

Abstract
Polo-like kinase 1 (PLK1) plays crucial roles in multiple stages of cell division. Our previous studies suggest that global transcriptional regulation by PLK1 may contribute to its multiple functions. PLK1 depletion is associated with a decrease in cell viability and the induction of apoptosis; however, the underlying mechanisms are not completely understood. Here, we report that forkhead box protein O1 (FOXO1) is a novel physiological substrate of PLK1. FOXO1 is at the interface of crucial cellular processes, orchestrating programs of gene expression that regulate apoptosis, cell cycle progression, and oxidative-stress resistance. PLK1 interacts with and phosphorylates FOXO1, mainly at the G2/M phase of the cell cycle. PLK1-mediated phosphorylation leads to the impairment of FOXO1’s transcriptional activity in an Akt-independent manner. By immunofluorescence staining and subcellular fractionation, we demonstrate that PLK1-induced FOXO1 phosphorylation causes its nuclear exclusion. Furthermore, PLK1-mediated phosphorylation of FOXO1 negatively regulates its pro-apoptotic function and abrogates its ability to delay entry into and progression through G2/M transition. Therefore, our results suggest that PLK1 abrogates the inhibitory effects of FOXO1 on cell growth and survival to ensure timely cell cycle progression. This study not only reveals a novel and major regulatory mechanism of FOXO1 at the late phases of the cell cycle, but also provides new insight into the molecular mechanisms by which PLK1 inhibition leads to growth arrest and cell death.
Keywords: PLK1, FOXO1, phosphorylation, cell division, nuclear-cytoplasmic shuttling, apoptosis, regulation, transcription factor
Introduction
Mammalian Polo-like kinase 1 (PLK1) belongs to a family of 4 serine/threonine kinases.1 It regulates pivotal events in cell division, including mitotic entry, centrosome maturation, spindle assembly, chromatin segregation, mitotic exit, and cytokinesis.1,2 PLK1 contains a conserved N-terminal kinase catalytic domain and a C-terminal polo-box domain (PBD)—a phosphopeptide-binding motif. The PBD is required for the recruitment of PLK1 to substrates that have been primed by phosphorylation at specific docking sites, thereby directing the subcellular localization of PLK1.3 Although PBD-directed dynamic subcellular localization is believed to significantly contribute to the multi-faceted mitotic functions of PLK1, recent studies shed light on how PLK1 regulates the mitotic transcriptional network, thereby globally contributing to mitotic entry and progression. For example, we have recently demonstrated that during G2/M transition PLK1 directly interacts with and phosphorylates the mitotic transcription factor forkhead box protein M1 (FoxM1), activating its transcriptional activity.4 PLK1-mediated regulation of FoxM1 controls a large array of G2/M target genes, which are required for timely mitotic entry and progression.4 Previous studies have shown that loss of PLK1 expression can induce pro-apoptotic pathways and inhibit growth;5,6 however, the underlying mechanisms are not completely understood.
FOX-family transcription factors are DNA-binding proteins that are characterized by a conserved 110-amino acid winged-helix DNA binding domain.7 FOX genes are grouped into 19 subclasses (FOXA–FOXS) based upon homology with this region.8 FOXO factors belong to the “O” (“other”) class of the FOX superfamily, which include FOXO1 (previously known as FKHR), FOXO3a (previously known as FKHRL1), FOXO4 (previously known as AFX), and FOXO6 in mammals.9 Mounting evidence suggests that FOXO transcription factors are at the interface of crucial cellular processes, orchestrating programs of gene expression that regulate apoptosis, cell cycle progression, and oxidative-stress resistance.10 FOXO proteins induce cell cycle arrest at G1 by modulating the expression of the cyclin-dependent kinase (CDK) inhibitors p27KIP1 and p21WAF1, the retinoblastoma protein-related protein p130, and cyclin D1 and D211-15 and induce cell cycle arrest at G2 by modulating the expression of GADD45.16 Activation of FOXO proteins upregulates pro-apoptotic genes, such as Bim, Fas ligand (FasL), IGF binding protein-1 (IGFBP1), NIP3, tumor necrosis factor-related apoptosis-inducing ligand (TRAIL), and legumain.17-19
The importance of FOXO proteins is reflected by the fact that they are tightly regulated by several signaling pathways that control diverse biochemical processes to ensure that the transcription of specific target genes is responsive to environmental conditions. Regulation of subcellular localization and transcriptional activity is primarily achieved by posttranslational modifications, including phosphorylation, acetylation, and ubiquitination.20 Several phosphorylation sites in FOXO proteins have been identified. Akt-mediated phosphorylation of FOXO in response to insulin or growth factors is an important form of FOXO1 regulation. Phosphorylation of FOXO proteins (with the exception of FOXO6) at 3 conserved residues results in the export of FOXO factors from the nucleus to the cytoplasm, thereby inhibiting FOXO-dependent transcription.21-24
FOXO1 is the most abundant protein in the FOXO family. Although most of the evidence in the literature supports a role of FOXO1 in delaying G1/S transition, recent findings suggest that FOXO1 and its subfamily members may also be involved in the late phases of the cell cycle, including mitosis. FOXO3a mediates the transition from M to G1.25 Liu et al. report that CDK1 (the main regulator of G2/M transition) phosphorylates FOXO1 at serine 249, resulting in the inhibition of FOXO1 activity and the abolishment of the inhibitory effect of FOXO1 on cell growth and survival.26 Furthermore, expression of active FOXO1 causes a delay at G2/M.26 CDK1 has been reported to phosphorylate FOXO1 in cycling cells and postmitotic neurons.27 Recently, it has been shown that FOXO1 is responsible for the growth arrest at G2/M phase induced by Aurora A kinase inhibition in hepatocellular carcinoma cells.28 Together, these findings suggest that FOXO1 plays a role in the late stages of the cell cycle; however, the detailed functions and molecular mechanisms remain to be determined.
Interestingly, a genome wide RNAi screen of kinases and phosphatases to find regulators of dFoxO activity in Drosophila S2 cells identified Plks as a dFoxO regulator.29 This finding suggests that PLK1’s role in mitosis may also involve the regulation of FOXO1 in mammalian cells. In this report, we show that PLK1 binds to and phosphorylates FOXO1, which results in the nuclear exclusion of FOXO1 and, consequently, leads to the inhibition of FOXO1’s transcriptional activity in the late phases of the cell cycle. Enforced expression of FOXO1 induces pro-apoptotic pathways and delays G2/M transition, which can be abrogated by PLK1-mediated phosphorylation. Therefore, our results suggest that PLK1 inhibits the anti-proliferative functions of FOXO1 to ensure timely entry into and progression through G2/M.
Results
PLK1 interacts with FOXO1
A genome-wide RNAi screen of kinases and phosphatases identified Plks as a potential dFoxO regulator in Drosophila S2 cells.29 Thus, we examined whether PLK1 regulates FOXO1 in mammalian cells. We first investigated whether PLK1 interacts with FOXO1. We were able to co-immunoprecipitate ectopically expressed FOXO1 with endogenous PLK1 in 293T cells (Fig. 1A). Furthermore, we detected endogenous FOXO1/PLK1 complex in HeLa cells (Fig. 1B). Importantly, FOXO1 bound to PLK1 to a greater extent in G2/M-phase cells than in asynchronous cells (Fig. 1C), suggesting that PLK1-mediated regulation of FOXO1 occurs mainly during G2/M. To determine whether FOXO1 binds directly to PLK1, in vitro GST pull-down assays were performed using in vitro translated FOXO1 and GST-fused PLK1 or GST alone. In vitro translated FOXO1 associated with GST-fused PLK1, but not with GST (Fig. 1D), indicating that PLK1 binds directly to FOXO1.
