaneously, but mice lacking both genes develop tumors more rapidly and include a broader range of histological subtypes than in singly mutant animals.19,20 In an experimental model of skin cancer, the effect of Arf deficiency on early papilloma growth exceeds the effect of p53 deficiency, but the rate of progression from papilloma to malignancy is similar in the absence of either gene.21 Further, in the RIP–Tag2 model of pancreatic islet cell neoplasia, Arf loss accelerates the angiogenic switch by a mechanism that is not strictly p53-dependent.22 Notably, which of the above-mentioned p53-independent effects contribute to tumor suppression remains a mystery.
Beyond tumor surveillance, p19Arf plays an essential role for vision as it guides vascular involution in the vitreous space during late stages of mouse eye development.15,23,24 It accomplishes this by limiting the proliferation of perivascular cells flanking the hyaloid vascular system (HVS). Without Arf, primary vitreous hyperplasia and failed HVS regression in the newborn period render the animals sightless.24,25 This developmental function also cannot be attributed solely to p53, because most p53−/− mice have normal eyes.26 Genetic and biochemical studies show that p19Arf acts in a cell-intrinsic way to control the expression of Pdgfrβ mRNA and protein,27 and that Pdgfrβ is essential for primary vitreous hyperplasia that develops without Arf.15,28 Because p19Arf can repress Pdgfrβ protein expression in TKO MEFs, understanding how it can accomplish this will shed light on the p53-independent mechanisms used by p19Arf to control cell cycle arrest in development, and possibly also as a tumor suppressor.
microRNA species have emerged as factors that finely tune the expression of many proteins during development and in disease processes.29-31 It has recently been demonstrated that p19Arf controls the expression of microRNAs in a cell type-specific manner,32 or more broadly by repressing the translation of Drosha, an RNase III endonuclease required for microRNA processing.18 Following this lead, we used a candidate-based approach to determine whether microRNAs might contribute to p53-independent cell cycle control and Pdgfrβ repression by p19Arf. Our results reveal a previously unrecognized capacity for p19Arf, acting without p53, to guide the expression of miR-34 family microRNAs previously linked to p53.33-37 Using complementary cell culture-based and mouse models, we demonstrate that Arf status dictates miR-34a, b, and c expression in cells and in vivo; that miR-34a is sufficient to block Pdgfrβ expression; and that this microRNA is required for Arf-driven, p53-independent cell cycle arrest and Pdgfrβ repression. These findings help to clarify certain confusing aspects of miR-34 biology, increase our understanding of miR-34 regulation, and underline the role that miR-34a can play as a p53-independent effector of p19Arf in normal development and disease.
Results
Arf regulates expression of certain miRNAs independently of p53
We recently demonstrated that p19Arf uses p53-independent mechanisms to block Pdgfrβ protein expression without influencing the level of its mRNA. Considering whether microRNAs might play a role in the post-transcriptional regulation of this protein, we used a candidate-based approach to prioritize potential microRNA regulators of Pdgfrβ. We employed TargetScan and miRDB to generate a list of microRNAs that may target the 3' untranslated region (UTR) of Pdgfrβ. We narrowed the list by only focusing on those microRNAs that are highly conserved or expressed in the developing eye (Fig. 1A).38 We utilized quantitative real-time PCR (qRT-PCR) to measure how the expression of 9 of these changed when p19Arf was ectopically expressed in TKO MEFs. Ectopic p19Arf expression significantly induced 3 microRNAs—miR-29a, miR-34a, and miR-34b—whereas one, miR-222, decreased in Arf-expressing TKO MEFs (Fig. 1B). We focused further studies on miR-34a, because it exhibited the greatest p53-independent induction by p19Arf and had previously been implicated as a tumor suppressor.39

Figure 1. p19Arf regulates miRNAs independently of p53. (A) Schematic diagram depicting candidate miRNAs expressed in the developing eye with predicted binding sites in mouse Pdgfrβ 3′UTR. Pursued candidates are in bold. (B) Quantitative analysis of expression of candidate microRNAs, measured by qRT-PCR, in TKO MEFs transduced with retrovirus encoding p19Arf or GFP (control). (*P < 0.05)
miR-34a is required for Pdgfrβ repression and cell proliferation arrest by p19Arf
p19Arf can block cell proliferation stimulated by Pdgf-B in cultured MEFs, including TKO MEFs lacking endogenous Arf, Mdm2, and p53.15,28 In order to understand if miR-34a is necessary for p19Arf to blunt Pdgfrβ expression, we utilized TKO MEFs transduced with retrovirus vectors expressing p19Arf or Gfp as a control. In this context, p19Arf substantially decreased Pdgfrβ; however, transfection of an anti-microRNA to miR-34a reversed this effect (Fig. 2A).

Figure 2. miR-34a is required for p19Arf driven repression of Pdgfrβ. (A) Representative western blot showing Pdgfrβ, p19Arf and HSC-70 protein expression in lysates prepared from cells transduced with p19Arf retrovirus or control and plus or minus miR-34a hairpin inhibitor. Quantification, on right, is normalized to HSC-70. (B and C) Change in S-phase fraction in TKO MEFs, stimulated by Pdgf-B, is assessed by propidium iodide staining and FACs (B) or by BrdU incorporation (C) in cells transduced with p19Arf or GFP control (+ and −, respectively); and miR-34a hairpin inhibitor or control (+ and −, respectively). (*P < 0.05, when compared with baseline control)
As an additional functional test of miR-34a importance in the p53-independent effects of p19Arf, we considered whether it was needed for Arf expression to overcome cell cycle progression stimulated by Pdgf-B. Similar to previous work,15 exogenous Pdgf-B increased the S-phase fraction—measured by either propidium iodide staining or BrdU incorporation—in cultured TKO MEFs, and ectopic p19Arf expression blunted that effect (Fig. 2B and C, left 2 lanes). Transfection of an anti-microRNA targeting miR-34a nullified the ability of p19Arf to block Pdgf-B-driven cell proliferation (Fig. 2B and C, right 2 lanes). These results indicate that p53-independent Pdgfrβ repression and cell cycle arrest imposed by p19Arf both depend on miR-34a.
