

Abstract
Amongst the human papillomaviruses (HPVs), the genus Alphapapillomavirus contains HPV types that are uniquely pathogenic. They can be classified into species and types based on genetic distances between viral genomes. Current circulating infectious HPVs constitute a set of viral genomes that have evolved with the rapid expansion of the human population. Viral variants were initially identified through restriction enzyme polymorphisms and more recently through sequence determination of viral fragments. Using partial sequence information, the history of variants, and the association of HPV variants with disease will be discussed with the main focus on the recent utilization of full genome sequence information for variant analyses. The use of multiple sequence alignments of complete viral genomes and phylogenetic analyses have begun to define variant lineages and sublineages using empirically defined differences of 1.0–10.0% and 0.5–1.0%, respectively. These studies provide the basis to define the genetics of HPV pathogenesis.
Keywords: HPV, Human papillomavirus variants, Alphapapillomaviruses, HPV variant lineages, HPV evolution
Introduction and background
Papillomaviruses are small circular double-stranded DNA viruses. They are highly species specific and preferentially infect cutaneous or mucocutaneous epithelium. Papillomavirus genomes have been isolated and characterized from reptiles (Herbst et al., 2009), birds (Terai et al., 2002), marsupials (Bennett et al., 2010) and multiple other mammalian species (for recent review see Bernard et al. (2010)) suggesting an evolutionary history spanning more than 300 million years (Herbst et al., 2009). Papillomaviruses replicate their genomes using the host enzymatic machinery, ensuring a high degree of proof reading with low mutation rates (Rector et al., 2007). As their genomes have evolved, mutations, insertions/deletions (indels) and rearrangements have been selected and/or fixed through host isolation into discrete entities (called types), with the intermediary genomes generally lost (Bernard et al., 2006; Gottschling et al., 2007). More recent evolutionary events are responsible for the large heterogeneity of related viral variants detected in the modern era.
Papillomavirus evolution has been predominantly asexual, although extremely rare recombination events cannot be excluded. This implies that multiple mutations/variations occurring in papillomavirus genomes are not related to genetic distance as in recombining genomes, i.e., linkage disequilibrium, but to sequential accumulation of genetic changes. This process of speciation has been termed lineage fixation (Chen et al., 2005). That is, groups of single nucleotide polymorphisms and/or insertions/deletions (indels) tend to become fixed within viral lineages. Over time, the quantity of these lineage-defining variations increases eventually leading to speciation (entities referred to as types). For papillomaviruses this has been defined when approximately 10% or more of the nucleotide positions differ between 2 genomes that share a most recent common ancestor; this takes millions of years (Chen et al., 2009; Rector et al., 2007).
A distinct human papillomavirus (HPV) “type” is established and curated when the DNA sequence of the L1 open reading frame (ORF) of the cloned viral genome differs from that of any other characterized type by at least 10% (Bernard et al., 2010; de Villiers et al., 2004). Within the PV research community, isolates of the same HPV type are referred to as variants or subtypes when the nucleotide sequences of the L1 ORF differ by less than 10%. The criterion for HPV types has proven extremely stable and useful for basic researchers, clinicians, epidemiologists and immunologists.
Over 160 HPV types have been fully characterized (see 〈http://www.hpvcenter.se/html/refclones.html〉); approximately 60 of these are predominantly detected in mucosal epithelia and sort to the genus Alphapapillomavirus (alpha-PV) (Bernard et al., 2010; de Villiers et al., 2004). Human alpha-PV infections are involved in the development of both benign and malignant disease, e.g., condylomata acuminatum/respiratory papillomatosis and cervical/anal/head and neck cancers, respectively. Cervical cancer is the most common gynecologic malignancy and one of the leading causes of cancer mortality in women worldwide (Jemal et al., 2011). Most oncogenic or high-risk (HR) types associated with invasive cervical cancer (Li et al., 2011; Munoz et al., 2003; Smith et al., 2007) are clustered in one clade of the alpha-HPVs that contains species groups alpha-5, alpha-6, alpha-7 and alpha-9 (Burk et al., 2009; Schiffman et al., 2005) and account for ∼90% of all cervical cancers worldwide (Li et al., 2011; Smith et al., 2007). Despite phylogenetic relatedness, HPV variants can differ in pathogenicity (Berumen et al., 2001; Burk et al., 2003; Chan et al., 2013; Cornet et al., 2013b; Giannoudis and Herrington, 2001; Hecht et al., 1995; Sabol et al., 2012; Schiffman et al., 2010; Villa et al., 2000; Xi et al., 2007). Because of their medical importance, the predominant research focus on papillomaviruses is their association with pathologic and oncogenic lesions. Thus, this review will focus exclusively on human papillomaviruses and predominantly on the HPVs within the genus Alphapapillomavirus.
In this review, we update knowledge of variant lineages and sublineages using currently available complete genome sequences of human Alphapapillomavirus from species groups: alpha-3 (HPV61), alpha-5 (HPV26, 51, 69 and 82), alpha-6 (HPV30, 53, 56, 66), alpha-7 (HPV18, 39, 45, 59, 68, 70, 85 and 97), alpha-9 (HPV16, 31, 33, 35, 52, 58 and 67), alpha-10 (HPV6 and 11), alpha-11 (HPV34 and 73) and alpha-13 (HPV54). We summarize the rationale for the grouping of related HPV types and highlight HPV16 lineages as an example of the increased complexity in a dichotomized classification into European and non-European groups by reviewing the use of an alphanumeric nomenclature. The establishment of a coherent classification and nomenclature system for HPV variant lineages will facilitate comparisons amongst studies that directly determine HPV sequences, especially as next-generation sequencing of PCR products and complete sample DNA (metagenomics) is rapidly being applied in new studies (Barzon et al., 2011; Conway et al., 2012; Ekstrom et al., 2011; Mokili et al., 2013). Moreover, specific variants occur on lineages that are stable and have correlated changes and diagnostic polymorphisms throughout the genome that can be used by all investigators (Alizon et al., 2011; Chen et al., 2005). Without a system for naming variant lineages, investigators have had to rely on referring to specific changes at nucleotide positions.