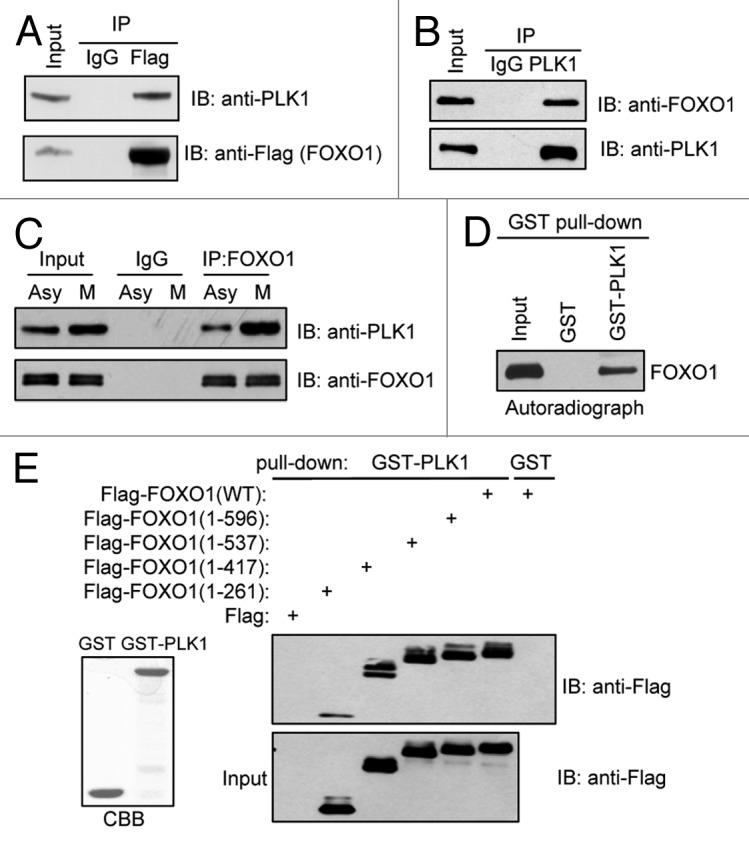
Figure 1. PLK1 interacts with FOXO1 in vitro and in vivo. (A) 293T cells were transfected with constructs encoding Flag-tagged FOXO1, and co-immunoprecipitation assays were performed. (B) Cell extracts were prepared from HeLa cells and subjected to immunoprecipitation using anti-PLK1 antibodies. (C) Cell extracts were prepared from asynchronous (Asy) or mitotic (M) HeLa cells and subjected to immunoprecipitation using anti-FOXO1 antibodies. (D) In vitro translated FOXO1 was used in a pull-down assay with either GST or GST-PLK1 immobilized on agarose beads. (E) 293T cells were transfected with plasmids encoding Flag-tagged C-terminal truncated FOXO1. Cell lysates were subjected to pull-down assays using beads coated with GST-PLK1. Beads coated with GST alone were used as a negative control. Loading controls are shown on the left (CBB, Coomassie blue staining).
FOXO1 proteins contain a forkhead DNA-binding domain (FKD) in the middle region and a transactivation domain (TAD) at the C terminus.30 We used a series of Flag-tagged C-terminal-truncated FOXO1 constructs31 to determine the region(s) of FOXO1 that is responsible for its interaction with PLK1. 293T cells were transfected with the deletion mutants and subjected to GST pull-down assays (Fig. 1E). Only the smallest FOXO1 N-terminal fragment (residues 1–261) showed much weaker binding to PLK1 than wild-type (WT) FOXO1, suggesting that the region of residues 262–417 and, to a lesser extent, the N-terminus (residues 1–261), are involved in FOXO1’s interaction with PLK1 (Fig. 1E).
PLK1 phosphorylates FOXO1 in the late phases of the cell cycle
To determine whether PLK1 directly phosphorylates FOXO1, we performed an in vitro kinase assay using GST–FOXO1 fusion proteins as substrates, and constitutively active (TD) PLK1 and kinase-defective (KD) PLK1 as kinase sources. Since initial priming phosphorylation of PLK1 substrates is required for the recruitment and subsequent activation of PLK1, we used PLK1 TD, instead of PLK1 WT, to “skip” the priming step. Notably, FOXO1 was phosphorylated by PLK1 TD, but not by the PLK1 KD (Fig. 2A). We next sought to map the major PLK1 phosphorylation sites on FOXO1. A series of FOXO1 deletion mutants were tested for their ability to be phosphorylated by PLK1 in vitro (Fig. 2B).31 The FOXO1 fragments FO1–1 (residues 1–167) and FO1–3 (residues 211–419) showed strong phosphorylation by PLK1 (Fig. 2C). We next generated smaller fragments of FO1–1, as shown in Figure 2D. Excitingly, only fragment FO1–1b showed strong phosphorylation by PLK1 (Fig. 2E). We then mutated serine and/or threonine residues within that region to identify the target residue(s). Mutation of serine 75 to alanine abolished the PLK1-mediated phosphorylation of FO1–1b (Fig. 2F), suggesting the serine 75 is the major PLK1 phosphorylation site on the FO1–1 fragment. To identify phosphorylation sites on the FO1–3 fragment, single- and double-site mutants were generated, and we examined their ability to be phosphorylated by PLK1 in vitro. As shown in Figure 2G, single mutation of serine 218 or serine 329 reduced PLK1-dependent phosphorylation of FOXO1, whereas phosphorylation by PLK1 was almost abolished when the S218A/S329A double mutant was used. Taken together, we have successfully identified all of the major PLK1 phosphorylation sites (serein 75, serine 218, and serine 329) on FOXO1. Importantly, all 3 PLK1-dependent phosphorylation sites, as well as their surrounding amino acids, are highly conserved among vertebrates (Fig. 2I), suggesting that phosphorylation of these sites may have an evolutionarily conserved role in regulating FOXO1 activity.

Figure 2. PLK1 phosphorylates FOXO1 in vitro. (A) PLK1 phosphorylates FOXO1 in vitro. Bacterially expressed full-length FOXO1 was subjected to in vitro kinase assays. Constitutively active PLK1 (TD) or a kinase-defective (KD) PLK1 were purified from insect cells and used as kinase sources. Loading controls are shown in the bottom panel. (B) Schematic diagram of the 5 deletion mutants of GST-FOXO1, FO1–1 to FO1–5. The forkhead DNA binding domain (FKD), nuclear localization signal domain (NLS), nuclear exclusion signal domain (NES), and transactivation domain (TAD) are indicated. (C) In vitro kinase assays using GST-fused FOXO1 deletion mutants as described in (B) as substrates (upper panel). Equal loading of kinase substrates is indicated by CBB (lower panel). (D) Schematic diagram of the 3 deletion mutants of GST-FO1–1, FO1–1a to FO1–1c. (E) In vitro kinase assays using GST-fused FO1–1 deletion mutants as substrates (upper panel). Equal loading of kinase substrates is indicated by CBB (lower panel). (F) FO1–1b segments harboring single mutation were used as substrates for in vitro kinase assay. (G) FO1–3 segments harboring single or double mutations were used as substrates for in vitro kinase assay. (H) 293T cells were transfected with plasmids encoding Flag-tagged full-length FOXO1 wild-type (WT), or mutant (S218A, S329A, or S218A/S329A). Cell lysates were subjected to pull-down assays using beads coated with GST-PLK1. Beads coated with GST alone were used as a negative control. (I) Comparison of serine 75, 218, and 329 motifs in the FOXO1 coding sequence in multiple species. Three major phosphorylation sites are highlighted.
Notably, serine 218 and serine 329 are within the regions of FOXO1 that interact with PLK1 (Fig. 1E). When we mutated these 2 sites to alanine, the interaction between FOXO1 and PLK1 was abolished (Fig. 2H). PLK1 phosphorylates these 2 serine residues on FOXO1, suggesting that PLK1 serves as a priming kinase to create docking sites at serine 218 and 329 on FOXO1, allowing subsequent binding. In addition to self-priming, some of PLK1 substrates are subject to non-self-priming by CDK1.3 However, mutation of the CDK1 phosphorylation site at serine 249 on FOXO1 to alanine (to prevent phosphorylation) showed no effect on its binding to PLK1, suggesting that serine 249 is not a docking site on FOXO1, and that CDK1 is not a priming kinase for FOXO1 (data not shown.)
