

Abstract
Kaposi sarcoma-associated herpesvirus (KSHV) is a tumor virus encoding several proto-oncogenes. However, the roles of these viral genes in KSHV-induced tumorigenesis have not been defined. In this study, we used a recently developed model of KSHV-induced cellular transformation and tumorigenesis combining with a reverse genetic system to examine the role of a KSHV latent gene vCyclin (ORF72), a cellular Cyclin D2 homolog, in KSHV-induced oncogenesis. Deletion of vCyclin did not affect cell proliferation and cell cycle progression at a low-density condition, when cells were at an active proliferation state. However, vCyclin mutant cells were contact-inhibited and arrested at G1 phase at a high-density condition. As a result, vCyclin mutant cells formed less and smaller colonies in soft agar assay. Nude mice inoculated with vCyclin mutant cells had reduced tumor incidence and extended tumor latency and survival compared with mice inoculated with wild-type (WT) virus-infected cells. WT but not mutant virus effectively induced Cyclin-dependent kinase inhibitor p27/Kip1 Ser10 phosphorylation and cytoplasmic relocalization. shRNA knockdown of p27 released the blockage of the mutant cells from cell cycle arrest at G1 phase at a high-density condition. Together, these results indicate that vCyclin primarily functions to enhance cellular transformation and tumorigenesis by promoting cell cycle progression and cell proliferation at a contact-inhibited condition.
Keywords: Kaposi sarcoma, KSHV, vCyclin/ORF72, cell proliferation, cell cycle progression, contact inhibition, p27/Kip1, cellular transformation, tumorigenesis
Introduction
Infection by Kaposi sarcoma-associated herpesvirus (KSHV) is required for the development of Kaposi sarcoma (KS), a vascular malignancy of proliferative endothelial cells.1 KSHV is also associated with several other lymphoproliferative malignancies, including primary effusion lymphoma (PEL) and a subset of multicentric Castleman disease (MCD). Despite intensive studies in the last 2 decades, the mechanism of KSHV-induced oncogenesis remains largely unclear, which is, in part, due to the lack of a KSHV-induced cellular transformation and tumorigenesis system. The recent development of such a system should facilitate the delineation of the mechanism of KSHV-induced oncogenesis and identification of viral and cellular factors that are essential for this process.2
KSHV encodes over 2 dozen genes that manifest regulatory functions in cell growth, proliferation, and survival.1 While most of KSHV-regulatory genes are expressed during viral lytic replication, a number of them, including vFLIP (ORF71), vCyclin (ORF72), LANA (ORF73), and a cluster of microRNAs (miRNA), all of which are located at the latent locus of the viral genome, are expressed during the latent phase of KSHV life cycle.3 In KS lesions, most tumor cells are latently infected by KSHV, with only a small number of them undergoing lytic replication.3 Thus, viral latent infection and latent genes are presumably essential for the development of KSHV-induced malignancies.3 Indeed, our recent study has shown that KSHV-encoded miRNAs are required for KSHV-induced cellular transformation and tumorigenesis.4 However, the functions of other viral genes in tumorigenesis in the context of KSHV infection have not been defined so far.
Among the KSHV latent genes, vCyclin is a cellular Cyclin homolog, sharing 54% homology and 32% identity with Cyclin D2.5,6 vCyclin binds to several Cyclin-dependent kinases (CDK) including CDK2, CDK4, CDK6, and CDK9 in vitro.5-9 However, only vCyclin interactions with CDK6 and CDK9 have been shown to be functional in cells so far. The vCyclin–CDK6 complex has a large range of substrates including pRb, Bcl-2, histone H1, ORC1, CDC6, nucleophosmin, p53, p21/Cip1, and p27/Kip1.5,7,9-14 In PEL cell lines, phosphorylation by the vCyclin–CDK6 complex leads to p21 inactivation as well as p27 inactivation, cytoplasmic sequestration, and degradation, resulting in accelerated G1/S phase transition.11,15,16 Single gene overexpression also causes vCyclin-dependent p27 degradation.17,18 While the activities of cellular Cyclin–CDK complexes are highly regulated by both inhibitory and activating phosphorylation mainly through interactions with Cyclin-dependent kinase inhibitors (CKIs), the vCyclin-CDK6 enzymatic activity does not require an activating phosphorylation, nor is its activity inhibited by Cip (p21), Kip (p27), and Arf (p19) CKIs, though there are conflicting reports on the ability of p16/Ink CKI to inhibit vCyclin–CDK6 kinase activity.8,10,18-20 Although the activating phosphorylation is not necessary for the vCyclin–CDK6 enzymatic activity, it is required for its resistance to inhibition by CKIs.8,21,22 Without inhibition by CKIs, the vCyclin–CDK6 complex is free to promote an accelerated G1/S transition in an unrestrained manner, presumably through phosphorylation of pRb and release of E2F. Inactivation of p21 and p27 by the vCyclin–CDK6 complex further removes the inhibition on the Cyclin E–CDK2 enzymatic activity, allowing both vCyclin– and Cyclin E–CDK complexes to promote the G1/S transition.8,13,18
Single gene overexpression of vCyclin in a transgenic model has been shown to induce tumors.19,23 However, the function of vCyclin in KSHV-induced cellular transformation and tumorigenesis has not been investigated. In this study, using the newly developed KSHV-induced cellular transformation and tumorigenesis system,2 we have surprisingly found that deletion of vCyclin does not affect cell proliferation at a low-density proliferating condition; however, it decreases cell proliferation at a high-density, contact-inhibited condition. While vCyclin is not required for KSHV immortalization of primary cells, it promotes cellular transformation and tumorigenesis. Furthermore, vCyclin’s pro-oncogenic effect is mediated by p27 pSer10 phosphorylation and cytoplasmic sequestration.
Results
Generation of KSHV vCyclin mutant and its revertant
To determine the role of vCyclin in KSHV-induced cellular transformation and tumorigenesis, we generated a vCyclin deletion mutant and its genetic revertant by homologous recombination.24 A kanamycin-resistance cassette (Kan) fragment flanked by 2 LoxP sites and a fragment of 57-bp of KSHV sequence immediately outside the vCyclin coding frame was generated (Fig. 1A). Homologous recombination between the wild-type KSHV genome (WT) and the targeting fragment led to the generation of a vCyclin intermediate mutant. Cre-mediated recombination led to the excision of the Kan, resulting in the generation of the vCyclin deletion mutant, which is called “Mutant” or “Mut” in this study (Fig. 1A). We further generated a revertant recombinant virus (“Revertant” or “Rev”) using a similar strategy. A targeting fragment containing the vCyclin coding frame and a zeocin-resistance cassette (Zeo) fragment was generated. Homologous recombination between the mutant and targeting fragment resulted in the generation of the intermediate revertant. Cre-mediated homologous recombination led to the excision of the Zeo and generation of the revertant (Fig. 1A).