miR-34a directly targets Pdgfrβ
miR-34a has a number of protein targets that are known to be important for cell cycle regulation, including Cdk4/6, N-Myc and c-Met (reviewed in Hermeking et al.40). Previous mRNA-based surveys for miR-34a targets did not find Pdgfrβ;37 however, the 3′UTR of mouse Pdgfrβ actually contains 2 miR-34a target sequences (Fig. 1A). To determine whether this transcript could be a direct miR-34a target, we sub-cloned wild-type and mutant versions of 1 of the putative miR-34a targets from the mouse Pdgfrβ 3′UTR (Fig. 3A) into the pMIR-REPORT plasmid. Ectopic expression of miR-34a by transient transfection of HEK293T cells demonstrated that this microRNA blunted expression of the Luciferase cDNA containing the Pdgfrβ wild-type 3′UTR target, but not the mutated sequence (Fig. 3B).

Figure 3. Pdgfrβ is a direct target of miR-34a. (A) miR-34a target sequence from Pdgfrβ 3' UTR. Seed sequence is in bold, and asterisks highlight mutated nucleotides. (B) Quantitative analysis of luciferase reporter in HEK293T cells, transiently transfected with expression plasmids for miR-34a (PIG-34a) or control (PIG), showing that miR-34a represses the reporter linked to the wild-type (WT) but not mutant (MUT) 3' UTR of Pdgfrβ. Luciferase activity, normalized to β-galactosidase from co-transfected reporter, is presented relative to control for each reporter. (C) Quantitative analysis by qRT-PCR for miR-34a (left) or Pdgfrβ mRNA (right) expression upon transduction of TKO MEFs with retroviral vectors expressing miR-34a or empty vector control, as indicated. (D) Representative western blot (left panel) and quantitative analysis, relative to HSC70, (right panel) for Pdgfrβ protein expression in lysates from TKO MEFs transduced as described in (C). (*P < 0.05, compared with baseline control)
To understand if miR-34a expression is sufficient to diminish endogenous Pdgfrβ expression, we examined changes in Pdgfrβ mRNA and protein following retroviral expression of miR-34a in TKO MEFs. Although Pdgfrβ mRNA expression was not altered by miR-34a, western blot revealed Pdgfrβ protein to be significantly lower (Fig. 3C and D). Taken together, these data indicate that miR-34a is sufficient to repress Pdgfrβ translation.
miR-34a expression depends on p19Arf in cells and in the developing mouse eye
If miR-34a acts downstream of p19Arf to mediate its anti-proliferative activities without p53, we considered whether endogenous levels of p19Arf could influence miR-34a expression in cultured cells and in vivo. We addressed this using wild-type MEFs, as well as Arf-deficient MEFs derived from ArfGfp/Gfp41 or ArflacZ/lacZ mice.42 In both of these lines, the first exon in Arf is replaced by cDNA encoding 1 of the 2 reporters. At passages 1, 3, and 5, miR-34a expression was significantly lower in Arf-deficient MEFs as compared with the wild-type cells (Fig. 4A). p53 is known to regulate miR-34a expression in human colorectal33 and lung carcinoma cells,34,35 and in MEFs.36 We considered, then, whether decreased miR-34a in the absence of Arf might simply reflect the decreased p53 activity.5,7 That miR-34a expression was even lower in TKO MEFs than Arf-deficient MEFs supports the fact that p53 does play a role in our model (Fig. 4A, right panel). However, examining miR-34a expression in p53−/− MEFs expressing shRNA directed at Arf allowed us to determine whether a component of miR-34a regulation was independent of p53. With approximately 60% reduction of endogenous Arf mRNA in p53−/− MEFs, we observed a quantitatively similar decrease in miR-34a (Fig. 4B). Of note, miR-34b and c were similarly diminished when Arf expression was knocked down in these MEFs (Fig. 4C). Hence, endogenous Arf drives the expression of miR-34 family microRNAs even in MEFs lacking p53.

Figure 4. miR-34a expression correlates with p19Arf status. (A) Quantitative analysis of miR-34a expression, measured by qRT-PCR, in MEFs that are derived from WT, ArfGfp/Gfp (ARF G/G), ArflacZ/lacZ (ARF L/L), and Arf−/−, p53−/−, MDM2−/− (TKO) mice and cultivated for 1, 3, or 5 passages. (B and C) Quantitative RT-PCR analyses of Arf, and miR-34a, b, and c, mRNA expression, as indicated, in p53−/− MEFs transduced with retroviral vectors targeting p19Arf (Sh-Arf) or control (Sh-Ctrl). In each case, data are expressed relative to baseline control. (*P < 0.05, when compared with baseline control)
In order to investigate whether miR-34a is controlled by p19Arf in vivo, we utilized laser capture microdissection (LCM) to isolate discrete compartments of the mouse eye at embryonic day (E) 13.5 (Fig. 5A), when Arf is first robustly expressed in the vitreous.15 At this point in development, we already observe Arf-dependent alterations in Pdgfrβ mRNA and increased cell proliferation, but there are few other anatomic differences between the wild-type and Arf−/− animal;15 as such, any Arf-dependent change in miR-34a would not be obscured by dramatic ocular pathology. By utilizing qRT-PCR, we demonstrated that miR-34a was most highly expressed in the primary vitreous compartment (Fig. 5B), which is the only anatomic site where Arf is measurably expressed.23,28 Broadening the scope of our query, we noted that other members of the miR-34 microRNA family were more highly expressed in the vitreous than miR-29a or miR-221, 2 other microRNAs predicted to target the Pdgfrβ 3′UTR (Figs. 1A and 5C). We then inquired if these microRNAs were influenced by the presence or absence of Arf by comparing their expression in the vitreous in eyes taken from E13.5 wild-type and ArfGfp/Gfp embryos. Consistent with our analyses of MEFs, the absence of Arf significantly diminished the expression of miR-34a, b, and c, as well as miR-29a, which was also induced by ectopic p19Arf in MEFs. The expression of miR-221 was not significantly influenced by Arf expression in vivo (Fig. 5D) or in cultured MEFs (Fig. 1B). The Arf-dependent expression in the vitreous of the miR-34 microRNAs supports their candidacy as physiological regulators of Pdgfrβ during mouse eye development.