Historical perspective and previous reviews of HPV variants
This review builds upon the pioneering work of early investigators in the field that appreciated the significance of studying HPV variants. The earliest work in the field predated PCR and relied upon restriction enzyme digestion polymorphisms and/or Southern blot hybridization to recognize related viral variants (de Villiers et al., 1981; Mounts et al., 1982). Previous reviews of viral variants can be found from the era when sequencing DNA was cumbersome, time consuming and technically challenging (Bernard, 1994; Bernard et al., 1994). The lab of Bernard was the first to recognize the association of HPV16 (Ho et al., 1993) and HPV18 (Ong et al., 1993) viral variants with human population migrations and continent of origin (see Bernard (1994)) for review of this topic). The lab of CM Wheeler provided some of the early evidence for the intratype variation of HPV types associated with cervical neoplasia and HPV16 and HPV18 heterogeneity (Arias-Pulido et al., 2005; Stewart et al., 1996; Yamada et al., 1997, 1995). With the demonstration of an association of HPV16 variants and cervical cancer (Berumen et al., 2001; Cornet et al., 2013b; Hildesheim et al., 2001; Sichero et al., 2007; Xi et al., 2006), many of the studies on variants explored this aspect of the biology of variants as described in a number of excellent reviews (Bernard et al., 2006; Giannoudis and Herrington, 2001; Hildesheim and Wang, 2002; Lizano et al., 2009; Sichero and Villa, 2006). This current review focuses predominantly on the study of alpha-HPV variants using complete genome analyses.
The study of HPV variants from other genera provided evidence for significant intratype variation of HPV5 and HPV8 from individuals with epidermodysplasia verruciformis (Deau et al., 1993, 1991). In fact, the intratype heterogeneity of HPVs from the genera Betapapillomavirus and Gammapapillomavirus has remained basically unexplored, and there is currently a significant effort to characterize novel types from these genera (Bottalico et al., 2011; Kohler et al., 2011; Li et al., 2012). With implementation of Next-Gen sequencing (Barzon et al., 2011; Ekstrom et al., 2011), it is anticipated that there will be rapid advances in studying these genome variants (Bernard, 2013), particularly if associated with pathologic lesions.
Although the co-evolution of human populations and HPV16 and HPV18 variants is well supported, the geographic associations for variants of other types remains unresolved (Calleja-Macias et al., 2004, 2005; Chan et al., 1997; Heinzel et al., 1995; Matos et al., 2013; Prado et al., 2005). Global studies of HPV variant lineages from worldwide populations are needed to better understand the relationship between HPV and the recent and past evolution and dispersion of their human hosts.
HPV variants and cervical cancer pathogenesis
The natural history of HPV and subsequent development of cervical cancer follows a set of stages from sexual exposure to an oncogenic HPV (Bouvard et al., 2009), persistence of infection, development of a precancerous lesion and progression of the precancerous lesion to invasive disease (for recent review see Schiffman and Wentzensen (2013)). Each stage of a pernicious papillomavirus infection leading to cancer (Burk, 1999) may be influenced by both the host and viral genome variations and environmental factors (Hildesheim and Wang, 2002; Wheeler, 2008). Since the outcome of cervical cancer for any specific HPV exposure is extremely rare, most studies have used case-control designs to investigate the association of specific variants with precancer and cervix cancer. Histologic diagnosis is the end point of choice, although many studies have used cytologic outcomes. The most robust histologic “biomarker” of precancer is cervical intraepithelial neoplasia grade 3 lesions (CIN3) (Luhn et al., 2013), nevertheless studies also utilize CIN2 to enhance power by forming a high-grade class. Thus, if we only consider oncogenic HPV types, the contribution of viral variants to cancer development could be related to (1) acquisition of infection given exposure (differences in infectivity), (2) long-term persistence (e.g., greater than 1 or 2 years and could reflect viral differences in immune clearance or other properties), (3) development of precancer usually given persistence (differences in viral dysregulation of cellular differentiation and/or accumulation of somatic cellular changes in the premalignant or stromal cells) and lastly, (4) invasion of HPV containing cells. It is assumed that most invasive cervix cancer goes through all stages, however there are significant differences between the more common squamous cell carcinoma and the rarer adenocarcinoma of the cervix (Kim et al., 2013; Schiffman et al., 2007; Schiffman and Wentzensen, 2013).
Analysis of the pathogenic affects of HPV variants for this review encompassed evaluation of over 50 manuscripts spanning nearly 20 years. The topic turned out to be extremely complicated because of the complexity of the interaction of the virus with differences in host genetics (de Araujo Souza et al., 2009; Hildesheim and Wang, 2002; Xi et al., 2006) and prevalence of circulating HPV variants in different populations and/or geographic regions. In addition, description or characterization of the variants was not always simple to convert to a “categorical” description, such as has been previously designated or summarized in this review (see Table 1 and Figs. 1–5). No studies on relative infectivity were identified, thus point 1 above has not been evaluated for different variants. A summary of the current data is presented below.
Table 1.
Representative genomes for viral variant lineages and sublineages.