We generated phospho-specific anti-FOXO1 antibodies that recognize phosphorylated serine 75 and serine 329, respectively, and subsequently validated their specificity. We mutated FOXO1 serine 75 and serine 329 to alanine to abrogate phosphorylation at these residues. As shown in Figure 3A, the anti-p75 antibody immunoreacted with WT FOXO1, but not the FOXO1 S75A mutant. Similarly, the anti-p329 antibody recognized WT, but not the S329A mutant (Fig. 3B). Furthermore, neither antibody detected λ-phosphatase-treated WT FOXO1. Thus, the anti-p75 and anti-p329 antibodies are highly specific and only recognize their corresponding targets.

Figure 3. PLK1-mediated FOXO1 phosphorylation mainly occurs at the late stages of the cell cycle. (A and B) Characterization of the anti-phospho-FOXO1-specific antibodies, anti-p75 (A) and anti-p329 (B). 293T cells were transfected with the indicated plasmids. Ectopically expressed FOXO1 proteins were immunoprecipitated with anti-Flag antibody. Immunoprecipitates were mock treated or treated with lambda phosphatase and then subjected to immunoblot with the indicated antibodies against phosphorylated serine residues of FOXO1. (C) PLK1-mediated FOXO1 phosphorylation mainly occurs in the late phases of the cell cycle. HeLa cells were synchronized at G1 (by treating with 0.5 mM mimosine for 24 h), S (by treating with 4 mM HU for 40 h), or G2/M (by treating with 100 ng/mL nocodazole for 16 h). The phosphorylation status of FOXO1 was examined by western blot using anti-p75 and anti-p329 antibodies. Total FOXO1 was detected with anti-FOXO1 antibodies. The expression of cell cycle markers were determined by western blot using anti-cyclin B1, -cyclin A, and -pH3 antibodies. Cell cycle distributions were determined by flow cytometry. (D) HeLa cells were infected with PLK1shRNA lentivirus and synchronized at G2/M using nocodazole treatment. Cell extracts were subjected to western blot using the indicated antibodies. The phosphorylation status of FOXO1 was examined by western blot using anti-p75, anti-p329 FOXO1 antibodies. Total FOXO1 was detected with anti-FOXO1 antibody. (E) HeLa cells were synchronized by a thymidine–aphidicolin arrest and release protocol, and cells were collected at the indicated time points after release. Cell extracts were subjected to western blot using the indicated antibodies. The results were from duplicate blots. Cell cycle distributions were determined by flow cytometry.
We next used the phospho-specific antibodies to determine whether FOXO1 is phosphorylated in vivo and at which cell cycle phase these phosphorylation events occur. We arrested cells at G1 phase (by mimosine treatment), S phase (by hydroxyurea [HU] treatment), or G2/M phase (by nocodazole treatment). In agreement with our results in Figure 1C, phosphorylation of endogenous FOXO1 at serine 75 and serine 329 was easily detected in G2/M, but not in the other (G1 and S) phases (Fig. 3C), indicating that these phosphorylation events mainly occur in the late phases of the cell cycle. To assess whether PLK1 is required for these phosphorylation events, we depleted endogenous PLK1 using PLK1 shRNA and synchronized the cells at G2/M by nocodazole treatment. Compared with control cells (with control shRNA knockdown), PLK1 depletion led to a dramatic reduction of FOXO1 phosphorylation at serine 75 and serine 329 (Fig. 3D), suggesting that PLK1 phosphorylates FOXO1 in vivo.
We next examined the kinetics of PLK1-mediated phosphorylation of FOXO1 during the late stages of the cell cycle progression. HeLa cells were synchronized at the G1/S boundary by a double thymidine/aphidicolin block and sampled at various time points after release. Cell extracts were subjected to western blot analysis using the indicated antibodies. Phosphorylation of FOXO1 at serine 329 was initiated as early as the G2 phase, whereas phosphorylation at serine 75 occurred later, mainly at M phase (Fig. 3E), suggesting that FOXO1 undergoes an initial priming phosphorylation, which allows its subsequent phosphorylation; PLK1 is responsible for both phosphorylation events. Interestingly, CDK1-mediated phosphorylation of FOXO1 at serine 249 started to decline upon entry into M phase, at which time PLK1-mediated phosphorylation (at serine 75) peaks (Fig. 3E).
The transcriptional activity of FOXO1 is inhibited by PLK1-mediated phosphorylation
FOXO1 functions primarily as a transcription factor. To study the functional significance of the PLK1-mediated interaction and phosphorylation of FOXO1, we tested whether PLK1 regulates FOXO transcriptional activity using a FOXO1-luciferase reporter plasmid. Intriguingly, PLK1 WT, but not PLK1 KD, significantly attenuated the transcriptional activity of FOXO1 in a dose-dependent manner (Fig. 4A), suggesting a direct inhibition of FOXO1’s activity by PLK1-dependent phosphorylation. Western bot analysis revealed that PLK1 had no effect on the abundance of FOXO1 protein (Fig. 4A).

Figure 4. The transcriptional activity of FOXO1 is inhibited by PLK1-mediated phosphorylation. (A) PLK1 inhibits FOXO1 transcriptional activity. U2OS cells were co-transfected with the plasmid encoding FOXO1, the reporter plasmid FOXO1-responsive IGFBP-1 and FasL promoter luciferase reporter constructs in conjunction with SV40-Renilla to control for transfection efficiency, and increasing amounts of plasmids (20, 40, 60 ng) encoding either wild-type (WT) or kinase-defective (KD) PLK1. Luciferase activities were measured 24 h after transfection. Luciferase levels were normalized to Renilla luciferase activity. The data were expressed as percentage of FOXO1 activity (mean s.d. of 3 separate experiments in triplicate). FOXO protein levels were measured by immunoblotting using anti-FOXO1 antibodies. *P < 0.05 vs. control. (B) HeLa cells were transfected with the FOXO1 reporter plasmid and plasmids encoding for either empty vector, Flag-tagged FOXO1 WT, or a mutant (S75A, S75D, or S218A/S329A). Luciferase activities were measured 24 h after transfection. *P < 0.05 vs. WT. (C) The mRNA of FOXO1 target genes (GADD45, Bim, and FasL) in U2OS cells expressing FOXO1 WT or mutants were analyzed by real-time RT-PCR as described in “Materials and Methods”. *P < 0.05 vs. control. (D) HeLa cells were transfected with the indicated plasmids. Dual luciferase assays were performed 24 h after transfection. Cell lysates were subjected to western blot analysis. * and **, P < 0.05. (E) HeLa cells were transfected with the indicated plasmids and treated for 30 min with 20 μM wortmannin before cell lysates were prepared. The activity of Akt in these cells was evaluated by western blot using anti-phospho-AKT (serine 473) antibody. *P < 0.05.
To investigate whether PLK1-mediated phosphorylation of FOXO1 inhibits the transcriptional activity of FOXO1, we generated a series of FOXO1 mutants by mutating PLK1 phosphorylation sites to alanine or aspartate—to either block or mimic PLK1 phosphorylation—and examined their effects on FOXO1 transcriptional activity. Compared with WT FOXO1, the unphosphorylatable mutant FOXO1-S75A showed significant enhancement in transcriptional activity, whereas the phospho-mimicking mutant FOXO1-S75D exhibited decreased transcriptional activity (Fig. 4B). When the interaction between FOXO1 and PLK1 was abolished by mutating the PLK1 priming sites at serine 218 and serine 329 to alanine, the transcriptional activity of the FOXO1 significantly increased compared with WT FOXO1 (Fig. 4B). To provide an in vivo correlation, we next examined the expression of endogenous FOXO1 target genes in these cells (Fig. 4C). Indeed, consistent with previous reports,17-19 ectopic expression of WT FOXO1 induced the expression of its target genes including GADD45, FasL, and Bim. The mRNA levels of these target genes were further upregulated in cells expressing the FOXO1-S75A mutant or the FOXO1-S218A/S329A mutant. In contrast, the FOXO1-S75D mutant had no effect on expression of these target genes (Fig. 4C). These results suggest that PLK1-dependent phosphorylation of FOXO1 has an inhibitory effect on FOXO1’s transcriptional activity.