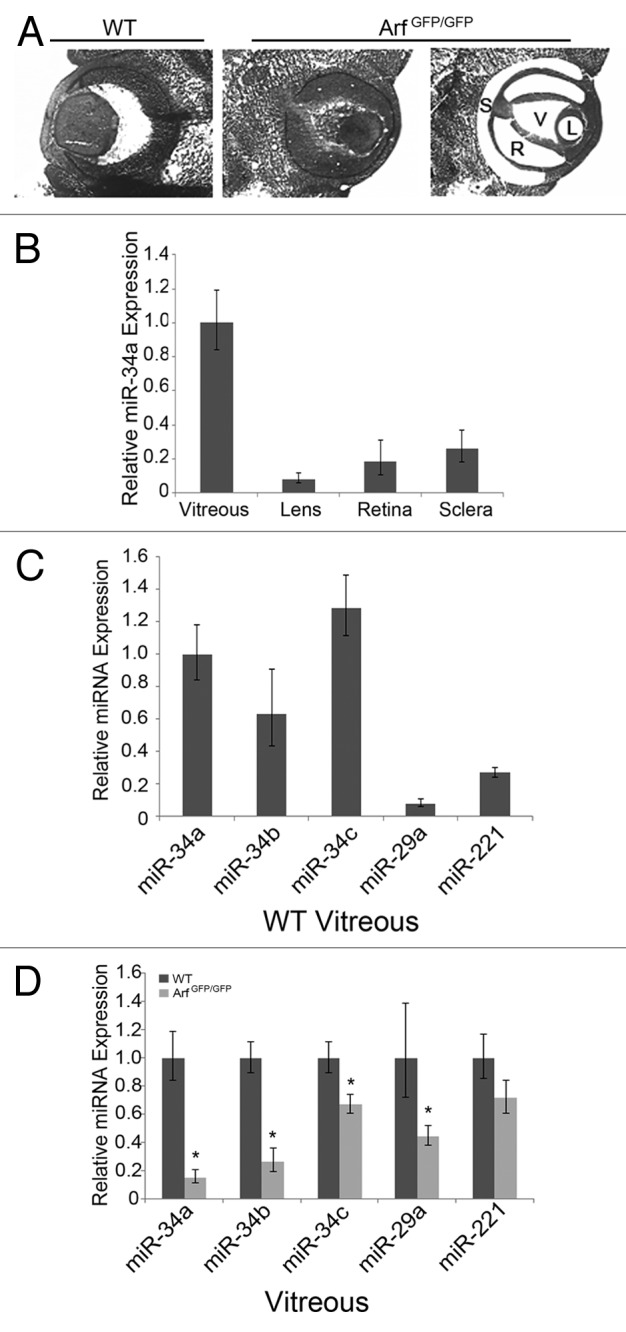
Figure 5. miR-34a expression in vivo is dependent upon p19Arf. (A) Representative photomicrographs of E13.5 mouse embryo eye from wild-type (WT) or ArfGfp/Gfp embryo, as indicated, showing laser capture microdissection (LCM) of sclera (S), retina, (R), vitreous (V), and lens (L). (B) Quantitation of mature miR-34a expression, measured by qRT-PCR, in LCM specimens from different parts of the wild-type E13.5 mouse eye. Expression is normalized to U6 snRNA and arbitrarily displayed relative to that in the vitreous fraction. (C and D) Quantitative analysis shows relative expression of different microRNAs in the vitreous of E13.5 wild-type (WT) and ArfGfp/Gfp mouse embryos. In each case, expression is normalized to U6 snRNA and shown relative to miR-34a expression in the wild-type vitreous (C) or relative to each microRNA expression in wild-type embryos (D). (*P < 0.05, when compared with wild-type embryos)
Discussion
Although initially described as a tumor suppressor that acts through p53,5-7 p19Arf clearly has functional capabilities that do not depend on this downstream effector. Such activities include its capacity to interfere with rRNA processing,10 perhaps by translational repression of Drosha;18 inhibition of signaling to NFκB;14 blockade of Myc-driven transcriptional activation;12,13 and fostering sumoylation of Mdm2 and other nuclear proteins.16,17 Perhaps the best example supporting the in vivo relevance of p53-independent biochemical activities relates to repression of Pdgfrβ by p19Arf. In this instance, deregulated proliferation of perivascular cells in the primary vitreous of Arf−/− mice leads to severe ocular developmental defects and blindness; that the primary vitreous hyperplasia associated with this phenotype is reversed in Arf−/−, Pdgfrβ−/− animals28 demonstrates that this biochemical pathway is crucial for a normal developmental process. Such a developmental role for p19Arf cannot be easily explained by its activation of p53, because p53−/− mice usually have normal eyes (see more below).23,26 Further, cell culture-based studies indicate that Arf expression represses Pdgfrβ independently of p53.28 Given that in vivo relevance is already established, understanding how Pdgfrβ repression is accomplished becomes an important matter.