| Species | Type | Lineage | Sublineage | Variant genome ID | GenBank accession no. | Other names* | ||
|---|---|---|---|---|---|---|---|---|
| Alpha-9 | HPV16 | A | A1 | Ref | K02718 | European (E) | ||
| A2 | W0122 | AF536179 | European (E) | |||||
| A3 | AS411 | HQ644236 | E | |||||
| A4 | W0724 | AF534061 | Asian, E(As) | |||||
| B | B1 | W0236 | AF536180 | African-1, Afr1a | ||||
| B2 | Z109 | HQ644298 | African-1, Afr1b | |||||
| C | R460 | AF472509 | African-2, Afr2a | |||||
| D | D1 | QV00512 | HQ644257 | North American (NA)1 | ||||
| D2 | QV15321 | AY686579 | Asian–American (AA)2 | |||||
| D3 | QV00995 | AF402678 | Asian–American (AA)1 | |||||
| HPV31 | A | A1 | Ref | J04353 | ||||
| A2 | IN221709 | HQ537675 | ||||||
| B | B1 | QV03876 | HQ537676 | |||||
| B2 | QV17700 | HQ537680 | ||||||
| C | C1 | QV03136 | HQ537682 | |||||
| C2 | QV14043 | HQ537684 | ||||||
| C3 | QV00693 | HQ537685 | ||||||
| HPV33 | A | A1 | Ref | M12732 | A | |||
| A2 | QV34060 | HQ537698 | B | |||||
| A3 | LZcc12 | EU918766 | ||||||
| B | QV23819 | HQ537705 | C | |||||
| C | BF375 | KF436865 | ||||||
| HPV35 | A | A1 | Ref | X74477 | ||||
| A2 | RW128 | HQ537727 | ||||||
| HPV52 | A | A1 | Ref | X74481 | ||||
| A2 | QV15145 | HQ537739 | ||||||
| B | B1 | QV03594 | HQ537740 | |||||
| B2 | IN141070 | HQ537743 | ||||||
| C | C1 | QV05867 | HQ537744 | |||||
| C2 | QV00615 | HQ537746 | ||||||
| D | QV02575 | HQ537748 | ||||||
| HPV58 | A | A1 | Ref | D90400 | ||||
| A2 | QV15606 | HQ537752 | ||||||
| A3 | QV00961 | HQ537758 | ||||||
| B | B1 | BF134 | HQ537762 | |||||
| B2 | RW937 | HQ537764 | ||||||
| C | QV13816 | HQ537774 | ||||||
| D | D1 | QV03841 | HQ537768 | |||||
| D2 | RW697 | HQ537770 | ||||||
| HPV67 | A | A1 | Ref | D21208 | ||||
| A2 | QV22701 | HQ537780 | ||||||
| B | QV25738 | HQ537783 | ||||||
| Alpha-7 | HPV18 | A | A1 | Ref | AY262282 | AsAi, AA, E1 | ||
| A2 | Qv16306 | EF202146 | AsAi, AA, E1 | |||||
| A3 | Qv15586 | EF202147 | E, E2 | |||||
| A4 | Qv02876 | EF202151 | E, E2 | |||||
| A5 | CU11 | GQ180787 | E | |||||
| B | B1 | Qv04924 | EF202155 | Af, Af1 | ||||
| B2 | BF172 | KC470225 | Af | |||||
| B3 | Qv17199 | EF202152 | Af, Af2 | |||||
| C | Qv39775 | KC470229 | Af | |||||
| HPV39 | A | A1 | Ref | M62849 | ||||
| A2 | Qv21219 | KC470239 | ||||||
| B | RW72 | KC470247 | ||||||
| HPV45 | A | A1 | Ref | X74479 | ||||
| A2 | Qv27565 | EF202157 | ||||||
| A3 | BF134 | KC470256 | ||||||
| B | B1 | Qv00550 | EF202161 | |||||
| B2 | Qv25000 | EF202164 | ||||||
| HPV59 | A | A1 | Ref | X77858 | ||||
| A2 | Qv25652 | KC470261 | ||||||
| A3 | Qv23880 | KC470263 | ||||||
| B | Qv25808 | KC470264 | ||||||
| HPV68 | A | A1 | Ref | X67161 | A | |||
| A2 | Qv01017 | KC470269 | A | |||||
| B | Qv18016 | KC470270 | A | |||||
| C | C1 | ME180 | FR751039 | B | ||||
| C2 | Qv33015 | KC470274 | B | |||||
| D | D1 | Qv00677 | KC470275 | B | ||||
| D2 | Qv30285 | KC470276 | B | |||||
| E | Qv17725 | KC470277 | B | |||||
| F | F1 | Qv25395 | KC470279 | B | ||||
| F2 | Rw826 | KC470281 | ||||||
| HPV70 | A | Ref | U21941 | |||||
| B | Qv05102 | KC470287 | ||||||
| HPV85 | A | Ref | AF131950 | |||||
| HPV97 | A | Ref | EF202168 | |||||
| Alpha-5 | HPV26 | A | Ref | X74472 | ||||
| HPV51 | A | A1 | Ref | M62877 | ||||
| A2 | QV00891 | KF436870 | ||||||
| A3 | INJP06405 | KF436873 | ||||||
| A4 | QV09427 | KF436875 | ||||||
| B | B1 | QV10565 | KF436883 | |||||
| B2 | BF366 | KF436886 | ||||||
| HPV69 | A | A1 | Ref | AB027020 | A | |||
| A2 | QV29573 | KF436859 | A | |||||
| A3 | QV35103 | KF436861 | A | |||||
| A4 | QV31811 | KF436863 | B | |||||
| HPV82 | A | A1 | Ref | AB027021 | ||||
| A2 | QV34571 | KF436787 | ||||||
| A3 | QV28313 | KF436793 | ||||||
| B | B1 | RW17 | KF436794 | |||||
| B2 | P5 860 | KF444055 | ||||||
| C | C1 | IS39/AE2 | AF293961 | |||||
| C2 | RW606 | KF436800 | ||||||
| C3 | RW74 | KF436801 | ||||||
| C4 | RW15 | KF436802 | ||||||
| C5 | QV28248 | KF436803 | ||||||
| Alpha-6 | HPV30 | A | A1 | Ref | X74474 | |||
| A2 | QV00510 | KF436842 | ||||||
| A3 | RW060 | KF436844 | ||||||
| B | QV31356 | KF436850 | ||||||
| HPV53 | A | Ref | X74482 | A | ||||
| B | RW866 | KF436818 | ||||||
| C | QV28044 | EF546477 | B | |||||
| D | D1 | QV31688 | EF546482 | B | ||||
| D2 | QV28877 | KF436823 | B | |||||
| D3 | TJ43 | GQ472849 | ||||||
| D4 | QV22707 | EF546479 | B | |||||
| HPV56 | A | A1 | Ref | X74483 | A | |||
| A2 | QV22608 | EF177179 | B | |||||
| B | QV26762 | EF177176 | C | |||||
| HPV66 | A | Ref | U31794 | |||||
| B | B1 | QV25111 | EF177188 | |||||
| B2 | QV25260 | EF177187 | ||||||
| Alpha-11 | HPV34 | A | A1 | Ref | X74476 | |||
| A2 | QV24615 | KF436808 | ||||||
| B | QV12026 | KF436810 | ||||||
| C | C1 | QV34920 | KF436812 | |||||
| C2 | QV29446 | KF436816 | ||||||
| HPV73 | A | A1 | Ref | X94165 | ||||
| A2 | QV23185 | KF436829 | ||||||
| B | QV00749 | KF436834 | ||||||
| Alpha-10 | HPV6 | A | Ref | X00203 | ||||
| B | B1 | CAC306 | FR751337 | |||||
| B2 | CAC301 | FR751328 | ||||||
| B3 | 6a | L41216 | ||||||
| HPV11 | A | A1 | Ref | M14119 | ||||
| A2 | CAC86 | FN907962 | ||||||
| Alpha-13 | HPV54 | A | Ref | U37488 | ||||
| B | AE9 | AF436129 | ||||||
| C | QV18028 | KF436894 | ||||||
| Alpha-3 | HPV61 | A | A1 | Ref | U31793 | |||
| A2 | RW062 | KF436853 | ||||||
| B | QV02384 | KF436856 | ||||||
| C | RW940 | KF436858 | ||||||
The DNA sequences of complete HPV genomes used in this report are listed and represent a “reference genome” for a given lineage or sublineage.