It has been well established that Akt is a key regulator of FOXO1 function.22 There is frequent interplay of this canonical regulatory pathway with FOXO1 regulatory mechanisms.32 We next assessed whether the PLK1-mediated regulation of FOXO1 occurs independently of the Akt-mediated regulation of FOXO1. We co-transfected FOXO1-A3 (a constitutively active mutant of FOXO1 that harbors 3 alanine substitutions at the Akt-phosphorylation sites20) along with PLK1 WT, TD, or KD mutant. The transcriptional activity of FOXO1-A3 was attenuated by PLK1 WT and, to a greater extent, by PLK1 TD (Fig. 4D). PLK1 KD had no effect on the transactivation of FOXO1-A3 (Fig. 4D). Furthermore, while wortmannin, a potent and specific inhibitor of PI3K, upregulated the transcriptional activity of FOXO1 WT, it failed to abrogate the inhibitory effect of PLK1 TD on FOXO1 WT (Fig. 4E). These results suggest that PLK1 inhibits the transcriptional activity of FOXO1 in an Akt-independent manner.
PLK1-mediated phosphorylation of FOXO1 leads to its nuclear exclusion
To investigate whether nuclear exclusion plays a role in FOXO1 transcriptional repression following PLK1-mediated phosphorylation, we performed immunofluorescence staining to visualize the subcellular localization of Flag-tagged FOXO1 WT and FOXO1 phosphorylation mutants in cells. Interestingly, the majority of the phosphorylation-resistant mutant FOXO1-S75A localized to the nucleus, while ~80% of FOXO1 WT resided in the cytoplasm (Fig. 5A and B). In contrast, the phospho-mimicking mutant FOXO1-S75D was mainly retained in the cytoplasm (Fig. 5A and B). Moreover, when the PLK1 docking sites on FOXO1 were abolished by mutating serine 218 and serine 329 to alanine, nuclear localization of FOXO1 resulted (Fig. 5B). These results were further confirmed by subcellular fractionation assays (Fig. 5C). Together, these results suggest that PLK1-mediated phosphorylation of FOXO1 induces translocation to the cytosol, thereby impairing FOXO1’s nuclear transcriptional activity.
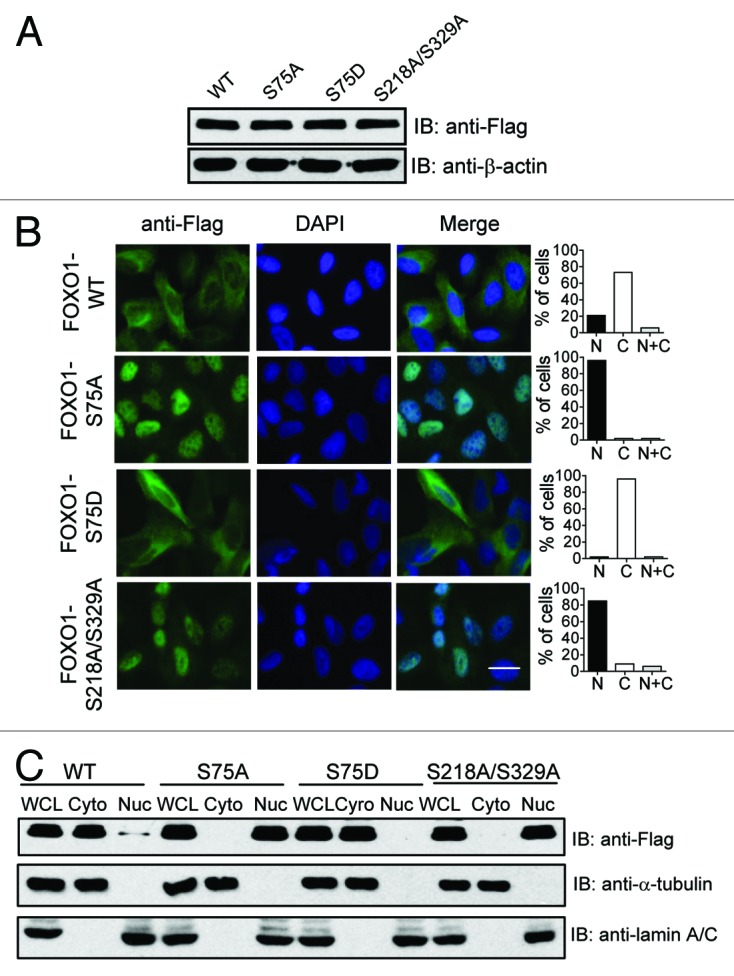
Figure 5. The effect of PLK1-mediated phosphorylation of FOXO1 on its subcellular localization. (A) U2OS cells were transfected with Flag-tagged FOXO1 wild-type (WT), or mutant (S75A, S75D, or S218A/S329A). Exogenous FOXO1 expression was detected by western blot using anti-Flag antibody. (B) The cellular localization of ectopically expressed FOXO1 WT or a FOXO1 phosphorylation mutant (S75A, S75D, or S218/S329A) was examined by immunofluorescence staining using anti-Flag antibody. Quantification of a representative experiment is shown in the bar graph. Similar results were obtained from 3 independent experiments. The scale bar is 10 μm. (C) An additional set of samples were subjected to subcellular fractionation. The levels of exogenous FOXO1 in nuclear and cytoplasmic fractions were determined by immunoblotting with anti-Flag antibody. The relative purity of the nuclear and cytoplasmic fractions was confirmed by sequential probing for the nuclear marker lamin A/C and the cytoplasmic marker α-tubulin.
The pro-apoptotic function of FOXO1 is inhibited by PLK1-mediated phosphorylation
FOXO1 is actively involved in promoting apoptosis by inducing the expression of pro-apoptotic factors such as Bim, FasL, NIP3, and legumain.33 To investigate whether PLK1-mediated phosphorylation of FOXO1 attenuates FOXO1-induced cell death, we transiently transfected U2OS cells with Flag-tagged FOXO1 WT or a FOXO1 phosphorylation mutant. The percentage of apoptotic cells was measured using flow cytometry after dual staining cells with Annexin V-FITC and propidium iodide (PI). Consistent with previous studies,34 ectopic expression of WT FOXO1 induced apoptosis (~14%) (Fig. 6A and B). The pro-apoptotic function of FOXO1 was further enhanced (almost 2-fold) by blocking PLK1 phosphorylation at serine 75 (S75A) (Fig. 6A and B). In contrast, a phospho-mimicking mutation at serine 75 (S75D) almost abolished the pro-apoptotic functions of FOXO1 (Fig. 6A and B). Results from 3 independent experiments are shown in Figure 6B. Western blot analysis of poly (ADP-ribose) polymerase (PARP) protein cleavage confirmed the induction of apoptosis (Fig. 6C). In agreement with our apoptotic results, forced expression of FOXO1 WT, S75A, or S218A/S329A induced the expression of Bim, a pro-apoptotic gene known to be a direct target gene of FOXO1 (Fig. 6C). However, such upregulation was not observed in cells harboring the FOXO1-S75D mutant (Fig. 6C). Together, these data suggest that PLK1-mediated phosphorylation of FOXO1 negatively regulates the pro-apoptotic function of FOXO1.

Figure 6. The pro-apoptotic function of FOXO1 is inhibited by PLK1-mediated phosphorylation. (A) HeLa cells were transfected with the indicated plasmids. Forty-eight hours post-transfection, cells were harvested, stained with Annexin V and PI solution, and then analyzed by flow cytometry. (B) Results from 3 independent experiments are shown. *P < 0.05 vs. control. (C) An additional set of samples was analyzed by western blot analysis for expression of Bim, PARP, and Flag-tagged FOXO1.