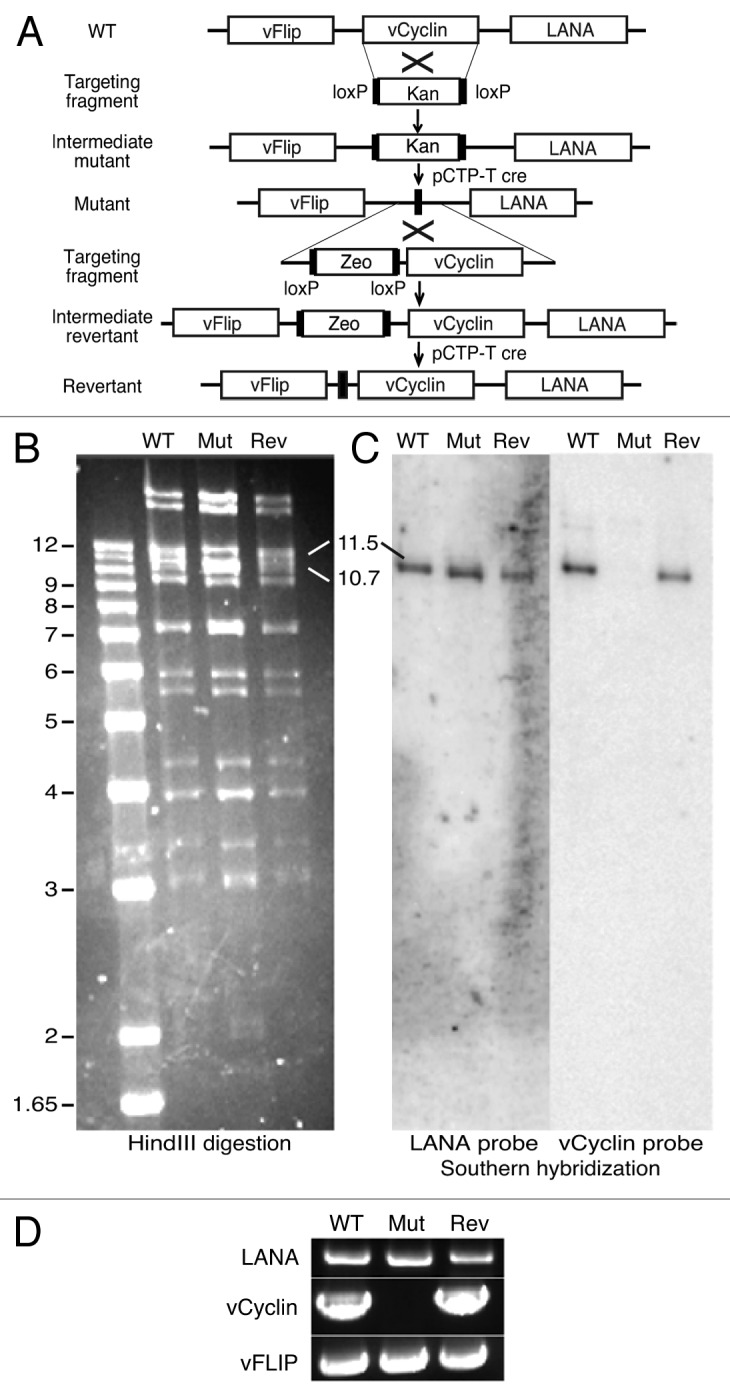
Figure 1. Generation of a KSHV vCyclin deletion mutant and its revertant. (A) Schematic illustration showing the strategy for generating a vCyclin mutant (Mutant) and its revertant (Revertant). A PCR fragment containing a kanamycin-resistance cassette (Kan) was introduced into the BAC36 genome by homologous recombination to replace vCyclin followed by cre-mediated excision of the Kan to generate the mutant. A similar strategy was employed to generate the revertant using a zeocin-resistance cassette (Zeo) as the selection marker. (B) HindIII restriction analysis of the WT, mutant (Mut), and revertant (Rev) genomes. (C) Southern blot hybridization with LANA and vCyclin as probes. (D) PCR detection of LANA, vCyclin and vFLIP from genomic DNA of different recombinant viruses to confirm the deletion of vCyclin and the presence of adjacent viral genes in the mutant.
To confirm the deletion of vCyclin in the mutant genome and the repair of vCyclin in the revertant genome as well as the lack of any disruptions in other parts of the viral genomes, we performed restriction enzyme digestion of the recombinant genomes followed by Southern blot hybridization with selected probes. Similar restriction patterns were observed for the WT, mutant, and revertant genomes following HindIII digestion, with the exception of the 11 491-bp vCyclin-containing fragment (Fig. 1B). Deletion of vCyclin resulted in the loss of a 771-bp fragment, shifting the 11 491-bp band to a new 10 718-bp band. Upon generation of the revertant, the 10.7-kb band was shifted back to the 11.5-kb band (Fig. 1B). Southern blot hybridization with vCyclin as a probe confirmed the deletion of the vCyclin gene in the mutant (Fig. 1C). Using LANA as a probe, we confirmed the presence of the LANA in all recombinant viruses (Fig. 1C). PCR amplification of the viral genome further showed that vFLIP and LANA were present in all recombinant viruses, while vCyclin was only absent in the mutant genome (Fig. 1D). DNA sequencing also showed that there were not any other unexpected genomic alterations and mutations in the latent locus in both the mutant and revertant genomes.
To determine the effect of vCyclin deletion in the context of KSHV infection, we first characterized primary rat metanephric mesenchymal precursor (MM) cells latently infected by the WT, mutant, and revertant viruses. We did not observe any differences in cell morphology among mock, WT, mutant, and revertant cultures, except that the mock cells were slightly larger than the other 3 types of cells (Fig. 2A). Immunofluorescence staining revealed similar LANA speckle pattern in WT, mutant and revertant cell cultures (Fig. 2B). Deletion of vCyclin did not affect the number of speckle dots per cells and the staining intensities. As expected, PCR amplification of the viral genomes showed that vFLIP and LANA were present in cultures of all recombinant viruses, while vCyclin was only absent in the mutant culture (Fig. 2C). Consistent with the results of immunofluorescence staining, genomic qPCR analysis indicated that there was no difference of KSHV genome copy numbers per cell in all KSHV-infected cells (data not shown). Western blot showed that deletion of vCyclin did not affect the expression levels of LANA protein and transcript (Fig. 2D and E). Since we were not able to obtain an antibody that can detect the expression of vFLIP protein in KSHV-infected cells, we performed RT-qPCR to detect the expression of vFLIP transcript. As shown in Figure 2F, the expression level of vFLIP transcript was not affected following the deletion of vCyclin. As expected, vCyclin protein and transcript were detected in the WT and revertant cells but not in the mutant cells (Fig. 2D and G). Together, these results indicated that vCyclin was successfully deleted in the mutant genome but repaired in the revertant genome without affecting other parts of the viral genome. The expression of adjacent viral latent genes LANA and vFLIP were not affected following the deletion of vCyclin from the KSHV genome.

Figure 2. Deletion of vCyclin does not affect the expression of other adjacent viral genes. (A) Morphology of rat metanephric mesenchymal precursor (MM) cells infected by different recombinant viruses. (B) Detection of LANA protein expression in mock, WT, mutant and revertant cells by immunofluorescence staining. (C) Detection of LANA, vCyclin, and vFLIP genomic DNAs in cells infected by different recombinant viruses by PCR. (D) Detection of LANA and vCyclin proteins in cells infected by different recombinant viruses by western blotting. (E–G) Detection of mRNAs of LANA (E), vFLIP (F), and vCyclin (G) in cells infected by different recombinant viruses by RT-qPCR.
vCyclin is not required for KSHV cellular immortalization but enhances cellular transformation by promoting cell proliferation at contact-inhibited condition
We determined the effect of vCyclin deletion on cellular immortalization. Cells were passaged and counted every 3 d, and the cumulative total numbers of cells in the cultures were calculated (Fig. 3A). As previously reported, mock cells underwent senescence after 23 passages.2 Infection by WT or revertant viruses led to the immortalization of the mock cells. Deletion of vCyclin did not affect the ability of KSHV to immortalize the MM cells. However, the cumulative total number of mutant cells was lower than those of WT and revertant cells (Fig. 3A), indicating a slower overall cell proliferation rate of the mutant cells.