Other common names for HPV16 are shown and can be found in the following references (Cornet et al., 2012; Ho et al., 1993; Smith et al., 2011; Yamada et al., 1995), as can other names for HPV18 (Arias-Pulido et al., 2005; Chen et al., 2009; Ong et al., 1993), previous assignments for lineages for HPV33/53/56/68/69 variants analyzed in Schiffman et al. (2010) that have changed are also listed above.
Fig.1.
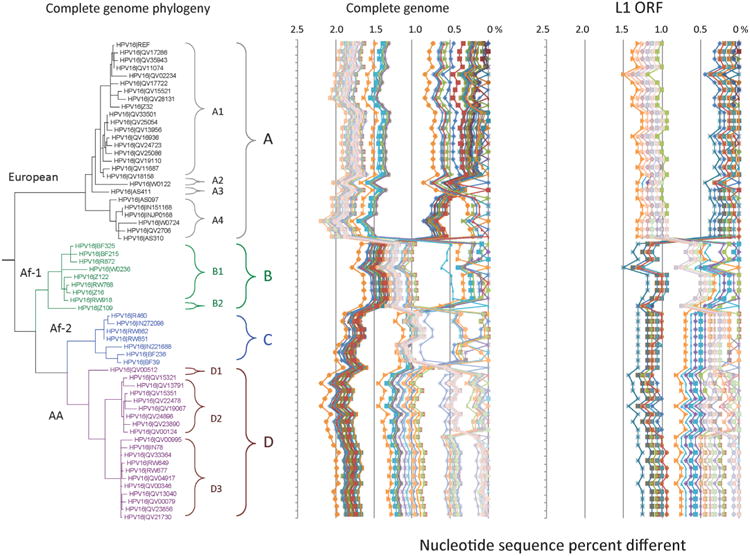
HPV16 variant tree topology and pairwise comparisons of individual complete genomes. A maximum likelihood (ML) tree was inferred from a global alignment of 62 complete genome nucleotide sequences of HPV16 using RAxML HPC v7.2.8 (Stamatakis, 2006). The 62 HPV16 genomes were previously described in Smith et al. (2011). Distinct variant lineages (i.e., termed A/B/C/D) and sublineages (e.g., termed A1/A2/A3/A4) were classified according to the topology and nucleotide sequence differences from > 1% to < 10%, and > 0.5% to < 1% ranges, respectively (Bernard et al., 2010; Chen et al., 2011). The p-distance method in MEGA5 (Tamura et al., 2011) was used to calculate pairwise nucleotide sequence differences for each isolate compared to all other isolates based on the complete genome nucleotide sequences and are shown in the middle panel, labeled “Complete genome” at the top of the figure. For comparison, the L1 ORF nucleotide sequence of each isolate was compared to that of all other isolates and is shown in the right panel, labeled “L1 ORF”. Values for each comparison of a given isolate are connected by lines and the comparison to self is indicated by the 0% difference point.
Fig. 5.
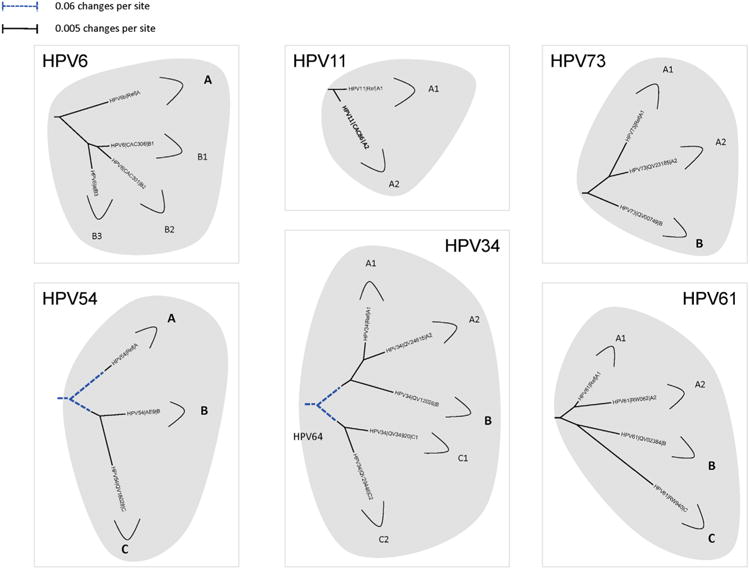
Phylogenetic trees showing representative types and variant lineages/sublineages. A maximum likelihood tree was constructed using RAxML HPC v7.2.8 (Stamatakis, 2006) inferred from the global alignment of complete genome nucleotide sequences using the program MUSCLE (Edgar, 2004) linearized at the first ATG of the E1 ORF. Representative genomes of each lineage and/or sublineage are listed in Table 1, with GenBank accession numbers. The bold part represents groupings of each type. The length of dashed and solid lines represents distance between isolates, although the number of changes is different for these two lines; the scale is indicated in the upper left corner of Figs. 2, 4 and 5; it is shown in the upper right corner of Fig. 3.
HPV16 variants and cervical pathogenesis
There is compelling data that HPV16 variants influence persistence, progression to precancer, development of cancer and histologic type of cervix cancer. Most studies implicate the non-European (NE) lineages (B/C/D, see Fig. 1) as being more pathogenic in comparison to isolates from the European (E) lineage (A).