PLK1-mediated phosphorylation overrides FOXO1-induced delay in the late phases of the cell cycle
FOXO1 is a negative regulator of the cell cycle. FOXO1 negatively regulates cell cycle progression at G1 by modulating p27KIP1 and cyclin D1 and D2.14 It has recently been reported that forced expression of FOXO1 causes a delay in G2/M transition,26 suggesting that FOXO1 negatively regulates the late phases of cell cycle progression. To smoothly progress through G2–M, such functions of FOXO1 need to be negatively regulated. We hypothesized that the inhibitory effect of FOXO1 on the late phases of cell cycle progression is relieved by PLK1-mediated phosphorylation. To test this hypothesis, we monitored cell cycle progression after releasing U2OS cells harboring FOXO1 WT or a FOXO1 phosphorylation mutant (S75A, S75D, or S218A/S329A) from a thymidine–aphidicolin double block (S-phase block). Flow cytometric analysis revealed that the majority (>50%) of cells expressing empty vector (pcDNA 3.1) or FOXO1-S75D mutant were at G2/M 7 h after release from the S-phase blockage (Fig. 7A and B). In contrast, 55–60% of cells expressing FOXO1 WT or FOXO1-S75A mutant reached G2/M at a later time point, around 9 h after the release (Fig. 7A and B). Results from 3 independent experiments are shown in Figure 7B.

Figure 7. PLK1-mediated phosphorylation of FOXO1 overrides FOXO1-induced delay in the late phases of the cell cycle. (A) FACS analysis of the effects of phosphorylation of FOXO1 on cell cycle progression. U2OS cell lines expressing Flag-tagged FOXO1 WT or a FOXO1 mutant (S75A, S75D, or S218AS329A) were synchronized at G1/S boundary by thymidine–aphidicolin double block, and released by culturing in aphidicolin-free medium. Cells were collected at different time points as indicated, fixed and stained with PI solution, and then analyzed by flow cytometry. (B) Results from 3 independent experiments are shown in a bar chart.
To determine whether the 4N cells are in G2 or M phase, we examined the phosphorylation of histone H3 on serine 10 (a mitotic marker) in above cells by immunofluorescence. As shown in Figure 8A and B, 7 h after releasing from G1/S boundary, the mitotic index was significantly lower in cells expressing FOXO1 WT, S75A, or S218A/S329A that in cells transfected with an empty vector. The mitotic index remained significantly lower 9 h after release, at which time the majority of cells had 4N DNA content (Fig. 8A and B), suggesting delayed entry into and progression through G2/M. Similar to results for FOXO1 WT, forced expression of FOXO1-S75A enhanced the level of endogenous GADD45 expression, a protein that has a critical function in inhibiting the transition from G2 to M phase (Fig. 8C). However, no such effect was observed in cells expressing FOXO1-S75D (Fig. 8C). Taken together, these data suggest that FOXO1 is an important negative regulator at the late phases of the cell cycle, and that PLK1-mediated phosphorylation of FOXO1 relieves this inhibitory effect to ensure normal cell cycle progression.

Figure 8. PLK1-mediated phosphorylation of FOXO1 attenuates FOXO1-induced G2 delay. (A) The effect of PLK1-mediated phosphorylation of FOXO1 on G2/M transition. U2OS cell lines expressing Flag-tagged FOXO1 WT or a FOXO1 mutant (S75A, S75D, or S218A/S329A) were synchronized at G1/S boundary by thymidine–aphidicolin double block, and released by culturing in aphidicolin-free medium. At 7 h and 9 h after release, immunofluorescence staining using anti-phospho-histone H3 serine 10 antibody (pH3; green) was performed to score mitotic cells. Three random fields were scored. Representative fields are shown. The scale bar is 10 μm. (B) Quantification results from 3 independent experiments were shown in bar graphs. Error bars indicate s.d. among 3 individual experiments. *P < 0.05 vs. control. (C) The expression of GADD45 protein was determined by western blot analysis.
Discussion
PLK1 is a master regulator of cell division and plays a key role in multiple steps of mitotic progression. The mechanisms of how a single protein executes so many functions have been actively investigated. It is well accepted that PBD-dependent recruitment of PLK1 to its substrates confers on PLK1 the ability to regulate various cell cycle events in an orderly temporal and spatial manner. Importantly, in an effort to understand how PLK1 functions as a global, multi-faceted regulator of cell division, we previously showed that PLK1 directly regulates FoxM1, which, in turn, controls the expression of a large array of G2/M regulators that are essential for timely mitotic entry and progression.4 This would suggest that global transcriptional regulation may contribute to multiple functions of PLK1. In our present study, we reveal another important layer of regulation by PLK1. We show that another member of the FOX family, FOXO1, is regulated by PLK1 in the late phases of the cell cycle. In contrast to FoxM1, PLK1-mediated phosphorylation negatively modulates FOXO1’s transcriptional activity, thereby inhibiting its anti-proliferative and pro-apoptotic function at G2/M. Therefore, PLK1 functions in an opposing manner—activating proliferative transcription factor (FoxM1) and inhibiting anti-proliferative transcription factor (FOXO1), both functions are required for proper cell proliferation.
PLK1 maximally accumulates in G2 and M phases, declines to a nearly undetectable level following mitosis and throughout G1 phase, and then begins to accumulate again during S phase, which is concomitant with the fluctuation of its enzymatic activity. Our results indicate that PLK1 binds to and phosphorylates FOXO1 at G2/M, at a time PLK1’s expression and activity peak. These data suggest that FOXO1 may have an important function at G2/M, which is subject to regulation by PLK1. Indeed, we show that ectopic expression of FOXO1 induces apoptosis and delays entry into and progression through G2/M. These anti-proliferative and pro-apoptotic effects were abrogated by PLK1-mediated phosphorylation, indicating that PLK1 functions as a negative regulator to keep FOXO1 inactive at the late phases of the cell cycle. Moreover, this regulatory pathway is independent of canonical Akt/FOXO1 signaling (Fig. 4D and E). This suggests that, unlike other kinases (such as casein kinase 1, which functions as a “fine-tuner” for FOXO1), PLK1, in parallel to Akt signaling, is another major FOXO1 regulator. Previous studies reported that serine 249 of FOXO1 is phosphorylated by CDK1, which abolishes the inhibitory effect of FOXO1 on cell growth and survival.26 In this study, we carefully examined the timing of CDK1-mediated phosphorylation, and showed that it mainly occurred at G2 and started to decline at G2/M transition (Fig. 3E). In contrast, PLK1-mediated phosphorylation (at serine 75) initiated at G2/M transition and sustained throughout the M phase (Fig. 3E). These complementary phosphorylation patterns suggest that FOXO1 is differentially regulated in the late phases of the cell cycle; initially by CDK1, and subsequently by PLK1.
Serine 329 on FOXO1 was previously identified as an in vivo phosphorylation site.35 Woods et al. also showed that phosphorylation of serine 329 was not modulated by stimulation with insulin-like growth factor or by transfection with 3-phosphoinosititide-dependent protein kinase-1.35 This is in line with our observation that PLK1 inhibits FOXO1’s transcriptional activity in an Akt-independent manner (Fig. 4D and E). However, the role of serine 329 phosphorylation in the regulation of FOXO1 has not been characterized.35 On the basis of results from in vitro experiments, it was suggested that DYRK1A phosphorylates serine 329 on FOXO1.35 Herein, we provide both in vitro and in vivo evidence supporting the notion that PLK1 phosphorylates FOXO1 at serine 329 (Figs. 2 and 3). We also demonstrate that serine 329 is one of the docking sites on FOXO1 responsible for binding to PLK1, and that this site plays an essential role in PLK1-mediated regulation of FOXO1. Our results suggest that in late G2 phase, FOXO1 is initially phosphorylated at serine 218 and serine 329 by PLK1, creating docking sites for the PBD of PLK1. This initial phosphorylation event is required for efficient PLK1 binding. Subsequently, the PBD of PLK1 binds to FOXO1 and further phosphorylates it at serine 75, leading to inhibition of FOXO1’s transcriptional activity.
In line with its paramount functions in cell division, PLK1 depletion is associated with a decrease in cell viability and the induction of apoptosis.5,6 However, the underlying mechanisms remain largely unknown. In this study, we demonstrate for the first time that PLK1 executes its anti-apoptotic function through negative regulation of FOXO1. In addition to abrogating cell cycle delay, PLK1-mediated phosphorylation of FOXO1 also overrides the pro-apoptotic functions of FOXO1 by inhibiting its transcriptional activity, and thereby blocking the transcription of its pro-apoptotic target genes, such as Bim and FasL. It is likely that PLK1 may also regulate other unidentified apoptotic regulators and/or pathways, which collectively suppress apoptotic pathways and promote survival. PLK1 depletion would relieve these inhibitory effects and consequently induce cell death.