Figure 3. Effect of vCyclin deletion on KSHV-mediated cell proliferation, (A) Deletion of vCyclin does not affect KSHV immortalization of primary cells. Cells were passaged every 3 days, and the cumulative total cell numbers were calculated. Mock cells underwent crisis at around passage 23, while WT, mutant, and revertant cells continued to grow without encountering any crisis. (B) vCyclin is required to maintain accelerated cell proliferation at contact-inhibited condition. Cells were seeded at 2 × 104 cells/well in 6-well plates and counted daily. (C) Deletion of vCyclin reduced cell saturation density. Cells were seeded at 2 × 105 cells/well in 6-well and cultured with daily change of medium. Total numbers of cells were counted at day 14 post-seeding. (D) Mutant cells form fewer and smaller foci than WT and revertant cells do in culture. Cells were cultured as described in (C) and foci were examined at day 14 post-seeding. (E–G) Mutant cells form fewer and smaller colonies than WT and revertant cells do in softagar assay as illustrated by images of the colonies (E), the numbers of colonies (F) calculated based on the plates described in (E), and a graph showing the distribution of colony sizes (G) calculated based on the plates described in (E).
We further investigated cell proliferation of cells infected by different KSHV recombinant viruses. Cells were seeded at an equal cell number at 20% confluency, and live cells were counted daily for 5 d. WT and revertant cells had an accelerated rate of cell proliferation compared with the mock cells as previously reported (Fig. 3B). Deletion of vCyclin did not affect cell proliferation before day 3 post-seeding. However, a decreased cell proliferation rate was observed in the mutant culture starting at day 3 post-seeding, when the culture reached 100% confluency (Fig. 3B). These results indicated that vCyclin promoted cell proliferation at a high-density condition, which imposed a contact-inhibition effect on the cells. Consistent with these results, WT and revertant cultures reached significantly higher saturation cell densities than mock and mutant cultures did (1.59 × 106 and 1.51 × 106 cells/well vs. 4.1 × 105 and 6.3 × 105 cells/well, respectively) (Fig. 3C). WT and revertant cells also formed many large foci following prolonged culture, while mutant cells only formed smaller and fewer foci (Fig. 3D). We did not observe any foci in the mock cell cultures. Furthermore, WT and revertant cells formed many large colonies in softagar assay (Fig. 3E). Mutant cells also formed colonies in the softagar assay. However, the numbers of colonies were significantly lower, and the sizes of the colonies were significantly smaller than those of WT and revertant cells (Fig. 3E–G). No colony was observed with the mock cells.
Together, the above results indicate that vCyclin is not required for KSHV-induced cellular immortalization and transformation. However, vCyclin enhances cellular transformation by promoting cell proliferation at contact-inhibited condition.
Deletion of vCyclin decreases tumor incidence and extends animal survival
We further examined the role of vCyclin in KSHV-induced tumorigenesis. Athymic nude mice were subcutaneously injected with mock, WT, mutant, and revertant cells. Tumor incidence and growth were monitored over time. WT and revertant cells induced tumors in 100% and 95% of the mice with tumor latencies of 34.9 ± 7.7 and 42.5 ± 10.5 d, respectively (Fig. 4A). Deletion of vCyclin reduced the tumor incidence to 45% and increased the tumor latency to 60.0 ± 7.1 d (P < 0.05 for tumors of mutant cells vs. tumors of both WT and revertant cells) (Fig. 4A). None of the mice inoculated with mock cells developed any tumors as previously reported.2 WT and revertant cells induced tumors with faster growth rates than mutant cells did (Fig. 4B). Mice inoculated with the mutant cells had extended survival rate compared with those inoculated with WT and revertant cells (P < 0.01 for tumors of mutant cells vs. tumors of both WT and revertant cells) (Fig. 4C). H&E staining showed that tumors from WT, mutant, and revertant cells displayed spindle-shape cells, which were positive for KSHV latent protein LANA (Fig. 4D). All tumors exhibited the slit-like structures, which were characteristic of KS tumors (Fig. 4D). These results indicate that vCyclin is not required for KSHV-induced tumorigenesis, but it promotes tumor formation and progression.

Figure 4. vCyclin promotes tumor incidence and growth. (A) Tumor incidence over time in nude mice inoculated with cells transformed by different KSHV recombinant viruses. The threshold of tumor volume was set as 0.2 cm3 or whenever the tumor was palpable. (B) Tumor growth curves showing average tumor sizes. (C) Kaplan–Meier survival curves. (D) Immunohistochemical staining of tumors. Tumors were stained for H&E and LANA. Tumor analyses were performed when the volume reached 1 cm3.
vCyclin promotes cell cycle progression by overriding contact inhibition but has minimal effect on apoptosis and senescence
Because vCyclin promoted cell proliferation at high-density but not at low-density conditions (Fig. 3), we further examined cell cycle progression at these conditions. Cells at proliferating 50–60% low-density and saturation high-density conditions were analyzed for cell cycle profiles. Deletion of vCyclin did not affect cell cycle progression under low-density condition. Under this condition, WT, mutant and revertant cultures had similar number of cells in S-phase but they all had significantly more cells in S-phase than the mock culture had (55%, 58%, and 58%, respectively, vs. 33%) (Fig. 5A and B). However, at a high-density condition, WT and revertant cultures had significantly more cells in S phase than the Mutant culture had (37% and 32%, respectively, vs. 20%) (Fig. 5C and D). Examination of BrdU incorporation showed that under a low-density condition, WT, mutant, and revertant cultures had similar BrdU incorporation rates at 42%, 43%, and 43%, respectively, which were significantly higher than that of the 33% rate of the mock culture had (Fig. 5E and F). However, at a high-density condition, WT and revertant cultures had significantly higher BrdU incorporation rates than that of the mutant culture had (52% and 53%, respectively, vs. 27%) (Fig. 5G and H). In fact, the BrdU incorporation rate of the mutant culture was more similar to the 20% rate of the mock culture. Thus, the decrease in cell proliferation at a high-density condition in the mutant culture was at least in part due to the slower G1/S phase transition.