In this report, we illustrate how p19Arf can block Pdgfrβ protein synthesis without affecting its mRNA level and without engaging Mdm2 and p53. First, we have established that Arf expression elevates miR-34a, among several others microRNAs with the predicted capacity to target Pdgfrβ. This can be achieved even in MEFs that lack Mdm2 and p53, which is novel, because miR-34a is largely recognized as a p53 target.33,34,36,37 Second, we show that miR-34a can directly target the 3′UTR of mouse Pdgfrβ. Although this does not diminish Pdgfrβ mRNA level, miR-34a expression is sufficient to decrease Pdgfrβ protein. The absence of a measurable effect on the transcript may help to explain why larger, RNA-based surveys did not identify Pdgfrβ as a miR-34a target.37 Third, an essential role for miR-34a in Arf-driven Pdgfrß repression is indicated, because anti-microRNA targeting miR-34a completely blocks the ability of p19Arf to repress Pdgfrβ and to blunt Pdgf-B-driven cell proliferation. Although it remains possible that other closely related miRNAs with the same seeding sequence may also contribute, our findings do discount the possible roles for microRNAs targeting other regions of the Pdgfrβ 3′UTR (see Fig. 1A). Finally, we demonstrate that miR-34 family members are highly expressed in the primary vitreous—the only region in the eye with detectable expression of Arf23,28—and they are repressed in Arf−/− embryos. This represents the first in vivo evidence in which expression of miR-34a is directly correlated with a developmental disease. Further, providing in vivo evidence for Arf regulation of this microRNA supports the validity of our analyses of cultured MEFs.
Our new findings allow us to pose a model by which p19Arf controls Pdgfrß expression by 2 mechanisms: p53-dependent transcriptional repression and post-transcriptional repression via miR-34a, a process that can be separated from p53 (Fig. 6). This model offers new insight into the confusing role that p53 seems to play during mouse embryo eye development, which is heavily influenced by mouse genetic background.23,26,43 In most genetic backgrounds, p53−/− mice have normal eyes. When bred into pure C57BL/6 and pure BALB/c backgrounds, p53−/− mice can develop primary vitreous hyperplasia, mimicking that observed without Arf.23,26,43 However, in a mixed C57BL/6 × 129/Sv lineage, the eyes are usually normal.23 We previously speculated that this might be due to a p53-independent capacity for p19Arf to block Pdgfrβ translation, and its capacity to do so might vary with genetic background.28 We can now attribute that capacity to Arf-dependent regulation of miR-34a and related microRNAs in the eye.
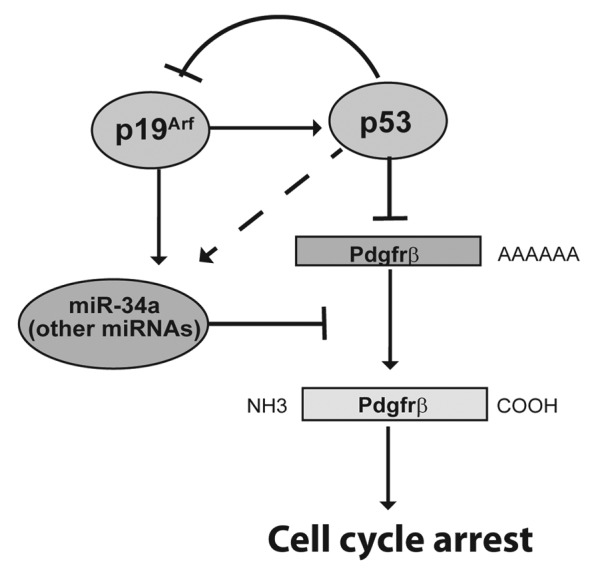
Figure 6. Schematic diagram shows dual mechanisms for Arf dependent control of Pdgfrβ expression. p19Arf mediates transcriptional repression of Pdgfrβ by stabilizing p53 and post-transcriptional repression of Pdgfrβ protein levels via induction of miR-34a and potentially other microRNAs. Although Arf expression can induce miR-34a and other microRNAs independently of p53 in cultured MEFs (solid arrow), p53 may contribute to miR-34a expression in the developing mouse eye (dashed arrow).
Our model also helps to resolve another surprising paradox. Although miR-34a is well-known to be a transcriptional target of p53,33,34,36 and it exerts anti-proliferative effects in a number of tumor models,35,39 mice lacking all 3 members of the miR-34 microRNA family are seemingly normal, with no overt developmental defects or cancer susceptibility.44 One might expect the eyes of miR-34a (and miR-34a, b, c)-null animals to be normal, because Arf-driven, p53-dependent transcriptional repression of Pdgfrβ mRNA will still be intact (Fig. 6). However, loss of miR-34 family members might unmask an eye development defect in certain lines of p53-deficient mice.
Based on current understanding, we can propose at least 2 reasonable mechanisms by which p19Arf regulates miR-34a without p53. First, it is possible that in vivo, like in cultured MEFs, its expression is dually controlled by p53 and p19Arf. Interestingly, we previously showed that Arf mRNA increases in the vitreous of p53−/− animals.28 As such, effects of p19Arf on miR-34a should be magnified without p53. A second possibility stems from a recent report by Kuchenreuther and Weber demonstrating that p19Arf represses Drosha expression in cultured MEFs, and loss of the Arf-Drosha pathway substantially increases or decreases a number of microRNAs.18 miR-34a was not listed among them, but their analysis focused only on a subset of 147 expressed more highly. Here, Arf might normally repress Drosha, enhancing miR-34a expression and blocking primary vitreous hyperplasia.