Persistence
The magnitude of the effect (NE vs. E) is approximately 2-fold (Schiffman et al., 2010; Villa et al., 2000); although this biologic affect can be modified by host genetic makeup (Xi et al., 2006). Amongst European populations with predominantly HPV16 isolates of the A lineage, variation at HPV16 EUR-350T vs. −350G, imparted an approximately 2 fold risk of persistence (Cornet et al., 2013a; Gheit et al., 2011).
Precancer
Consistent results implicate the NE lineages compared to E isolates, with a 2–4 fold increase for cervical neoplasia in general and CIN3, specifically, in many studies (Hildesheim et al., 2001; Schiffman et al., 2010; Sichero et al., 2007; Villa et al., 2000; Xi et al., 2007; Zuna et al., 2009). Complexities of differences in HPV16 variant distributions in specific populations have indicated that in Taiwan, the E(As) or A4 lineage imparts a 10-fold risk for CIN3+ compared to the E lineage (A1). This study demonstrated that within a specific lineage there can be definable viral genetic markers, as had been suggested for the AA (D) lineage (Smith et al., 2011).
Cancer and histologic subtype
There is a strong evidence that HPV16 NE variants have elevated risks for cancer, although much of the affect is related to the increased risk for the AA (D) lineage (Berumen et al., 2001; Burk et al., 2003; Zuna et al., 2009), and there appears to be geographic complexity (Cornet et al., 2013b). There is also multiple studies that indicate the HPV16 AA (D) lineage compared to the E (A) lineage is disproportionately (4–35 fold increased) associated with adenocarcinoma (AdCa) vs. squamous cell carcinoma (SCC) (Berumen et al., 2001; Burk et al., 2003; Quint et al., 2010; Zuna et al., 2011). Nevertheless, a study from Indonesian, Surinamese and Dutch cancer cases did not replicate this finding (De Boer et al., 2005).
HPV18 variants and cervical pathogenesis
HPV18 is the second most common HPV type associated with cervix cancer, but it is much less prevalent than HPV16 in the population limiting study of this interesting oncogenic HPV. In fact, there is a lack of compelling evidence for HPV18 variants role in different stages of the pathogenesis.
Persistence
There are conflicting studies on whether non-African (European) A lineage isolates or Af variants (B/C) are associated with persistence (Schiffman et al., 2010; Sichero et al., 2007). These analyses could be confounded by genetic make-up of the host (Xi et al., 2006).
Precancer
There are few studies on precancer, in part, because it is well established that HPV18-associated precancerous lesions are infrequently detected compared to HPV18-related cancer (Guan et al., 2012). At least one study indicates that the European and AsAi variants are approximately 2-fold risk factors compared to the Af variants for 2-year cumulative incidence of CIN3 (Xi et al., 2007).
Cancer and histologic subtype
There is no compelling evidence that different HPV18 variants are associated with risk of cancer (Arias-Pulido et al., 2005). However, one out of three studies suggests that the AsAi lineage (A1/A2) is 4-fold more common in AdCa than the E lineage (A3/A4) (Arias-Pulido et al., 2005; Burk et al., 2003; De Boer et al., 2005).
HPV31 variants and cervical pathogenesis
Two recent studies provide consistent data that HPV31 lineage C is more persistent that A and/or B (Schiffman et al., 2010; Xi et al., 2013). Enigmatically, it appears that HPV31 lineages A/B are more commonly associated with and development of, CIN3 (Schiffman et al., 2010; Xi et al., 2012). In particular, lineage B was associated with CIN3 compared to lineage C with an aOR of 2.7 (95% CI: 1.2–5.7) (Xi et al., 2012).
HPV52 variants and cervical pathogenesis
At least 2 groups have reported that HPV52 variants are associated with precancer (Chang et al., 2011; Formentin et al., 2013; Schiffman et al., 2010). The most convincing association was reported from a large study in Taiwan that demonstrated variants from lineage C compared to B were associated with a 7.6 fold risk (95% CI: 1.3–44) in women with HPV52 as a single infection (Chang et al., 2011). In contrast, a study from Canada indicated the A1 lineage was more common in CIN2/3 (OR=10.5, 95% CI: 1.2–96) (Formentin et al., 2013).
HPV58 variants and cervical pathogenesis
The complexity of HPV58 heterogeneity has complicated compiling the few studies that have investigated these variants (Chan et al., 2013; Chang et al., 2011; Schiffman et al., 2010). A study from Taiwan reported that in single HPV58 infections, lineage A1 compared to A3 was associated with CIN3+ in a nested case-control study (OR=2.4, 95% CI: 0.5–12.1). In contrast, another study indicated A3 was a risk factor for CIN3+(OR=4.4, 95% CI: 1.5–13) (Chan et al., 2013). Lastly, comparison of A vs. B/C/D suggested the former lineage was associated with persistence and possibly CIN3+ (Schiffman et al., 2010).
Future studies investigating the association and/or risks of infection with specific variants should consider grouping variants by categorical states, such as lineages or sublineages as the first level of testing hypothesis about natural history or disease states. This recommendation is based on the fact that most nucleotide changes within lineages are highly correlated and analysis by lineage takes this into consideration. However, there are also non-lineage specific changes (e.g., SNPs) that should be analyzed independently (Smith et al., 2011). With implementation of Next-Gen sequencing in large studies, future work will illuminate the role of variants in the pathogenesis of cervix cancer. Consideration of population stratification and other risk factors will require large collaborative studies.