Nuclear–cytoplasmic shuttling plays a key role in regulating FOXO1’s activity and function.20 We demonstrate that PLK1-mediated phosphorylation of FOXO1 regulates its cellular trafficking. Phosphorylation of FOXO1 at serine 75 or docking sites (serine 218 and serine 329) are sufficient for its nuclear exclusion. However, the underlying mechanisms of this nuclear–cytoplasmic shuttling remain to be determined. Chromosomal region maintenance protein 1 (CRM1) and 14-3-3 proteins are components of the nuclear transport machinery that have been shown to mediate FOXO1 transport from nucleus to the cytoplasm.22,36,37 We plan to examine the effects of PLK1-mediated phosphorylation on the recruitment of components of the nuclear transport machinery. Furthermore, multiple mechanisms may contribute to PLK-dependent suppression of transactivation by FOXO1. For instance, it has been reported that the Akt-phosphorylation site in the FKD of FOXO1 protein is linked to the regulation of DNA binding.38 Given that one of the FOXO1 docking sites, serine 218, is located in the FKD, it is tempting to speculate that PLK1-mediated priming and subsequent binding to FOXO1 interferes with its DNA binding, thereby inhibiting its transactivation. Future studies are warranted to address these questions.
In conclusion, we provide evidence that FOXO1 is a physiological substrate of PLK1. PLK1 negatively regulates FOXO1’s functions at the late phases of the cell cycle by directly interacting with and phosphorylating FOXO1, which, in turn, leads to nuclear exclusion and transactivation inhibition. The growth-inhibitory effect of FOXO1 (apoptosis and cell cycle arrest) is abrogated by PLK1-mediated regulation during the late phases of the cell cycle to ensure efficient execution of the mitotic program. This study not only reveals a novel and major regulatory mechanism of FOXO1, but also provides new insight into the molecular mechanism by which inhibition of PLK1 leads to growth arrest and cell death.
Materials and Methods
Plasmids, short hairpin RNA (shRNA), and chemicals
Flag-tagged WT FOXO1, the Akt-phosphorylation-resistant mutant of FOXO1 (FOXO1-A3, in which 3 Akt-phosphorylation sites [threonine 24, serine 256, and serine 319] were converted to alanine), and the luciferase reporter construct (3xIRS, which contains 3 copies of the FOXO response element in the promoter of the IGFBP1 gene) were kindly provided by K Guan.24 Expression vectors for the truncated forms of FOXO1 were kindly provided by H Huang (Mayo Clinic at Rochester).31,39 Plasmids for amino acid substitution mutants of Flag-tagged FOXO1-S75A (serine 75 was converted to alanine), FOXO1-S75D (serine 75 was converted to aspartate), and FOXO1-S218A/S329A (serine 218 and serine 329 were converted to alanine) were generated by PCR-based mutagenesis (Stratagene). Human full-length Plk1 WT, constitutively active (TD), and catalytically inactive (KD) mutants in the pRc/CMV vector was a gift from EA Nigg (Max Planck Institute of Biochemistry). Baculovirus encoding human Plk1 WT, TD, and KD mutants were generous gifts from RL Erikson (Harvard University). Lentivirus-based plasmids expressing shRNA to target PLK1 (pLKO.1-PLK1), mimosine, thymidine, aphidicolin, and nocodazole were purchased from Sigma.
Cell culture, transfection, and synchronization
HeLa and 293T cells were grown in RPMI 1640 medium (Invitrogen) supplemented with 10% fetal bovine serum, 100 U/mL penicillin, and 100 mg/mL streptomycin. U2OS cells were grown in Dulbecco modified Eagle medium (DMEM; Invitrogen) containing 10% fetal bovine serum, 100 U/mL penicillin, and 100 mg/mL streptomycin. Transient transfection of U2OS, HeLa, and 293T cells was performed with Lipofectamine 2000 according to the manufacturer’s instructions. To establish stable U2OS-derived cell lines, transfected cells were diluted and grown in media containing 800 μg/mL G418 for 14 d. Individual clones were then selected and maintained in media containing 400 μg/mL G418. For G1-phase arrest, cells were treated with 0.5 mM mimosine for 24 h; for S-phase arrest, cells were treated with 4 mM hydroxyurea for 40 h; for G2/M arrest, cells were treated with 100 ng/mL nocodazole for 16 h. To synchronize HeLa and U2OS cells at G1/S boundary, cells were treated with 2 mM thymidine for 18 h, released for 10 h, and then treated with 2 μg/mL aphidicolin for 18 h.
Antibodies, immunoblotting, and immunoprecipitation
Anti-Cyclin B1, anti-Cyclin A (Santa Cruz Biotechnology), anti-FOXO1, anti-GADD45 (Cell signaling) antibodies were used at dilutions of 1:1000. Anti-Flag, anti-β-actin (Sigma) antibodies were used at a dilution of 1:5000. Anti-phospho-Histone H3 (Upstate) was used at a dilution of 1:3000. Rabbit polyclonal antibodies recognizing phosphorylated serine 75 (anti-p75), phosphorylated serine 329 (anti-p329) of FOXO1 were raised by immunizing rabbits with the phosphorylated peptides DFMSNLpSLLEESEDC and TISGRLpSPIMTEC, respectively, and affinity-purified using their corresponding peptide columns. Cells were lysed in NETN lysis buffer (150 mM NaCl, 1 mM EDTA, 20 mM Tris pH 8, 0.5% NP-40) containing protease and phosphatase inhibitors for 20 min at 4 °C. Cell lysates were centrifuged for 10 min at 4 °C. For immunoprecipitation or co-immunoprecipitation experiments, cleared lysates (1–2 mg protein) were immunoprecipitated with the indicated polyclonal antibodies and protein A-Sepharose for 1 h. For phosphatase-treated sample, FOXO1 immunoprecipitates were treated with 400 U of λ phosphatase in the accompanying buffer (New England Biolabs) for 1 h at 30 °C. Proteins were separated on 7.5% SDS-PAGE, transferred to Immobilon P membranes and immunoblotted with the indicated antibodies.
Subcellular protein fractionation
Cells were lysed in hypotonic buffer (10 mM HEPES-KOH, 1.5 mM MgCl2, 10 mM KCl, 0.5 mM DTT, 0.2 mM PEFA 1023, pH 7.9, 0.5% NP-40). Cell lysates were centrifuged for 10 s at 16 000 g at 4 °C. The supernatants were collected (cytoplasmic extracts), and the pellets were washed twice with hypotonic buffer, and lysed with high-salt buffer (450 mM NaCl, 1 mM PMSF, 50 mM Tris pH 7.4, 0.2 mM Na3VO4, 5 mM β-glycerophosphate, 20% glycerol, 2 mM DTT, 1% NP-40), following incubation for 10 min on an end-over-end rotator at 4 °C. Cell lysates were centrifuged for 15 min at 16 000 g at 4 °C, and the supernatants were collected (nuclear extracts).
GST pull-down assays
Human FOXO1 was transcribed and translated in vitro using TNT quick-coupled transcription/translation system (Promega). GST-PLK1 or GST control bound to glutathione–sepharose resin was incubated with in vitro translated and labeled FOXO1 protein for 1 h at 4 °C, and washed complexes were boiled in SDS-PAGE sample buffer. Alternatively, glutathione-sepharose resin-coupled GST-PLK1 or the GST control was incubated with cell lysates containing Flag-tagged WT or mutant FOXO1 for 1 h at 4 °C. The resins were washed 4 times using NETN buffer. Resin-bound complexes were eluted by boiling, separated by SDS-PAGE, and analyzed by autoradiography or western blotting.
Immunostaining
Cells grown on coverslips were fixed for 15 min with 3% paraformaldehyde solution and then permeabilized with 0.5% Triton X-100 for 5 min. Slides were incubated with primary antibodies for 20 min at 37 °C, and then Fluorescein isothiocyanate-conjugated secondary antibody (Jackson ImmunoResearch) for 20 min at 37 °C. All antibodies were diluted in 5% goat serum. Cells were counterstained with 4', 6-diamidino-2-phenylindole (DAPI) dye (EMD Millipore) for 30 s.