Finally, our findings shed light on p53-independent tumor suppression by Arf. Differences in tumor susceptibility in mice lacking either Arf or p53 and in Arf−/−, p53−/− double knockout mouse lines underscore the importance of an alternative pathway for p19Arf to block tumor formation.19,20 Tumorigenesis studies in the RIP-Tag2 mouse model,22 as well as earlier studies of intestinal adenomas arising in Arf, Ink4a double knockout mice bred into the Min mouse,45 indicate that p19Arf might influence tumor vascular biology. In neither case is a molecular explanation clear. Indeed, Ulanet and Hanahan performed a broad array of molecular analyses, including RNA-based analysis of a panel of angiogenesis-related genes, and they found no significant differences.22 Given our findings that Arf induction of miR-34a can influence Pdgfrβ protein without influencing its mRNA, and recent findings that other angiogenesis-related proteins like Pdgf-B and Vegf-A are controlled post-transcriptionally by p19Arf,46 protein-based assays might be more revealing as one explores possible anti-angiogenic effects and p53-independent tumor suppression by p19Arf.
Materials and Methods
Plasmids and other recombinant DNA reagents
Anti-miR-34a and scrambled controls for in vitro studies (obtained from Dhamacon) were used at a final concentration 200 nM and transfected into MEFS using the Dharmafect transfection reagent. Retroviral plasmids, pMSCV-PIG and pMSCV-PIG-34A, were provided by Joshua Mendell at UT Southwestern Medical Center. MSCV-based bicistronic retrovirus vectors mouse Arf and/or Gfp or Rfp were prepared and used as previously described.15 Cells were infected with retrovirus shRNA targeting p19Arf47 on day 1 and day 2, and then selected with 2 μg/mL puromycin for 6 d prior to harvest. Transduction efficiency was monitored by GFP expression.
Mouse models and cell lines
Arf Gfp/Gfp mice41 and Arf lacZ/lacZ42 were maintained in a mixed C57BL/6 × 129/Sv genetic background. Experimental protocols were approved by the Institutional Animal Care and Use Committee at UT Southwestern Medical Center.
Primary MEFs from ArflacZ/lacZ; ArfGfp/Gfp; p53−/−, Mdm2−/−; and p53−/−, Arf−/−, Mdm2−/− (TKO) mutant and wild-type mice were cultivated as previously described.15 TKO MEFs were provided by G Zambetti (St. Jude Children’s Research Hospital).9
Laser-capture microdissection (LCM)
LCM was performed as previously described.28 Briefly, mouse embryos were harvested at E13.5, and heads were immediately embedded in OCT freezing medium without fixation. Fourteen-μm sections were cut on a CryoStar NX70 cryostat, mounted on PEN Membrane Metal Slides (Applied Biosystems), and stained with hematoxylin and eosin (Molecular Machines and Industries AG). LCM was performed on the Arcturus Veritas Microdissection System. At least 10 micro-dissected sections from the vitreous, lens, retina, and sclera were pooled from each embryo.
Quantitative RT-PCR (qRT-PCR)
Total RNA was extracted from MEFs using the miRNeasy mini kit (Qiagen). For qRT-PCR, 1 µg of total RNA was reverse transcribed using NCode miRNA First-Strand Synthesis (Invitrogen) and Fast SYBR Green Master Mix (Applied Biosystem). qRT-PCR was performed in a 96-well plate using ABI 7900HT instrument. The PCR program consisted of 20 s at 95 °C, followed by 40 cycles of 95 °C for 15 s and 60 °C for 20 s. Primer quality was analyzed by dissociation curves. The expression of miRNAs and Pdgfr-β was normalized to U6 and Hprt, respectively.
Luciferase reporter assay
The 362 bp 3′UTR of Pdgfrβ gene was amplified by PCR with primers 5′-GTACTAGTCT CTGGCTGAAG CAGAGGAC and 5′-CGAAGCTTAC CACCGTACAG TCGTGGAT. The PCR product was digested by SpeI/HindIII and inserted into pMIR-REPORT vector (Ambion). To create this mutant we replaced the seed sequence CACTGCC with ACGCGTC through site-directed mutagenesis using QuickChange kit (Stratagene). HEK293T cells were transiently transfected using Fugene 6 reagent (Roche) in 96-well plates, as previously reported.48 pCMV-LacZ was used as a control to monitor transfection efficiency of the luciferase reporter assay.
Western blotting
Protein expression was examined by Western-blotting according to a standard procedure. The following antibodies were used: anti-p19Arf (Ab80, Abcam, 1:1000), anti-Pdgfrß (AF1042, R&D, 1:1000), anti-Hsc70 (Sc-1059, Santa Cruz, 1:5000). Band intensity was quantified using NIH ImageJ software.