The era of whole genome analyses
HPV variant classification: lineage and sublineage taxons
The parameters for classifying related isolates of a given type were previously reported using the large dataset for HPV isolates (HPV16, HPV31, HPV33, HPV35, HPV52, HPV58 and HPV67) from the alpha-9 species group (Burk et al., 2011; Chen et al., 2011; Smith et al., 2011). A table of each genome compared to all others was created from the multiple sequence matrix. The distribution of percent differences between variants revealed a trimodal pattern (Fig. S1A). This trimodal distribution of pairwise comparisons indicated at least 3 discrete categories of relatedness, suggesting a natural taxonomy once the underlying relationship of each distribution was elucidated. To sort out the distinctions between variants, pairwise comparisons within each variant lineage (i.e., intra-lineage) or between variant lineages for each of the seven HPV types (i.e., inter-lineage)(Fig. S1B) were plotted. Previous examination of phylogenies for each of the alpha-9 types combined with an approximate cut-off of 1.0% difference between complete genomes was/is used to define major variant lineages based on lineage similarity plots as shown in Fig. 1, middle panel (Chen et al., 2011, 2005). Each major lineage was/is named using an alphanumeric, with the “A” clade always containing the reference genome for each type. The overlap between inter- and intra-lineage distributions (0.7–0.9%) indicates that a fixed value cannot simply be used to distinguish variant lineages (Fig. S1B). A conservative 1.0% difference or more has been used to define lineages, with the caveat that no classification system can exactly categorize the process of evolution. Two distributions are discernable between and within the genome comparisons of sublineages for each HPV type (Fig. S1C). Differences between genomes in the 0.5–1% range are designated as sublineages (e.g., A1, A2, etc.). These criteria to classify variants of HPV types from Alphapapillomavirus were based on available complete genomes in PubMed as part of an ongoing project to decipher HPV viral evolution and oncogenicity (Burk et al., 2009).
Subtypes
The use of the term “subtype”, although commonly used, at one time meant a difference in restriction digestion pattern of a papillomavirus (Mounts et al., 1982), with a current definition of genomes that are 2% to < 10% different in their L1 ORFs (Bernard et al., 2010; de Villiers et al., 2004). Because of the large continuum of differences between existing and potential subtypes, we favor the use of the term variants to include subtypes. The rationale for the use of complete genome (CG) sequences to define viral lineages instead of a specific region is well illustrated when comparing the L1 ORF and CG sequence differences between subtypes. As examples, subtypes within the alpha-HPV taxon demonstrate how different the two measurements, complete genome and L1 ORF comparisons are: HPV44 vs. HPV55 (CG: 6.2%, L1: 6.7%), HPV68 vs. ME180 (CG: 6.7%, L1: 7.9%), HPV34 vs. HPV64 (CG: 4.7%, L1: 5.4%), HPV82 vs. AE2 (CG: 10.2%, L1: 7.7%), HPV54 vs. AE9 (CG: 6.5%, L1: 4.6%), HPV52 lineage A vs. lineage D (CG: 2.2%, L1: 2.2%), HPV58 lineage A vs. D (CG: 1.7%, L1: 2.2%).
Analyses of complete genome variants of HPV16
Since HPV16 is the most pathogenic and medically important HPV type, we review the relationships amongst HPV16 variant complete genomes. Prior phylogenetic analyses of HPV16 variants (Chen et al., 2005; Ho et al., 1991; Yamada et al., 1997) identified 4 major intratypic variant lineages: European (E), Asian–American (AA), African-1 (Af-1) and African-2 (Af-2). Fig. 1 depicts a whole-genome phylogenetic tree (left panel, labeled “Complete genome phylogeny”) and a plot of the pairwise differences between all HPV16 complete-genome sequences (middle panel, labeled “Complete genome”); a plot of the differences based on the L1 ORF nucleotide sequence is shown for comparison (right panel, labeled “L1 ORF”). A global alignment of the 62 complete genomes resulted in a matrix that is 7916 bp in length with a maximum of 10 insertions/deletions (indels) across all sequences (Smith et al., 2011). All indels were within the highly variable non-coding region between E5 and L2 (NCR, positions 4102–4236) of the HPV16 genome. Comparison between all sequences identified a total of 540 single nucleotide polymorphisms (SNPs) with any pair of sequences having a maximum of 180 (2.3% of genome) differences. In addition to these genomes, Sabol et al. (2012) have reported a 63 bp duplication within the E1 ORF (HPV16 (E1).1374_1436dup, also listed as E1-1374^63nt duplication) and 76 complete HPV16 genomes were sequenced from central China (Sun et al., 2012), although the heterogeneity was limited to the A lineages. The most striking observation about HPV16 variants is the presence of genetic heterogeneity in what is called the “non-European” lineages (i.e., lineages B/C/D) in contrast to the relative homogeneity of the A (European) lineage. In addition to the differences between the major variant lineages, there appears to be sublineage structure; a feature notably well developed within the D (AA) lineage. The tree demonstrates excellent support (i.e., all partitions have posterior probability of 1.0) for the placement of all named lineages and sublineages. Sublineages refer to groups of genome sequences with 0.5–1.0% differences between their genomes that cluster in the phylogenetic tree (Chen et al., 2011). Sublineages are indicated by brackets within distinct blocks of similar color in Fig. 1. Taken together, these analyses suggest the presence of evolutionarily fixed sublineages at a deeper taxonomic level than the 4 commonly recognized variant clades (i.e., E, Af-1, Af-2 and AA; renamed A, B, C and D, respectively) (Chen et al., 2005). Of note is the loss of clear resolution between lineages and sublineages when only the L1 ORF nucleotide sequence is compared (compare Fig. 1. middle panel to right-side panel). This observation provides a concrete example for using complete HPV genomes to define variant taxons, whereas the L1 ORF sequence is sufficient and robust for distinguishing HPV types (Bernard et al., 2010).
Human alpha-PV species groups variant lineages
In this review, we present archetypal HPV genome isolates that represent a specific variant lineage or sublineage as defined above. The originally defined HPV type genome is always assigned to lineage “A” or when sublineages are present, “A1”. It is appreciated that with continued characterization of HPV complete genomes throughout the world there will be additional isolates identified that define future lineages and sublineages. The list of HPV genomes used in Figs. 2–5 is provided in Table 1. These figures show the phylogenetic relationships between representative isolates selected to provide a simplified topology for nomenclature; the species groups are presented by clinical importance based on association with cancers. Fig. 2 displays types constituting species group alpha-9 (Chen et al., 2011; Smith et al., 2011); HPV16, 52 and 58 each has 4 variant lineages and multiple sublineages. HPV35 is the least heterogeneous. Fig. 3 shows the topology of alpha-7 variant lineages and sublineages (Chen et al., 2013). HPV68 contains 2 distantly related clades that are also referred to as subtypes formed by isolates related to either the prototype HPV68 (lineages A and B) or the variant ME180 (lineages C/D/E/F). Relatively rare types, HPV85 and HPV97 have insufficient numbers of isolates detected to say with confidence that there is little or no viral heterogeneity. Fig. 4 shows the clade containing alpha-5 and alpha-6. HPV82 shows significant diversity and like HPV68, has 2 clades also referred to as subtypes (i.e., HPV82 prototype, lineages A/B; and AE, lineage C). Fig. 5 displays the topology of HPV isolates of types from species groups: alpha-11 (HPV34 and HPV73), alpha-10 (HPV6 and HPV11), alpha-13 (HPV54) and alpha-3 (HPV61). HPV34 and HPV54 isolates show deep bifurcations that represent subtypes. The characterization and nomenclature of HPV variants of HPV6 and HPV11 were recently reviewed (Burk et al., 2011).