Figure 5. vCyclin is required to maintain accelerated G1/S transition at contact-inhibited condition. (A and B) Deletion of vCyclin does not affect cell cycle progression at low-density as shown by representative histograms (A) and results of averages from three repeats (B). Cells seeded at a low-density at 2.5 × 105 cells/flask in T25 flasks overnight were analyzed for cell cycle. There was no difference in cell cycle profiles among WT, mutant, and revertant cells. However, all 3 KSHV-transformed cells had more cells in S phase and less cells in G1 phase than mock cells had. (C and D) Deletion of vCyclin compromises cell cycle progression at a high-density as shown by representative histograms (C) and results of averages from 3 repeats (D). Cells seeded at a high-density at 106 cells/flask in T25 flasks overnight were analyzed for cell cycle. WT and revertant cells had more cells in S-phase and less cells in G1-phase than mutant and mock cells had. (E and F) Deletion of vCyclin does not affect BrdU incorporation at low-density as shown by representative FACS images (E) and results of averages from 3 repeats (F). Cells seeded at a low-density at 2.5 × 105 cells/flask in T25 flasks overnight were pulsed with BrdU and analyzed by FACS. There was no difference in BrdU incorporation among WT, mutant, and revertant cells. However, all 3 KSHV-transformed cells had more BrdU incorporation than mock cells had. (G and H) Deletion of vCyclin decreases BrdU incorporation at high-density as shown by representative FACS images (G) and results of averages from 3 repeats (H). Cells seeded at a high-density at 106 cells/flask in T25 flasks overnight were pulsed with BrdU and analyzed by FACS. WT and revertant cells had more BrdU incorporation than mutant and mock cells had. (I and J) Deletion of vCyclin has no effect on apoptosis at a low-density (I) or a high-density condition (J). Cells were seeded at 2.5 × 105 cells/flask (I) or 106 cells/flask (J) in T25 flasks overnight and analyzed for apoptosis by Annexin V staining.
Previous studies have shown that overexpression of vCyclin can cause cell apoptosis and senescence.12,25 At a low-density condition, there was no difference in the number of apoptotic cells among the mock, WT, mutant and revertant cultures, with apoptotic rates at 1.6%, 1.8%, 1.7%, and 1.4%, respectively (Fig. 5I). At a high-density condition, the numbers of apoptotic cells of WT, mutant, and revertant cultures were increased to 3.1%, 3.2%, and 3.2%, respectively, while that of mock cells remained significantly lower at 1.8% (Fig. 5J). Examination of senescence cells indicated that all 4 types of cells had low rates of senescence cells at both low-density (<0.1%) and high-density (<1%) conditions (data not shown). Taken together, these results show that vCyclin has minimal effect on apoptosis and senescence in the context of KSHV infection.
vCyclin increases the protein expression levels of Cyclins D1, E and A1, as well as CKI p27 at contact-inhibited condition
Because our results so far have shown that vCyclin primarily promotes cell proliferation and transformation by regulating cell cycle progression and G1/S phase transition at a contact-inhibited condition, we examined the expression of Cyclins known to regulate cell cycle progression. Western blots showed that Cyclins D3, E, and A1 proteins had similar expression levels among all the cell types at a low-density condition (Fig. 6A). Under this condition, Cyclin D1 protein expression level was slightly higher in WT and revertant cells than in mutant cells, while it was not detectable in mock cells (Fig. 6A). At a high-density condition, higher levels of Cyclins D1, E and A1 were detected in WT and revertant cells than in mutant cells (Fig. 6A). Under this condition, Cyclin D3 was weakly detected in all cultures. Together, these results show that the protein expression levels of Cyclins are largely consistent with cell proliferation rates of the KSHV-transformed cells.

Figure 6. Detection of total and phosphorylation protein levels of p27/Kip1 and other cell cycle regulators in different KSHV-transformed cells by western blotting. (A) Detection of Cyclins D1, D3, E, and A1 proteins. (B) Detection of total p27 protein, and Ser10 and Thr187 phosphorylated p27 proteins.
The activities of Cyclin–CDK complexes are regulated by CKIs.26 Among them, CKI p27 regulates G1/S phase transition and is implicated in mediating contact inhibition. Previous studies have shown that vCyclin can interact with p27 to cause its inactivation.8,17,18 Therefore, we examined the effect of vCyclin deletion on the levels of p27 protein in the KSHV-transformed cells. At a low-density condition, the total p27 protein level was higher in all KSHV-transformed cells than in mock cells; however, it was slightly higher in WT and revertant cells than in mutant cells (Fig. 6B). Phosphorylation at Thr187 targets p27 for ubiquitination and degradation while phosphorylation at Ser10 relocates p27 protein to the cytoplasm, which stabilizes and inactivates p27 protein.27,28 Overexpression of vCyclin has been shown to cause stabilization and inactivation of p27.11,15,16,18 Indeed, we detected low levels of p27 Ser10 phosphorylation in WT and revertant cells but not in mock and mutant cells at a low-density condition (Fig. 6B). At a high-density condition, the total p27 protein levels were increased in all KSHV-transformed cells, with both WT and revertant cells maintaining higher levels of p27 proteins than the mutant cells had (Fig. 6B). Consistent with these results, under this condition, the levels of p27 Ser10 phosphorylation were dramatically increased in WT and revertant cells, while only a weak level of p27 Ser10 phosphorylation was detected in the mutant cells. In immunofluorescence staining, both WT and revertant cells had overall stronger total p27 protein staining than the mutant cells had (Fig. 7A). In particular, a fraction of the WT and revertant cells had strong total p27 protein staining in the cytoplasm. To confirm that the high levels of p27 Ser10 phosphorylation in WT and revertant cells were correlated with cytoplasmic relocalization, we performed immunofluorescence staining for p27 Ser10 phosphorylation. Indeed, a portion of the WT and revertant cells had strong cytoplasmic staining for p27 Ser10 phosphorylation (Fig. 7B). Together, these results indicate that vCyclin can cause cytoplasmic sequestration of the p27 protein.

Figure 7. Detection of total and pSer10 phosphorylation of p27 protein in different KSHV-transformed cells by immunofluorescence staining. (A) Detection of total p27 protein. (B) Detection of pSer10 phosphorylation of p27 protein.
shRNA knockdown of p27 releases the blockage on cell proliferation of mutant cells in a contact-inhibited condition
The above results have shown that in the presence of vCyclin, p27 is sequestrated to cytoplasm and presumably inactivated resulting in the promotion of cell cycle progression at a contact-inhibited condition. To determine if inactivation of p27 by vCyclin was required for cell proliferation at a high-density condition, we performed shRNA knockdown of p27. Compared with cells transduced with a control shRNA, we observed knockdown of p27 protein in the range of 50–90% among all 4 types of the cells at day 3 following transduction with lentiviruses expressing p27-specific shRNAs (Fig. 8A). At a low-density condition in the first 2 days post-seeding of the cells, knockdown of p27 had minimal effects on cell proliferation in all 4 types of cells (Fig. 8B). At a high-density condition after day 3 post-seeding of the cells, knockdown of p27 also did not affect cell proliferation in the mock cells (Fig. 8B). However, a slight increase of cell proliferation in WT and revertant cells were observed after day 4 post-seeding, indicating that p27 exerted a slight inhibitory effects on cell proliferation, and that the expression of vCyclin protein from the KSHV genome had not completely abolished the function of p27 in these cells. The most significant effect was observed with the mutant cells, where knockdown of p27 significantly increased cell proliferation, increasing the rate by up to 2-fold, reaching the level close to those of WT and revertant cells (Fig. 8B). Consistent with cell proliferation, knockdown of p27 increased the numbers of cells in S-phase in WT, revertant, and mutant cells at a contact-inhibited condition (Fig. 8C). These results indicate that p27 represents a block to cell proliferation of KSHV-transformed cells at a contact-inhibited condition.