Cell cycle analysis
Cell cycle analysis was assessed in TKO MEFs transduced with p19Arf-expressing retrovirus, with or without transfection with anti-miR-34a reagent (Dharmacon), and with or without stimulation using Pdgf-B (50 ng/mL) (R&D) for 12 h. Relative change in S-phase fraction was assessed in 2 ways: first, propidium iodide (PI) (sigma) staining was performed after cells were harvested by trypsin-EDTA and fixed in 70% ethanol. Fixed cells were washed in PBS and centrifuged at 1200 rpm for 5 min. Cells were resuspended in 0.3 ml PBS and RNaseA (Sigma) was added to the suspension to final concentration of 0.5 mg/mL. After 1 h of incubation at 37 °C, PI was added to the suspension to final concentration of 10 μg/mL. PI-stained cells were analyzed for DNA content with a BD Calibur flow cytometer. Cell sorting results were analyzed with FlowJo Software using a cell cycle platform and Watson Pragmatic Model to calculate the distribution of cells in G1, S, and G2 cell cycle phases. In some experiments, cells were incubated with BrdU (10 μM) for 6 h prior to fixing with 2% paraformaldehyde. BrdU was assessed by immunofluorescence staining using a FITC-conjugated anti-BrdU antibody (BD 347583, BD Biosciences, 1:100) and quantified by determining the fraction of DAPI-positive with detectable BrdU in at least 5 fields from replicate plates.
Statistical analysis
Data are expressed as mean ± SD. Statistical analysis was determined using a 2-tailed Student t test. A P value of < 0.05 was considered significant. We conducted each experiment in triplicate.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We gratefully acknowledge excellent technical assistance from C Devitt, research technician in the Skapek Lab, for assistance with mouse breeding, tissue harvest, and preparation for LCM; J Mendell for the pMSCV-PIG and pMSCV-PIG-miR34-a plasmids; CJ Sherr (St. Jude Children’s Research Hospital) for providing ArfGfp/Gfp mice; J Sage (Stanford) for providing shRNA-p19Arf constructs; GP Zambetti (St. Jude Children’s Research Hospital) for providing TKO MEFs; J Elmquist for use of the Arcturus Veritas Micodissection system and the Flow Cytometry Core Facility at UT Southwestern Medical Center. This research is supported by grants to S.X.S. from the National Eye Institute (EY 014368 and EY 019942).
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/27725
References
- 1.Weinberg RA. The retinoblastoma protein and cell cycle control. Cell. 1995;81:323–30. doi: 10.1016/0092-8674(95)90385-2. [DOI] [PubMed] [Google Scholar]
- 2.Levine AJ. p53, the cellular gatekeeper for growth and division. Cell. 1997;88:323–31. doi: 10.1016/S0092-8674(00)81871-1. [DOI] [PubMed] [Google Scholar]
- 3.Kamb A, Gruis NA, Weaver-Feldhaus J, Liu Q, Harshman K, Tavtigian SV, Stockert E, Day RS, 3rd, Johnson BE, Skolnick MH. A cell cycle regulator potentially involved in genesis of many tumor types. Science. 1994;264:436–40. doi: 10.1126/science.8153634. [DOI] [PubMed] [Google Scholar]
- 4.Quelle DE, Zindy F, Ashmun RA, Sherr CJ. Alternative reading frames of the INK4a tumor suppressor gene encode two unrelated proteins capable of inducing cell cycle arrest. Cell. 1995;83:993–1000. doi: 10.1016/0092-8674(95)90214-7. [DOI] [PubMed] [Google Scholar]
- 5.Pomerantz J, Schreiber-Agus N, Liégeois NJ, Silverman A, Alland L, Chin L, Potes J, Chen K, Orlow I, Lee HW, et al. The Ink4a tumor suppressor gene product, p19Arf, interacts with MDM2 and neutralizes MDM2’s inhibition of p53. Cell. 1998;92:713–23. doi: 10.1016/S0092-8674(00)81400-2. [DOI] [PubMed] [Google Scholar]
- 6.Zhang Y, Xiong Y, Yarbrough WG. ARF promotes MDM2 degradation and stabilizes p53: ARF-INK4a locus deletion impairs both the Rb and p53 tumor suppression pathways. Cell. 1998;92:725–34. doi: 10.1016/S0092-8674(00)81401-4. [DOI] [PubMed] [Google Scholar]
- 7.Kamijo T, Weber JD, Zambetti G, Zindy F, Roussel MF, Sherr CJ. Functional and physical interactions of the ARF tumor suppressor with p53 and Mdm2. Proc Natl Acad Sci U S A. 1998;95:8292–7. doi: 10.1073/pnas.95.14.8292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ko LJ, Prives C. p53: puzzle and paradigm. Genes Dev. 1996;10:1054–72. doi: 10.1101/gad.10.9.1054. [DOI] [PubMed] [Google Scholar]
- 9.Weber JD, Jeffers JR, Rehg JE, Randle DH, Lozano G, Roussel MF, Sherr CJ, Zambetti GP. p53-independent functions of the p19(ARF) tumor suppressor. Genes Dev. 2000;14:2358–65. doi: 10.1101/gad.827300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sugimoto M, Kuo ML, Roussel MF, Sherr CJ. Nucleolar Arf tumor suppressor inhibits ribosomal RNA processing. Mol Cell. 2003;11:415–24. doi: 10.1016/S1097-2765(03)00057-1. [DOI] [PubMed] [Google Scholar]