Fig. 2.
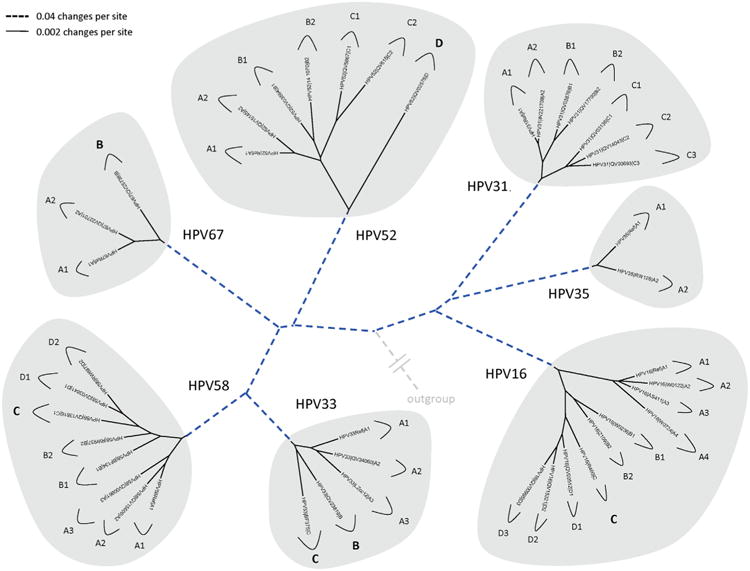
Phylogenetic trees showing representative types and variant lineages/sublineages of alpha-9. A maximum likelihood tree was constructed using RAxML HPC v7.2.8 (Stamatakis, 2006) inferred from the global alignment of complete genome nucleotide sequences using the program MUSCLE (Edgar, 2004) linearized at the first ATG of the E1 ORF. Representative genomes of each lineage and/or sublineage are listed in Table 1, with GenBank accession numbers. The bold part represents groupings of each type. The length of dashed and solid lines represents distance between isolates, although the number of changes is different for these two lines; the scale is indicated in the upper left corner of Figs. 2, 4 and 5; it is shown in the upper right corner of Fig. 3.
Fig. 3.
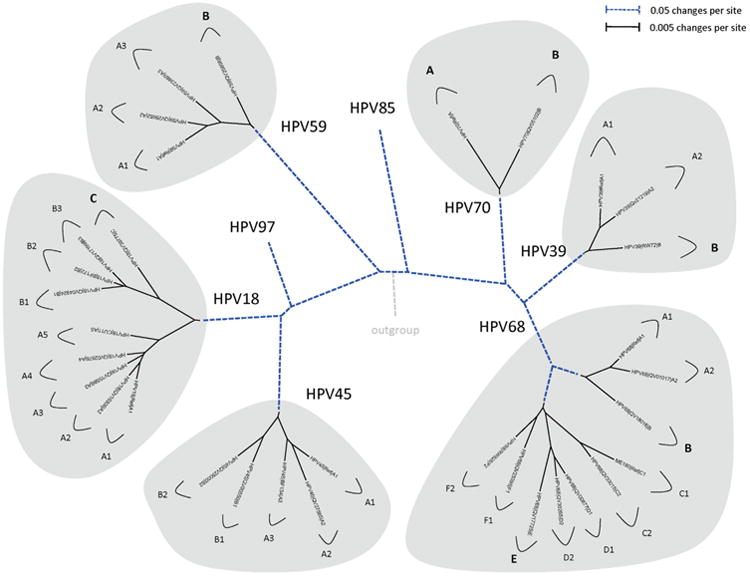
Phylogenetic trees showing representative types and variant lineages/sublineages of alpha-7. A maximum likelihood tree was constructed using RAxML HPC v7.2.8 (Stamatakis, 2006) inferred from the global alignment of complete genome nucleotide sequences using the program MUSCLE (Edgar, 2004) linearized at the first ATG of the E1 ORF. Representative genomes of each lineage and/or sublineage are listed in Table 1, with GenBank accession numbers. The bold part represents groupings of each type. The length of dashed and solid lines represents distance between isolates, although the number of changes is different for these two lines; the scale is indicated in the upper left corner of Figs. 2, 4 and 5; it is shown in the upper right corner of Fig. 3.
Fig. 4.
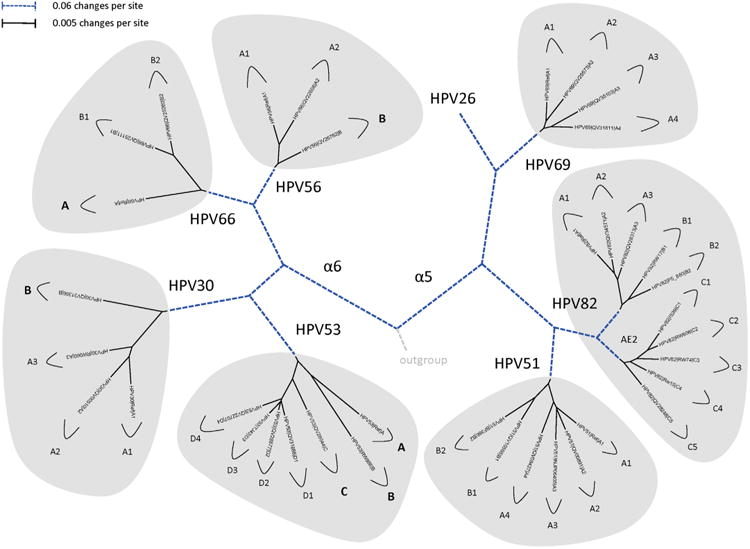
Phylogenetic trees showing representative types and variant lineages/sublineages. A maximum likelihood tree was constructed using RAxML HPC v7.2.8 (Stamatakis, 2006) inferred from the global alignment of complete genome nucleotide sequences using the program MUSCLE (Edgar, 2004) linearized at the first ATG of the E1 ORF. Representative genomes of each lineage and/or sublineage are listed in Table 1, with GenBank accession numbers. The bold part represents groupings of each type. The length of dashed and solid lines represents distance between isolates, although the number of changes is different for these two lines; the scale is indicated in the upper left corner of Figs. 2, 4 and 5; it is shown in the upper right corner of Fig. 3.