Protein kinase production and kinase assays
Human PLK TD and PLK1 KD were expressed in the baculovirus/insect cell system. GST-FOXO1 and its mutant proteins were expressed in Escherichia coli BL21 strain. Kinase and substrates (FOXO1 proteins) were incubated in kinase buffer (20 mM Hepes, pH 7.4, 150 mM KCl, 10 nM MgCl2, 1 mM EGTA, 0.5 mM dithiothreitol, 5 mM NaF, 10 μM ATP, 5 μCi γ-32P) for 30 min at 30 °C. Reactions were stopped by the addition of SDS sample buffer. Then samples were heated for 5 min to 95 °C before analysis by SDS-PAGE and autoradiography.
Luciferase assays
U2OS and HeLa cells were transfected with the indicated plasmids using Lipofectamine 2000 according to manufacturer’s instruction. Luciferase activity was determined 24 h after transfection using the dual-luciferase kit (Promega). Luciferase levels were normalized to Renilla luciferase activity. The promoter activity resulting from transfection with FOXO1 WT was set at 100%, and relative luciferase activity is shown. Experiments were performed in triplicate, and statistical analysis was performed using Microsoft Excel.
Cell cycle analysis by flow cytometry
Cells were collected, washed with PBS, and fixed with ice-cold 70% ethanol for at least 1 h. Cells were washed twice in PBS and treated for 30 min at 37 °C with RNase A at 5 μg/mL and PI at 50 μg/mL, and analyzed on a FACScan flow cytometer (Becton Dickinson). The percentage of cells in different cell cycle phases was calculated using ModFit LT for Mac (BD Biosciences).
Measure of apoptosis by annexin-V and PI staining
HeLa cells were transiently transfected with indicated plasmids. At 48 h, the entire cell population was subjected to double staining for FITC-annexin V and PI using FITC-annexin V apoptosis detection kit (BD Biosciences) and analyzed by flow cytometry for apoptotic events according to the manufacturer’s instructions.
Real-time quantitative PCR
Total RNA extraction and real-time PCR analysis were performed as described previously.40 Briefly, total RNA was extracted from cells expressing FOXO1 WT or a FOXO1 mutant using TRIzol reagent (Invitrogen) according to the manufacturer’s instructions. A 2-step real-time PCR was performed using cDNA prepared from RNA using a Superscript III First-Strand cDNA Synthesis Kit (Invitrogen) and a SYBR Green PCR Master Mix (Applied Biosystems) on an ABI 7900 instrument following the manufacturer’s instructions. The thermal cycling conditions were 50 °C for 2 min, 95 °C for 10 min, and 40 cycles of 95 °C for 15 s, then 60 °C for 1 min. The fold change in expression levels (using GAPDH as internal control) was determined by a comparative Ct method using the Equation 2−ΔΔCt, where Ct is the threshold cycle of amplification. The sequences of PCR primers were as following: Bim forward: tgccaggcct tcaacca, reverse: gttcagcctg cctcatgga; FasL forward: gcagcccttc aattacccat, reverse: cagaggttgg acagggaaga a; GADD45 forward: tgctcagcaa agccctgagt, reverse: gcaggcacaa caccacgtta; GAPDH forward: acctgacctg ccgtctagaa, reverse: tccaccaccc tgttgctgta.
Statistics
All experiments were performed at least 3 times in triplicates for each group. The results are presented as the mean ± s.d. Statistical significance was determined using Student t test, and the level of significance was set at P < 0.05.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This work was supported by a grant from the US Department of Defense Prostate Cancer Research Program (PC094435 to ZF) and an NCI Cancer Center Support Grant (P30CA016059 to VCU Massey Cancer Center).
Glossary
Abbreviations:
- PLK1
Polo-like kinase 1
- FoxM1
forkhead box protein M1
- FOXO1
forkhead box protein O1
- CDK
cyclin-dependent kinase
- PBD
Polo-box domain
- FasL
Fas ligand
- IGFBP1
IGF binding protein-1
- TRAIL
tumor necrosis factor-related apoptosis-inducing ligand
- FKD
forkhead DNA-binding domain
- WT
wild-type
- KD
kinase defective
- TD
constitutively active
- TAD
transactivation domain
- HU
hydroxyurea
- PI
propidium iodide
- PARP
poly (ADP-ribose) polymerase
- DMEM
Dulbecco modified Eagle medium
- DAPI
4’,6-diamidino-2-phenylindole
- CBB
coomassie blue staining
- NLS
nuclear localization signal domain
- NES
nuclear export signal domain
- shRNA
short hairpin RNA
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/27727
References
- 1.Barr FA, Silljé HH, Nigg EA. Polo-like kinases and the orchestration of cell division. Nat Rev Mol Cell Biol. 2004;5:429–40. doi: 10.1038/nrm1401. [DOI] [PubMed] [Google Scholar]
- 2.Nigg EA. Mitotic kinases as regulators of cell division and its checkpoints. Nat Rev Mol Cell Biol. 2001;2:21–32. doi: 10.1038/35048096. [DOI] [PubMed] [Google Scholar]
- 3.Park JE, Soung NK, Johmura Y, Kang YH, Liao C, Lee KH, Park CH, Nicklaus MC, Lee KS. Polo-box domain: a versatile mediator of Polo-like kinase function. Cell Mol Life Sci. 2010;67:1957–70. doi: 10.4161/cc.27727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fu Z, Malureanu L, Huang J, Wang W, Li H, van Deursen JM, Tindall DJ, Chen J. Plk1-dependent phosphorylation of FoxM1 regulates a transcriptional programme required for mitotic progression. Nat Cell Biol. 2008;10:1076–82. doi: 10.1038/ncb1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cogswell JP, Brown CE, Bisi JE, Neill SD. Dominant-negative Polo-like kinase 1 induces mitotic catastrophe independent of cdc25C function. Cell Growth Differ. 2000;11:615–23. [PubMed] [Google Scholar]
- 6.Mundt KE, Golsteyn RM, Lane HA, Nigg EA. On the regulation and function of human Polo-like kinase 1 (PLK1): effects of overexpression on cell cycle progression. Biochem Biophys Res Commun. 1997;239:377–85. doi: 10.1006/bbrc.1997.7378. [DOI] [PubMed] [Google Scholar]
- 7.Kops GJ, de Ruiter ND, De Vries-Smits AM, Powell DR, Bos JL, Burgering BM. Direct control of the Forkhead transcription factor AFX by protein kinase B. Nature. 1999;398:630–4. doi: 10.1038/19328. [DOI] [PubMed] [Google Scholar]
- 8.Kaestner KH, Knochel W, Martinez DE. Unified nomenclature for the winged helix/forkhead transcription factors. Genes Dev. 2000;14:142–6. [PubMed] [Google Scholar]
- 9.Greer EL, Brunet A. FOXO transcription factors at the interface between longevity and tumor suppression. Oncogene. 2005;24:7410–25. doi: 10.1038/sj.onc.1209086. [DOI] [PubMed] [Google Scholar]
- 10.Huang H, Tindall DJ. Dynamic FoxO transcription factors. J Cell Sci. 2007;120:2479–87. doi: 10.1242/jcs.001222. [DOI] [PubMed] [Google Scholar]
- 11.Kops GJ, Medema RH, Glassford J, Essers MA, Dijkers PF, Coffer PJ, Lam EW, Burgering BM. Control of cell cycle exit and entry by protein kinase B-regulated forkhead transcription factors. Mol Cell Biol. 2002;22:2025–36. doi: 10.1128/MCB.22.7.2025-2036.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Medema RH, Kops GJ, Bos JL, Burgering BM. AFX-like Forkhead transcription factors mediate cell-cycle regulation by Ras and PKB through p27kip1. Nature. 2000;404:782–7. doi: 10.1038/35008115. [DOI] [PubMed] [Google Scholar]