Figure 8. p27/Kip1 mediates the arrest of cell proliferation and cell cycle progression. (A) Detection of p27 protein by western blotting following shRNA knockdown. Cells transduced with p27 shRNAs or a control shRNA for 3 d were collected and examined for the expression of p27 protein. (B) Knockdown of p27 abolished the arrest of cell proliferation imposed by contact inhibition in the mutant cells. Cells transduced with p27 shRNAs or a control shRNA for 48 h were reseeded at 2 × 104 cells/well in 6-well plates and counted daily. (C) Knockdown of p27 abolished the arrest of cell cycle progression imposed by contact inhibition in the mutant cells. Cells transduced with p27 shRNAs or a control shRNA for 48 h were reseeded at high-density at 106 cells/flask in T25 flasks for 48 h and analyzed for cell cycle.
Discussion
Infection by KSHV is required for the development of several malignancies including KS, PEL, and a subset of MCD. Despite intensive studies, the mechanism by which KSHV induces these malignancies remains unclear.1 This is in part due to the lack of tractable experimental models of KSHV-induced cellular transformation and tumorigenesis. We have recently developed such as a model, in which KSHV efficiently infects and transforms primary rat mesenchymal precursor cells.2 Subcutaneous inoculation of KSHV-transformed cells into nude mice effectively induces tumors with pathological and virological features closely resembling KS. Using this novel model and a vCyclin mutant-generated with a reverse genetic system,24 in this study, we have examined the role of vCyclin in KSHV-induced cellular transformation and tumorigenesis. Our results show that vCyclin is not required for KSHV-induced immortalization, cellular transformation, and tumorigenesis. However, vCyclin promotes cellular transformation and tumorigenesis.
In single gene overexpression studies, vCyclin is one of the few KSHV latent genes that manifests an oncogenic function.5-10,13,15-19,23,25,29 vCyclin interacts with CDKs, primarily CDK6, to form functional vCyclin–CDK complexes, resulting in accelerated cell cycle progression.7,8,17 Our results show that, in the context KSHV infection, vCyclin has no effect on cell proliferation and cell cycle progression at a low-density proliferating condition. However, vCyclin promotes cell proliferation and cell cycle progression at a high-density contact-inhibited condition. As a result, vCyclin promotes foci formation in culture and colony formation in softagar assay. Uncontrolled cell proliferation at high cell density and contact-inhibited condition is a feature of transformed cells and is essential for tumor development.30 Our results show that vCyclin contributes to this essential cancer feature in KSHV-induced tumorigenesis.
Among the identified vCyclin–CDKs substrates, p27 is implicated in mediating contact inhibition.31,32 In cells, p27 binds to the Cyclin–CDK complexes, primarily Cyclin E–CDK2 and Cyclin D–CDK4 complexes, and prevents their activation of arresting cells at G1 phase. During cell proliferation, p27 is targeted for unbiquitination and degradation following phosphorylation at Thr187 and is inactivated following phosphorylation at Ser10 and cytoplasmic relocalization. vCyclin has been shown to inhibit the function of p27 by causing its phosphorylation at both Thr187 and Ser10.33,34 Interestingly, we did not detect p27 Thr187 phosphorylation in any of the KSHV-transformed cell types with or without vCyclin expression, indicating that vCyclin was not likely to cause Thr187 phosphorylation and p27 degradation in these cells. In fact, we detected higher levels of total p27 protein in WT and revertant cells than in mutant cells, particularly at the high-density condition (Figs. 6B and 7A). Similar results were also observed in PEL cells.16 In contrast to Thr187 phosphorylation, vCyclin mediated weak p27 Ser10 phosphorylation at a low-density condition, which was dramatically increased at a high-density condition (Fig. 6B). Importantly, knockdown of p27 released cell cycle arrest and increased cell proliferation of the mutant cells at a high-density condition. Thus, p27 exerts an inhibitory effect on cell cycle progression and cell proliferation of KSHV-transformed cells at a contact-inhibited condition, which is overridden by vCyclin. We conclude that vCyclin primarily functions to disable p27’s inhibitory effect on cell cycle progression by causing its cytoplasmic relocalization during KSHV-induced cellular transformation. Nevertheless, single gene studies have shown that vCyclin also targets other cell cycle checkpoint proteins including pRb, p53, and p21 in addition to p27.5-8,10,13 Thus, it is possible that vCyclin deregulation of other checkpoint proteins could also contribute to the enhanced cellular transformation and tumorigenesis phenotypes.
Consistent with vCyclin’s oncogenic function, overexpression of vCyclin can cause cell apoptosis and senescence.25,29,35 In primary mouse embryonic fibroblasts, overexpression of vCyclin induced growth arrest and apoptosis as a result of p53 activation.19 However, vCyclin-expressing cells continued to proliferate in p53-null cells, indicating that inactivation of p53 function synergizes with vCyclin’s oncogenic function. Indeed, overexpression of vCyclin under the control of a B cell-specific promoter cooperated with p53 loss, resulting in the induction of lymphomas.19,23 However, we did not detect any increase in apoptosis in the mutant cells in our study. The numbers of cells undergoing senescence were also low (<1%) in all types of cells in both low-density and high-density conditions. A number of KSHV genes, including LANA, vFLIP, and kaposins, as well as KSHV-encoded miRNAs are expressed during viral latent infection, some of which have anti-apoptosis and -senescence functions,36-44 which could cooperate with vCyclin to induce cellular transformation. Indeed, results from our recent study have shown that KSHV miRNAs regulate cell proliferation and survival and are required for KSHV-induced cellular transformation and tumorigenesis.4 It would be interesting to use this powerful model to define the functions of other individual viral products in KSHV-induced cellular transformation in the context of viral infection. It is anticipated that the interplay of viral genes and their regulated cellular pathways might ultimately result in KSHV-induced cellular transformation and tumorigenesis (Fig. 9).
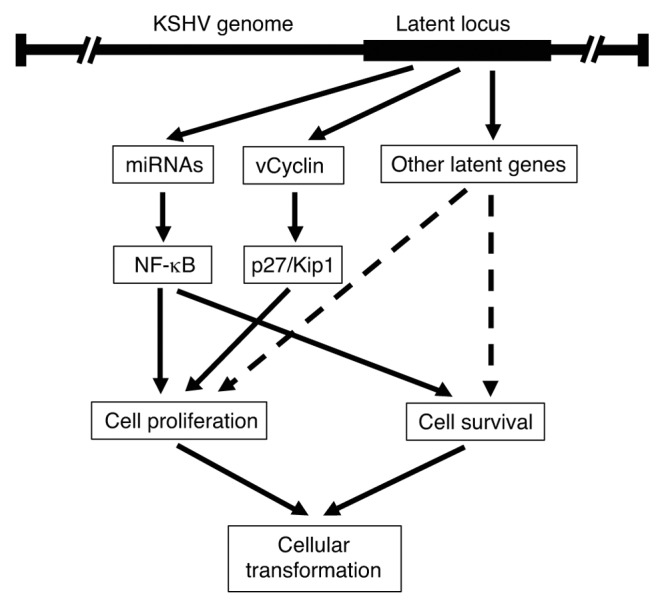
Figure 9. Model of KSHV-induced cellular transformation mediated by vCyclin and other KSHV latent products.