- 11.Eymin B, Karayan L, Séité P, Brambilla C, Brambilla E, Larsen CJ, Gazzéri S. Human ARF binds E2F1 and inhibits its transcriptional activity. Oncogene. 2001;20:1033–41. doi: 10.1038/sj.onc.1204220. [DOI] [PubMed] [Google Scholar]
- 12.Qi Y, Gregory MA, Li Z, Brousal JP, West K, Hann SR. p19ARF directly and differentially controls the functions of c-Myc independently of p53. Nature. 2004;431:712–7. doi: 10.1038/nature02958. [DOI] [PubMed] [Google Scholar]
- 13.Datta A, Nag A, Pan W, Hay N, Gartel AL, Colamonici O, Mori Y, Raychaudhuri P. Myc-ARF (alternate reading frame) interaction inhibits the functions of Myc. J Biol Chem. 2004;279:36698–707. doi: 10.1074/jbc.M312305200. [DOI] [PubMed] [Google Scholar]
- 14.Rocha S, Campbell KJ, Perkins ND. p53- and Mdm2-independent repression of NF-kappa B transactivation by the ARF tumor suppressor. Mol Cell. 2003;12:15–25. doi: 10.1016/S1097-2765(03)00223-5. [DOI] [PubMed] [Google Scholar]
- 15.Silva RL, Thornton JD, Martin AC, Rehg JE, Bertwistle D, Zindy F, Skapek SX. Arf-dependent regulation of Pdgf signaling in perivascular cells in the developing mouse eye. EMBO J. 2005;24:2803–14. doi: 10.1038/sj.emboj.7600751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rizos H, Woodruff S, Kefford RF. p14ARF interacts with the SUMO-conjugating enzyme Ubc9 and promotes the sumoylation of its binding partners. Cell Cycle. 2005;4:597–603. doi: 10.4161/cc.4.4.1588. [DOI] [PubMed] [Google Scholar]
- 17.Tago K, Chiocca S, Sherr CJ. Sumoylation induced by the Arf tumor suppressor: a p53-independent function. Proc Natl Acad Sci U S A. 2005;102:7689–94. doi: 10.1073/pnas.0502978102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kuchenreuther MJ, Weber JD. The ARF tumor-suppressor controls Drosha translation to prevent Ras-driven transformation. Oncogene. 2013 doi: 10.1038/onc.2012.601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kamijo T, Bodner S, van de Kamp E, Randle DH, Sherr CJ. Tumor spectrum in ARF-deficient mice. Cancer Res. 1999;59:2217–22. [PubMed] [Google Scholar]
- 20.Jacks T, Remington L, Williams BO, Schmitt EM, Halachmi S, Bronson RT, Weinberg RA. Tumor spectrum analysis in p53-mutant mice. Curr Biol. 1994;4:1–7. doi: 10.1016/S0960-9822(00)00002-6. [DOI] [PubMed] [Google Scholar]
- 21.Kelly-Spratt KS, Gurley KE, Yasui Y, Kemp CJ. p19Arf suppresses growth, progression, and metastasis of Hras-driven carcinomas through p53-dependent and -independent pathways. PLoS Biol. 2004;2:E242. doi: 10.1371/journal.pbio.0020242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ulanet DB, Hanahan D. Loss of p19(Arf) facilitates the angiogenic switch and tumor initiation in a multi-stage cancer model via p53-dependent and independent mechanisms. PLoS One. 2010;5:e12454. doi: 10.1371/journal.pone.0012454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McKeller RN, Fowler JL, Cunningham JJ, Warner N, Smeyne RJ, Zindy F, Skapek SX. The Arf tumor suppressor gene promotes hyaloid vascular regression during mouse eye development. Proc Natl Acad Sci U S A. 2002;99:3848–53. doi: 10.1073/pnas.052484199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Martin AC, Thornton JD, Liu J, Wang X, Zuo J, Jablonski MM, Chaum E, Zindy F, Skapek SX. Pathogenesis of persistent hyperplastic primary vitreous in mice lacking the arf tumor suppressor gene. Invest Ophthalmol Vis Sci. 2004;45:3387–96. doi: 10.1167/iovs.04-0349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gromley A, Churchman ML, Zindy F, Sherr CJ. Transient expression of the Arf tumor suppressor during male germ cell and eye development in Arf-Cre reporter mice. Proc Natl Acad Sci U S A. 2009;106:6285–90. doi: 10.1073/pnas.0902310106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ikeda S, Hawes NL, Chang B, Avery CS, Smith RS, Nishina PM. Severe ocular abnormalities in C57BL/6 but not in 129/Sv p53-deficient mice. Invest Ophthalmol Vis Sci. 1999;40:1874–8. [PubMed] [Google Scholar]
- 27.Thornton JD, Swanson DJ, Mary MN, Pei D, Martin AC, Pounds S, Goldowitz D, Skapek SX. Persistent hyperplastic primary vitreous due to somatic mosaic deletion of the arf tumor suppressor. Invest Ophthalmol Vis Sci. 2007;48:491–9. doi: 10.1167/iovs.06-0765. [DOI] [PubMed] [Google Scholar]
- 28.Widau RC, Zheng Y, Sung CY, Zelivianskaia A, Roach LE, Bachmeyer KM, Abramova T, Desgardin A, Rosner A, Cunningham JM, et al. p19Arf represses platelet-derived growth factor receptor β by transcriptional and posttranscriptional mechanisms. Mol Cell Biol. 2012;32:4270–82. doi: 10.1128/MCB.06424-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bentwich I, Avniel A, Karov Y, Aharonov R, Gilad S, Barad O, Barzilai A, Einat P, Einav U, Meiri E, et al. Identification of hundreds of conserved and nonconserved human microRNAs. Nat Genet. 2005;37:766–70. doi: 10.1038/ng1590. [DOI] [PubMed] [Google Scholar]