- 13.Nakamura N, Ramaswamy S, Vazquez F, Signoretti S, Loda M, Sellers WR. Forkhead transcription factors are critical effectors of cell death and cell cycle arrest downstream of PTEN. Mol Cell Biol. 2000;20:8969–82. doi: 10.1128/MCB.20.23.8969-8982.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ramaswamy S, Nakamura N, Sansal I, Bergeron L, Sellers WR. A novel mechanism of gene regulation and tumor suppression by the transcription factor FKHR. Cancer Cell. 2002;2:81–91. doi: 10.1016/S1535-6108(02)00086-7. [DOI] [PubMed] [Google Scholar]
- 15.Schmidt M, Fernandez de Mattos S, van der Horst A, Klompmaker R, Kops GJ, Lam EW, Burgering BM, Medema RH. Cell cycle inhibition by FoxO forkhead transcription factors involves downregulation of cyclin D. Mol Cell Biol. 2002;22:7842–52. doi: 10.1128/MCB.22.22.7842-7852.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tran H, Brunet A, Grenier JM, Datta SR, Fornace AJ, Jr., DiStefano PS, Chiang LW, Greenberg ME. DNA repair pathway stimulated by the forkhead transcription factor FOXO3a through the Gadd45 protein. Science. 2002;296:530–4. doi: 10.1126/science.1068712. [DOI] [PubMed] [Google Scholar]
- 17.Gilley J, Coffer PJ, Ham J. FOXO transcription factors directly activate bim gene expression and promote apoptosis in sympathetic neurons. J Cell Biol. 2003;162:613–22. doi: 10.1083/jcb.200303026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Modur V, Nagarajan R, Evers BM, Milbrandt J. FOXO proteins regulate tumor necrosis factor-related apoptosis inducing ligand expression. Implications for PTEN mutation in prostate cancer. J Biol Chem. 2002;277:47928–37. doi: 10.1074/jbc.M207509200. [DOI] [PubMed] [Google Scholar]
- 19.Dijkers PF, Medema RH, Lammers JW, Koenderman L, Coffer PJ. Expression of the pro-apoptotic Bcl-2 family member Bim is regulated by the forkhead transcription factor FKHR-L1. Curr Biol. 2000;10:1201–4. doi: 10.1016/S0960-9822(00)00728-4. [DOI] [PubMed] [Google Scholar]
- 20.Van Der Heide LP, Hoekman MF, Smidt MP. The ins and outs of FoxO shuttling: mechanisms of FoxO translocation and transcriptional regulation. Biochem J. 2004;380:297–309. doi: 10.4161/cc.27727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Biggs WH, 3rd, Meisenhelder J, Hunter T, Cavenee WK, Arden KC. Protein kinase B/Akt-mediated phosphorylation promotes nuclear exclusion of the winged helix transcription factor FKHR1. Proc Natl Acad Sci U S A. 1999;96:7421–6. doi: 10.4161/cc.27727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 1999;96:857–68. doi: 10.1016/S0092-8674(00)80595-4. [DOI] [PubMed] [Google Scholar]
- 23.Rena G, Guo S, Cichy SC, Unterman TG, Cohen P. Phosphorylation of the transcription factor forkhead family member FKHR by protein kinase B. J Biol Chem. 1999;274:17179–83. doi: 10.1074/jbc.274.24.17179. [DOI] [PubMed] [Google Scholar]
- 24.Tang ED, Nuñez G, Barr FG, Guan KL. Negative regulation of the forkhead transcription factor FKHR by Akt. J Biol Chem. 1999;274:16741–6. doi: 10.1074/jbc.274.24.16741. [DOI] [PubMed] [Google Scholar]
- 25.Alvarez B, Martínez-A C, Burgering BM, Carrera AC. Forkhead transcription factors contribute to execution of the mitotic programme in mammals. Nature. 2001;413:744–7. doi: 10.1038/35099574. [DOI] [PubMed] [Google Scholar]
- 26.Liu P, Kao TP, Huang H. CDK1 promotes cell proliferation and survival via phosphorylation and inhibition of FOXO1 transcription factor. Oncogene. 2008;27:4733–44. doi: 10.1038/onc.2008.104. [DOI] [PubMed] [Google Scholar]
- 27.Yuan Z, Becker EB, Merlo P, Yamada T, DiBacco S, Konishi Y, Schaefer EM, Bonni A. Activation of FOXO1 by Cdk1 in cycling cells and postmitotic neurons. Science. 2008;319:1665–8. doi: 10.1126/science.1152337. [DOI] [PubMed] [Google Scholar]
- 28.Lee SY, Lee GR, Woo DH, Park NH, Cha HJ, Moon YH, Han IS. Depletion of Aurora A leads to upregulation of FoxO1 to induce cell cycle arrest in hepatocellular carcinoma cells. Cell Cycle. 2013;12:67–75. doi: 10.4161/cc.22962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mattila J, Kallijärvi J, Puig O. RNAi screening for kinases and phosphatases identifies FoxO regulators. Proc Natl Acad Sci U S A. 2008;105:14873–8. doi: 10.1073/pnas.0803022105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Calnan DR, Brunet A. The FoxO code. Oncogene. 2008;27:2276–88. doi: 10.1038/onc.2008.21. [DOI] [PubMed] [Google Scholar]
- 31.Huang H, Regan KM, Lou Z, Chen J, Tindall DJ. CDK2-dependent phosphorylation of FOXO1 as an apoptotic response to DNA damage. Science. 2006;314:294–7. doi: 10.1126/science.1130512. [DOI] [PubMed] [Google Scholar]
- 32.Tzivion G, Dobson M, Ramakrishnan G. FoxO transcription factors; Regulation by AKT and 14-3-3 proteins. Biochim Biophys Acta. 2011;1813:1938–45. doi: 10.1016/j.bbamcr.2011.06.002. [DOI] [PubMed] [Google Scholar]
- 33.Fu Z, Tindall DJ. FOXOs, cancer and regulation of apoptosis. Oncogene. 2008;27:2312–9. doi: 10.1038/onc.2008.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huang H, Muddiman DC, Tindall DJ. Androgens negatively regulate forkhead transcription factor FKHR (FOXO1) through a proteolytic mechanism in prostate cancer cells. J Biol Chem. 2004;279:13866–77. doi: 10.1074/jbc.M314143200. [DOI] [PubMed] [Google Scholar]
- 35.Woods YL, Rena G, Morrice N, Barthel A, Becker W, Guo S, Unterman TG, Cohen P. The kinase DYRK1A phosphorylates the transcription factor FKHR at Ser329 in vitro, a novel in vivo phosphorylation site. Biochem J. 2001;355:597–607. doi: 10.1042/bj3550597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brunet A, Kanai F, Stehn J, Xu J, Sarbassova D, Frangioni JV, Dalal SN, DeCaprio JA, Greenberg ME, Yaffe MB. 14-3-3 transits to the nucleus and participates in dynamic nucleocytoplasmic transport. J Cell Biol. 2002;156:817–28. doi: 10.1083/jcb.200112059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brownawell AM, Kops GJ, Macara IG, Burgering BM. Inhibition of nuclear import by protein kinase B (Akt) regulates the subcellular distribution and activity of the forkhead transcription factor AFX. Mol Cell Biol. 2001;21:3534–46. doi: 10.1128/MCB.21.10.3534-3546.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang X, Gan L, Pan H, Guo S, He X, Olson ST, Mesecar A, Adam S, Unterman TG. Phosphorylation of serine 256 suppresses transactivation by FKHR (FOXO1) by multiple mechanisms. Direct and indirect effects on nuclear/cytoplasmic shuttling and DNA binding. J Biol Chem. 2002;277:45276–84. doi: 10.1074/jbc.M208063200. [DOI] [PubMed] [Google Scholar]
- 39.Huang H, Regan KM, Wang F, Wang D, Smith DI, van Deursen JM, Tindall DJ. Skp2 inhibits FOXO1 in tumor suppression through ubiquitin-mediated degradation. Proc Natl Acad Sci U S A. 2005;102:1649–54. doi: 10.4161/cc.27727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yuan C, Bu Y, Wang C, Yi F, Yang Z, Huang X, Cheng L, Liu G, Wang Y, Song F. NFBD1/MDC1 is a protein of oncogenic potential in human cervical cancer. Mol Cell Biochem. 2012;359:333–46. doi: 10.1007/s11010-011-1027-7. [DOI] [PubMed] [Google Scholar]