Materials and Methods
Generation of a vCyclin mutant and its revertant
We generated a KSHV mutant with the vCyclin-coding frame deleted (Mutant) and its genetic revertant with the deleted vCyclin repaired (Revertant) (Fig. 1A). The procedures were performed essentially as previously described.24 Briefly, a linear DNA fragment containing a Kan cassette flanked by loxP sites was PCR amplified with primers containing sequences with homology to the parts of KSHV genome outside of the vCyclin ORF. The primers are: 5′-GAGACCCTGA AACTCCAGGC TCTCAGGTAG GCCACATACG CTGCCACTCT ATATGGTGTA GGCTGGAGCT GCTTC-3′ (forward primer) and 5′-CCGCCTAAAC AAAATCACAA GCTTAATAGC TGTCCAGAAT GCGCAGATCA AAGTCCCATA TGAATATCCT CCTTAG-3′ (reverse primer). The resulting PCR fragment (targeting fragment) was electroporated into competent E. coli. containing the WT KSHV genome BAC36 and the pKD6 plasmid to facilitate recombination.45 After the first round of homologous recombination, the kanamycin- and chloramphenicol-resistant transformants containing the intermediate vCyclin deletion mutant were selected. The Kan cassette was then removed from the intermediate mutant by Cre-mediated recombination following transfection of the Cre recombinase-expressing plasmid pCTP-T into the E. coli containing the vCyclin deletion intermediate mutant. The resulting recombinant vCyclin mutant (Mutant) was verified by HindIII restriction digestion and Southern blot hybridization (Fig. 1B and C) and genomic PCR amplification (Fig. 1D).
The corresponding revertant of vCyclin mutant was generated with a similar strategy (Fig. 1A). The vCyclin ORF was amplified with the following primers: 5′-ATGGAACTGC CAATAACCCG-3′ (forward) and 5′-GTTAAGCTTT TAATAGCTGT CCACAATGC-3′ (reverse). A DNA fragment containing a Zeo cassette was amplified with the following primers: 5′-CATATGAATA CCTCCTTAG-3′ (forward) and 5′-CACATTCGAA CTGGAGCAAG-3′ (reverse). The amplified vCyclin and Zeo fragments were digested with HindIII restriction enzyme, ligated and gel purified. The product was amplified by PCR using the following primers: 5′- CCTGAAACTC CAGGCTCTAC AGGTAGGCCA CATCGCTCGC CACTCTATAT GGCAACTGCC AATAACCCG (forward) and 5′-CTTGTATATG TGAAGGCACC GATGTGGAAA AACAAAGGAA AATTTATTTT TCCGC CCTAAACAAA ATCACAAATT CATATGAA (reverse). Following gel purification, the resulting targeting fragment was electroporated into the E. coli containing the vCyclin mutant and the pKD46 plasmid. Upon homologous recombination, zeocin- and chloramphenicol-resistant colonies were selected. The Zeo was then removed by Cre-mediated recombination. The resulting vCyclin revertant was verified by HindIII restriction digestion, Southern blot hybridization (Fig. 1B and C), and genomic PCR amplification (Fig. 1D).
Restriction analysis and Southern-blot hybridization
Restriction digestion and Southern blot hybridization were performed as previously described.46
Cell lines and culture
MM cells were maintained in Dulbecco modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum. Cells infected by WT, mutant and revertant recombinant viruses were maintained in DMEM supplemented with 10% fetal bovine serum and 400 μg/mL hygromycin.
Transduction of shRNA lentiviruses and knockdown of p27
shRNAs against p27 cloned in pLenti-H1-shRNA vector containing a RFP-reporter and a Blasticidin selection marker were purchased from GenTarget. The shRNA sequences are shRNA1: 5′-CAAACTCTGA GGACCGGCAT T-3′ and shRNA2: 5′-GACCAAATGC CTGACTCGTC A-3′. A scrambled shRNA containing the sequence of 5′-GTCTCCACGC GCAGTACATT T-3′ was used a control. Cells were transduced with the lentivirus particles and analyzed at day 3 post-transduction. We obtained close to 100% transduction rates based on the expression of RFP.
Quantitative reverse transcription real-time PCR (RT-qPCR)
Total RNA was isolated with Trizol reagents as instructed by the manufacturer (Invitrogen). Reverse transcription was performed with total RNA using SuperScript III First Strand Synthesis System (Invitrogen). RT-qPCR was performed on the Eppendorf Real Plex 2 using primers specific for LANA, vFLIP, vCyclin, and β-actin as previously described.2,46
Immunofluorescence assay and western blotting
Immunofluorescence assay and western blotting were performed as previously described.47,48 LANA was detected with a rat monoclonal antibody (Abcam). A rabbit polyclonal antibody was used to detect vCyclin. Cyclin D1, D3, A1, and E, total p27 and p27 pSer10 phosphorylation were detected with rabbit polyclonal antibodies (Santa Cruz). pRb was detected with a mouse monoclonal antibody (Santa Cruz). A goat polyclonal antibody was used to detect p27 Thr187 phosphorylation (Santa Cruz).
Cell counting, cell cycle analysis, and BrdU incorporation
Cells were seeded in 24-well plates at a density of 20 000 cells/well. Cells were counted daily with a hemocytomter. For cell cycle, cells were seeded at either 2.5 × 104 (low density) or 1 × 106 cells (high density) per flask in T25 flasks. Cell cycle analysis was performed by propidium iodide staining and flow cytometry was performed in a FACSCanto System (BD Biosciences). Analysis was performed with FlowJo (Treestar). BrdU incorporation was performed by pulsing cells with 10 μM BrdU for 60 or 90 min and then stained with a Pacific Blue monoclonal antibody to BrdU (Invitrogen).
Apoptosis and senescence
Biotinylated anti-Annexin V antibody and APC-Cy7-streptavidin were used to detect apoptotic cells following the instructions of the manufacturer (BD Biosciences). Senescence cells were detected as previously described.49
Foci formation, saturation density, and soft agar assay
Cells were seeded at 106 cells per well in 6-well plates with daily change of medium and allowed to grow past confluency. At day 14, the wells were examined for the formation of foci and the total numbers of cells per well were counted. Soft agar assay was performed as previously described.2,50
Tumor growth in mice
This study was performed in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol was approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Texas Health Science Center at San Antonio (Animal Welfare Assurance Number: A3345-01). All surgery was performed under sodium pentobarbital anesthesia, and all efforts were made to minimize suffering.
Nude mice were subcutaneously injected with 107 cells per site. Two sites on the left and right flanks were inoculated for each mouse. Each site typically produced one tumor. Tumor volumes were measured twice weekly with a caliper using a 0.2 cm3 threshold or whenever the tumors were palpable. Tumors were allowed to grow up to 1 cm3, at which point the animals were sacrificed and the tumors harvested for analysis. Tumor analysis was performed as previously described.2
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank members of Dr Gao’s laboratory for technical assistance. This work was supported by grants from NIH (CA096512, CA124332, CA132637, and CA177377) to S.-J. Gao.