- 30.Ambros V, Lee RC, Lavanway A, Williams PT, Jewell D. MicroRNAs and other tiny endogenous RNAs in C. elegans. Curr Biol. 2003;13:807–18. doi: 10.1016/S0960-9822(03)00287-2. [DOI] [PubMed] [Google Scholar]
- 31.Lagos-Quintana M, Rauhut R, Lendeckel W, Tuschl T. Identification of novel genes coding for small expressed RNAs. Science. 2001;294:853–8. doi: 10.1126/science.1064921. [DOI] [PubMed] [Google Scholar]
- 32.Li C, Finkelstein D, Sherr CJ. Arf tumor suppressor and miR-205 regulate cell adhesion and formation of extraembryonic endoderm from pluripotent stem cells. Proc Natl Acad Sci U S A. 2013;110:E1112–21. doi: 10.1073/pnas.1302184110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chang TC, Wentzel EA, Kent OA, Ramachandran K, Mullendore M, Lee KH, Feldmann G, Yamakuchi M, Ferlito M, Lowenstein CJ, et al. Transactivation of miR-34a by p53 broadly influences gene expression and promotes apoptosis. Mol Cell. 2007;26:745–52. doi: 10.1016/j.molcel.2007.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tarasov V, Jung P, Verdoodt B, Lodygin D, Epanchintsev A, Menssen A, Meister G, Hermeking H. Differential regulation of microRNAs by p53 revealed by massively parallel sequencing: miR-34a is a p53 target that induces apoptosis and G1-arrest. Cell Cycle. 2007;6:1586–93. doi: 10.4161/cc.6.13.4436. [DOI] [PubMed] [Google Scholar]
- 35.Raver-Shapira N, Marciano E, Meiri E, Spector Y, Rosenfeld N, Moskovits N, Bentwich Z, Oren M. Transcriptional activation of miR-34a contributes to p53-mediated apoptosis. Mol Cell. 2007;26:731–43. doi: 10.1016/j.molcel.2007.05.017. [DOI] [PubMed] [Google Scholar]
- 36.He L, He X, Lim LP, de Stanchina E, Xuan Z, Liang Y, Xue W, Zender L, Magnus J, Ridzon D, et al. A microRNA component of the p53 tumour suppressor network. Nature. 2007;447:1130–4. doi: 10.1038/nature05939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bommer GT, Gerin I, Feng Y, Kaczorowski AJ, Kuick R, Love RE, Zhai Y, Giordano TJ, Qin ZS, Moore BB, et al. p53-mediated activation of miRNA34 candidate tumor-suppressor genes. Curr Biol. 2007;17:1298–307. doi: 10.1016/j.cub.2007.06.068. [DOI] [PubMed] [Google Scholar]
- 38.Karali M, Peluso I, Gennarino VA, Bilio M, Verde R, Lago G, Dollé P, Banfi S. miRNeye: a microRNA expression atlas of the mouse eye. BMC Genomics. 2010;11:715. doi: 10.1186/1471-2164-11-715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Welch C, Chen Y, Stallings RL. MicroRNA-34a functions as a potential tumor suppressor by inducing apoptosis in neuroblastoma cells. Oncogene. 2007;26:5017–22. doi: 10.1038/sj.onc.1210293. [DOI] [PubMed] [Google Scholar]
- 40.Hermeking H. The miR-34 family in cancer and apoptosis. Cell Death Differ. 2010;17:193–9. doi: 10.1038/cdd.2009.56. [DOI] [PubMed] [Google Scholar]
- 41.Zindy F, Williams RT, Baudino TA, Rehg JE, Skapek SX, Cleveland JL, Roussel MF, Sherr CJ. Arf tumor suppressor promoter monitors latent oncogenic signals in vivo. Proc Natl Acad Sci U S A. 2003;100:15930–5. doi: 10.1073/pnas.2536808100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Freeman-Anderson NE, Zheng Y, McCalla-Martin AC, Treanor LM, Zhao YD, Garfin PM, He TC, Mary MN, Thornton JD, Anderson C, et al. Expression of the Arf tumor suppressor gene is controlled by Tgfbeta2 during development. Development. 2009;136:2081–9. doi: 10.1242/dev.033548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Reichel MB, Ali RR, D’Esposito F, Clarke AR, Luthert PJ, Bhattacharya SS, Hunt DM. High frequency of persistent hyperplastic primary vitreous and cataracts in p53-deficient mice. Cell Death Differ. 1998;5:156–62. doi: 10.1038/sj.cdd.4400326. [DOI] [PubMed] [Google Scholar]
- 44.Concepcion CP, Han YC, Mu P, Bonetti C, Yao E, D’Andrea A, Vidigal JA, Maughan WP, Ogrodowski P, Ventura A. Intact p53-dependent responses in miR-34-deficient mice. PLoS Genet. 2012;8:e1002797. doi: 10.1371/journal.pgen.1002797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gibson SL, Dai CY, Lee HW, DePinho RA, Gee MS, Lee WMF, Furth EE, Brensinger C, Enders GH. Inhibition of colon tumor progression and angiogenesis by the Ink4a/Arf locus. Cancer Res. 2003;63:742–6. [PubMed] [Google Scholar]
- 46.Kawagishi H, Nakamura H, Maruyama M, Mizutani S, Sugimoto K, Takagi M, Sugimoto M. ARF suppresses tumor angiogenesis through translational control of VEGFA mRNA. Cancer Res. 2010;70:4749–58. doi: 10.1158/0008-5472.CAN-10-0368. [DOI] [PubMed] [Google Scholar]
- 47.Sage J, Miller AL, Pérez-Mancera PA, Wysocki JM, Jacks T. Acute mutation of retinoblastoma gene function is sufficient for cell cycle re-entry. Nature. 2003;424:223–8. doi: 10.1038/nature01764. [DOI] [PubMed] [Google Scholar]
- 48.Mei J, Bachoo R, Zhang CL. MicroRNA-146a inhibits glioma development by targeting Notch1. Mol Cell Biol. 2011;31:3584–92. doi: 10.1128/MCB.05821-11. [DOI] [PMC free article] [PubMed] [Google Scholar]