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/27758
References
- 1.Greene W, Kuhne K, Ye F, Chen J, Zhou F, Lei X, Gao SJ. Molecular biology of KSHV in relation to AIDS-associated oncogenesis. Cancer Treat Res. 2007;133:69–127. doi: 10.1007/978-0-387-46816-7_3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jones T, Ye F, Bedolla R, Huang Y, Meng J, Qian L, Pan H, Zhou F, Moody R, Wagner B, et al. Direct and efficient cellular transformation of primary rat mesenchymal precursor cells by KSHV. J Clin Invest. 2012;122:1076–81. doi: 10.1172/JCI58530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ye F, Lei X, Gao SJ. Mechanisms of Kaposi Sarcoma-Associated Herpesvirus Latency and Reactivation. Adv Virol. 2011;2011 doi: 10.1155/2011/193860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Moody R, Zhu Y, Huang Y, Cui X, Jones T, Bedolla R, Lei X, Bai Z, Gao SJ. KSHV microRNAs mediate cellular transformation and tumorigenesis by redundantly targeting cell growth and survival pathways. PLoS Pathog. 2013;9:e1003857. doi: 10.1371/journal.ppat.1003857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chang Y, Moore PS, Talbot SJ, Boshoff CH, Zarkowska T, Godden-Kent, Paterson H, Weiss RA, Mittnacht S. Cyclin encoded by KS herpesvirus. Nature. 1996;382:410. doi: 10.1038/382410a0. [DOI] [PubMed] [Google Scholar]
- 6.Li M, Lee H, Yoon DW, Albrecht JC, Fleckenstein B, Neipel F, Jung JU. Kaposi sarcoma-associated herpesvirus encodes a functional Cyclin. J Virol. 1997;71:1984–91. doi: 10.1128/jvi.71.3.1984-1991.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Godden-Kent D, Talbot SJ, Boshoff C, Chang Y, Moore P, Weiss RA, Mittnacht S. The Cyclin encoded by Kaposi sarcoma-associated herpesvirus stimulates cdk6 to phosphorylate the retinoblastoma protein and histone H1. J Virol. 1997;71:4193–8. doi: 10.1128/jvi.71.6.4193-4198.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Swanton C, Mann DJ, Fleckenstein B, Neipel F, Peters G, Jones N. Herpes viral Cyclin/Cdk6 complexes evade inhibition by CDK inhibitor proteins. Nature. 1997;390:184–7. doi: 10.1038/36606. [DOI] [PubMed] [Google Scholar]
- 9.Chang PC, Li M. Kaposi sarcoma-associated herpesvirus K-Cyclin interacts with Cdk9 and stimulates Cdk9-mediated phosphorylation of p53 tumor suppressor. J Virol. 2008;82:278–90. doi: 10.1128/JVI.01552-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Child ES, Mann DJ. Novel properties of the Cyclin encoded by Human Herpesvirus 8 that facilitate exit from quiescence. Oncogene. 2001;20:3311–22. doi: 10.1038/sj.onc.1204447. [DOI] [PubMed] [Google Scholar]
- 11.Järviluoma A, Koopal S, Räsänen S, Mäkelä TP, Ojala PM. KSHV viral Cyclin binds to p27KIP1 in primary effusion lymphomas. Blood. 2004;104:3349–54. doi: 10.1182/blood-2004-05-1798. [DOI] [PubMed] [Google Scholar]
- 12.Ojala PM, Yamamoto K, Castaños-Vélez E, Biberfeld P, Korsmeyer SJ, Mäkelä TP. The apoptotic v-Cyclin-CDK6 complex phosphorylates and inactivates Bcl-2. Nat Cell Biol. 2000;2:819–25. doi: 10.1038/35041064. [DOI] [PubMed] [Google Scholar]
- 13.Laman H, Coverley D, Krude T, Laskey R, Jones N. Viral Cyclin-Cyclin-dependent kinase 6 complexes initiate nuclear DNA replication. Mol Cell Biol. 2001;21:624–35. doi: 10.1128/MCB.21.2.624-635.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sarek G, Järviluoma A, Moore HM, Tojkander S, Vartia S, Biberfeld P, Laiho M, Ojala PM. Nucleophosmin phosphorylation by v-Cyclin-CDK6 controls KSHV latency. PLoS Pathog. 2010;6:e1000818. doi: 10.1371/journal.ppat.1000818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Järviluoma A, Child ES, Sarek G, Sirimongkolkasem P, Peters G, Ojala PM, Mann DJ. Phosphorylation of the Cyclin-dependent kinase inhibitor p21Cip1 on serine 130 is essential for viral Cyclin-mediated bypass of a p21Cip1-imposed G1 arrest. Mol Cell Biol. 2006;26:2430–40. doi: 10.1128/MCB.26.6.2430-2440.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sarek G, Järviluoma A, Ojala PM. KSHV viral Cyclin inactivates p27KIP1 through Ser10 and Thr187 phosphorylation in proliferating primary effusion lymphomas. Blood. 2006;107:725–32. doi: 10.1182/blood-2005-06-2534. [DOI] [PubMed] [Google Scholar]
- 17.Ellis M, Chew YP, Fallis L, Freddersdorf S, Boshoff C, Weiss RA, Lu X, Mittnacht S. Degradation of p27(Kip) cdk inhibitor triggered by Kaposi sarcoma virus Cyclin-cdk6 complex. EMBO J. 1999;18:644–53. doi: 10.1093/emboj/18.3.644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mann DJ, Child ES, Swanton C, Laman H, Jones N. Modulation of p27(Kip1) levels by the Cyclin encoded by Kaposi sarcoma-associated herpesvirus. EMBO J. 1999;18:654–63. doi: 10.1093/emboj/18.3.654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Verschuren EW, Klefstrom J, Evan GI, Jones N. The oncogenic potential of Kaposi sarcoma-associated herpesvirus Cyclin is exposed by p53 loss in vitro and in vivo. Cancer Cell. 2002;2:229–41. doi: 10.1016/S1535-6108(02)00123-X. [DOI] [PubMed] [Google Scholar]
- 20.Kaldis P. The N-terminal peptide of the Kaposi sarcoma-associated herpesvirus (KSHV)-Cyclin determines substrate specificity. J Biol Chem. 2005;280:11165–74. doi: 10.1074/jbc.M408887200. [DOI] [PubMed] [Google Scholar]
- 21.Kaldis P, Ojala PM, Tong L, Mäkelä TP, Solomon MJ. CAK-independent activation of CDK6 by a viral Cyclin. Mol Biol Cell. 2001;12:3987–99. doi: 10.1091/mbc.12.12.3987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yoshioka H, Noguchi K, Katayama K, Mitsuhashi J, Yamagoe S, Fujimuro M, Sugimoto Y. Functional availability of gamma-herpesvirus K-Cyclin is regulated by cellular CDK6 and p16INK4a. Biochem Biophys Res Commun. 2010;394:1000–5. doi: 10.1016/j.bbrc.2010.03.110. [DOI] [PubMed] [Google Scholar]
- 23.Verschuren EW, Hodgson JG, Gray JW, Kogan S, Jones N, Evan GI. The role of p53 in suppression of KSHV Cyclin-induced lymphomagenesis. Cancer Res. 2004;64:581–9. doi: 10.1158/0008-5472.CAN-03-1863. [DOI] [PubMed] [Google Scholar]
- 24.Zhou FC, Zhang YJ, Deng JH, Wang XP, Pan HY, Hettler E, Gao SJ. Efficient infection by a recombinant Kaposi sarcoma-associated herpesvirus cloned in a bacterial artificial chromosome: application for genetic analysis. J Virol. 2002;76:6185–96. doi: 10.1128/JVI.76.12.6185-6196.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Koopal S, Furuhjelm JH, Järviluoma A, Jäämaa S, Pyakurel P, Pussinen C, Wirzenius M, Biberfeld P, Alitalo K, Laiho M, et al. Viral oncogene-induced DNA damage response is activated in Kaposi sarcoma tumorigenesis. PLoS Pathog. 2007;3:1348–60. doi: 10.1371/journal.ppat.0030140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Starostina NG, Kipreos ET. Multiple degradation pathways regulate versatile CIP/KIP CDK inhibitors. Trends Cell Biol. 2012;22:33–41. doi: 10.1016/j.tcb.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ishida N, Kitagawa M, Hatakeyama S, Nakayama K. Phosphorylation at serine 10, a major phosphorylation site of p27(Kip1), increases its protein stability. J Biol Chem. 2000;275:25146–54. doi: 10.1074/jbc.M001144200. [DOI] [PubMed] [Google Scholar]
- 28.Tsvetkov LM, Yeh KH, Lee SJ, Sun H, Zhang H. p27(Kip1) ubiquitination and degradation is regulated by the SCF(Skp2) complex through phosphorylated Thr187 in p27. Curr Biol. 1999;9:661–4. doi: 10.1016/S0960-9822(99)80290-5. [DOI] [PubMed] [Google Scholar]
- 29.Leidal AM, Cyr DP, Hill RJ, Lee PW, McCormick C. Subversion of autophagy by Kaposi sarcoma-associated herpesvirus impairs oncogene-induced senescence. Cell Host Microbe. 2012;11:167–80. doi: 10.1016/j.chom.2012.01.005. [DOI] [PubMed] [Google Scholar]
- 30.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 31.Zhang X, Wharton W, Donovan M, Coppola D, Croxton R, Cress WD, Pledger WJ. Density-dependent growth inhibition of fibroblasts ectopically expressing p27(kip1) Mol Biol Cell. 2000;11:2117–30. doi: 10.1091/mbc.11.6.2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Polyak K, Lee MH, Erdjument-Bromage H, Koff A, Roberts JM, Tempst P, Massagué J. Cloning of p27Kip1, a Cyclin-dependent kinase inhibitor and a potential mediator of extracellular antimitogenic signals. Cell. 1994;78:59–66. doi: 10.1016/0092-8674(94)90572-X. [DOI] [PubMed] [Google Scholar]
- 33.Ishida N, Hara T, Kamura T, Yoshida M, Nakayama K, Nakayama KI. Phosphorylation of p27Kip1 on serine 10 is required for its binding to CRM1 and nuclear export. J Biol Chem. 2002;277:14355–8. doi: 10.1074/jbc.C100762200. [DOI] [PubMed] [Google Scholar]
- 34.Boehm M, Yoshimoto T, Crook MF, Nallamshetty S, True A, Nabel GJ, Nabel EG. A growth factor-dependent nuclear kinase phosphorylates p27(Kip1) and regulates cell cycle progression. EMBO J. 2002;21:3390–401. doi: 10.1093/emboj/cdf343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ojala PM, Tiainen M, Salven P, Veikkola T, Castaños-Vélez E, Sarid R, Biberfeld P, Mäkelä TP. Kaposi sarcoma-associated herpesvirus-encoded v-Cyclin triggers apoptosis in cells with high levels of Cyclin-dependent kinase 6. Cancer Res. 1999;59:4984–9. [PubMed] [Google Scholar]
- 36.Abend JR, Uldrick T, Ziegelbauer JM. Regulation of tumor necrosis factor-like weak inducer of apoptosis receptor protein (TWEAKR) expression by Kaposi sarcoma-associated herpesvirus microRNA prevents TWEAK-induced apoptosis and inflammatory cytokine expression. J Virol. 2010;84:12139–51. doi: 10.1128/JVI.00884-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chugh P, Matta H, Schamus S, Zachariah S, Kumar A, Richardson JA, Smith AL, Chaudhary PM. Constitutive NF-kappaB activation, normal Fas-induced apoptosis, and increased incidence of lymphoma in human herpes virus 8 K13 transgenic mice. Proc Natl Acad Sci U S A. 2005;102:12885–90. doi: 10.1073/pnas.0408577102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fakhari FD, Jeong JH, Kanan Y, Dittmer DP. The latency-associated nuclear antigen of Kaposi sarcoma-associated herpesvirus induces B cell hyperplasia and lymphoma. J Clin Invest. 2006;116:735–42. doi: 10.1172/JCI26190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gottwein E, Mukherjee N, Sachse C, Frenzel C, Majoros WH, Chi JT, Braich R, Manoharan M, Soutschek J, Ohler U, et al. A viral microRNA functions as an orthologue of cellular miR-155. Nature. 2007;450:1096–9. doi: 10.1038/nature05992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lei X, Bai Z, Ye F, Xie J, Kim CG, Huang Y, Gao SJ. Regulation of NF-kappaB inhibitor IkappaBalpha and viral replication by a KSHV microRNA. Nat Cell Biol. 2010;12:193–9. doi: 10.1038/ncb2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lei X, Zhu Y, Jones T, Bai Z, Huang Y, Gao SJ. A Kaposi sarcoma-associated herpesvirus microRNA and its variants target the transforming growth factor β pathway to promote cell survival. J Virol. 2012;86:11698–711. doi: 10.1128/JVI.06855-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Radkov SA, Kellam P, Boshoff C. The latent nuclear antigen of Kaposi sarcoma-associated herpesvirus targets the retinoblastoma-E2F pathway and with the oncogene Hras transforms primary rat cells. Nat Med. 2000;6:1121–7. doi: 10.1038/80459. [DOI] [PubMed] [Google Scholar]
- 43.Skalsky RL, Samols MA, Plaisance KB, Boss IW, Riva A, Lopez MC, Baker HV, Renne R. Kaposi sarcoma-associated herpesvirus encodes an ortholog of miR-155. J Virol. 2007;81:12836–45. doi: 10.1128/JVI.01804-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Suffert G, Malterer G, Hausser J, Viiliäinen J, Fender A, Contrant M, Ivacevic T, Benes V, Gros F, Voinnet O, et al. Kaposi sarcoma herpesvirus microRNAs target caspase 3 and regulate apoptosis. PLoS Pathog. 2011;7:e1002405. doi: 10.1371/journal.ppat.1002405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A. 2000;97:6640–5. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li Q, Zhou F, Ye F, Gao SJ. Genetic disruption of KSHV major latent nuclear antigen LANA enhances viral lytic transcriptional program. Virology. 2008;379:234–44. doi: 10.1016/j.virol.2008.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gao SJ, Kingsley L, Hoover DR, Spira TJ, Rinaldo CR, Saah A, Phair J, Detels R, Parry P, Chang Y, et al. Seroconversion to antibodies against Kaposi sarcoma-associated herpesvirus-related latent nuclear antigens before the development of Kaposi sarcoma. N Engl J Med. 1996;335:233–41. doi: 10.1056/NEJM199607253350403. [DOI] [PubMed] [Google Scholar]
- 48.Greene W, Zhang W, He M, Witt C, Ye F, Gao SJ. The ubiquitin/proteasome system mediates entry and endosomal trafficking of Kaposi sarcoma-associated herpesvirus in endothelial cells. PLoS Pathog. 2012;8:e1002703. doi: 10.1371/journal.ppat.1002703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, Medrano EE, Linskens M, Rubelj I, Pereira-Smith O, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci U S A. 1995;92:9363–7. doi: 10.1073/pnas.92.20.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gao SJ, Boshoff C, Jayachandra S, Weiss RA, Chang Y, Moore PS. KSHV ORF K9 (vIRF) is an oncogene which inhibits the interferon signaling pathway. Oncogene. 1997;15:1979–85. doi: 10.1038/sj.onc.1201571. [DOI] [PubMed] [Google Scholar]