Conclusions
This review summarizes the current classification of HPV variants and provides a framework for developing a common nomenclature as the papillomavirus fields embrace Next-Gen sequencing. Massively parallel high-throughput sequencing is anticipated to generate large amounts of data on papillomavirus isolates, where it will be commonplace to obtain the complete sequence in the matter of a few days. Implementation of the new technologies will enable defining the genotype-phenotype relationships between viral isolates and clinical outcomes, such as cervix cancer. The classification of variant genomes should be based on a set of complete genomes, whereas the classification of HPV types is based on the L1 nucleotide sequences (Bernard et al., 2010; de Villiers et al., 2004). The use of full genome sequences for variant classification is derived from the fact that isolates of the same type are closely related and the full extent of the sequence heterogeneity is best summed across the whole genome and recently evolved variant genomes have changes that are not always evenly distributed throughout the genome. Nevertheless, to define distinct variant lineages, a nucleotide sequence difference of approximately 1.0% or more between two or more variants of the same type is utilized. This value is derived from empiric data on the distribution of differences between isolate genomes of the same type from the alpha-9 species group (see Fig. S1). Similarly, differences across the genome of 0.5%–1.0% are used to identify sublineages. Each variant lineage has been classified and named with an alphanumeric value. The prototype or reference sequence (i.e., the cloned genome designated as the original type) is always designated variant lineage A and/or sublineage A1 (Chen et al., 2009, 2011 in press). Therefore, with classification of variants based on full genome sequences, it is thus possible to characterize and name isolates by the use of lineage-specific diagnostic single nucleotide polymorphisms (SNPs) or lineage-specific indels found in short sequence reads in various predefined regions of the genome.
To facilitate the consistent numbering of nucleotide positions, a summary table with key landmark nucleotide positions can be used as a reference to name specific nucleotide variations within human alpha-PV genomes. Nucleotide positions are based on the reference sequence for each type. At some point in the past, agreement arose within the PV community that the “A” of the first ATG in the E6 ORF should be designated position “1”. However, as shown in the Table S1, few of the human alpha-PVs used this criterion in naming position “1”. For instance, the sequence of HPV16 defines position “1” based on an alignment with the first 60 bp of HPV1a, HPV6b and BPV1 (Seedorf et al., 1985). Moreover, there were sequencing errors in some reference clones that have been corrected over time. For convenience, a table with both the first 8 nucleotides of the reference genome and the potential E6 start codon are provided. These 8 bp sequences can be used to search the genome to locate landmarks of interest. We recommend use of the E1 ORF ATG as a reasonably conserved site, at least, in the human alpha-PVs for multiple sequence alignments. Thus, the location of the E1 ATG established from the reference sequence numbering and the 8 bp subsequent to the E1 ATG are listed for quick identification by searching. Recent discussions amongst the Papillomavirus Working Group of the ICTV suggested that nucleotide position “1” of new papillomavirus genomes should be the first nucleotide after the stop codon in the L1 ORF. This recommendation is based on the fact that every papillomavirus has a unique L1 ORF stop codon, whereas there can be many 5′ ATGs in the E6 ORF that are not functional. As mentioned above, for multiple sequence alignment we recommend the E1 ORF start site, irrespective of nucleotide position.
The use of whole genomes for variant taxonomy provides HPV researchers a simple mechanism to discuss the properties of HPV variant lineages without having to describe sets of nucleotide changes to define a group of HPV variants. This will be particularly useful for future studies of the oncogenic HPV types. As discussed above, there is growing evidence that variants of HPV16, HPV31, HPV52 and HPV58 have different oncogenic properties (Berumen et al., 2001; Chan et al., 2013; Chang et al., 2011; Cornet et al., 2013b; Hildesheim et al., 2001; Schiffman et al., 2010; Smith et al., 2011; Xi et al., 2012).
In summary, both the foundations of HPV variant research and classification and nomenclature for HPV variants based on the sequences of complete genomes are reviewed. A summary of the genomes for each lineage and sublineage are listed that can be incorporated into analyses by investigators sequencing either the full genome or subgenomic regions for variant classifications. These variants should be useful for detailed studies addressing the genetic basis of the pathogenesis of specific HPVs, viral–host interactions and host evolution, amongst other applications and scientific inquiries. The question then becomes to define the specific nucleotides or combination of nucleotides that are associated with specific pathologic consequences. It is anticipated that multicenter studies and/or meta-analyses will be needed to validate the nucleotide level of pathogenesis and provide insights into the molecular basis of HPV-associated disease. This review provides the framework based on previous studies that should be instrumental in the next stages of HPV genome research.
Supplementary Material
Acknowledgments
This work was supported in part by the National Cancer Institute (CA78527) (RDB), the Einstein–Montefiore Center for AIDS funded by the NIH (AI-51519) and the Einstein Cancer Research Center (P30CA013330) from the National Cancer Institute.
Footnotes
Appendix A. Supporting information: Supplementary data associated with this article can be found in the online version at http://dx.doi.org/10.1016/j.virol.2013.07.018.
References
- Alizon S, Luciani F, Regoes RR. Epidemiological and clinical consequences of within-host evolution. Trends Microbiol. 2011;19:24–32. doi: 10.1016/j.tim.2010.09.005. [DOI] [PubMed] [Google Scholar]
- Arias-Pulido H, Peyton CL, Torrez-Martinez N, Anderson DN, Wheeler CM. Human papillomavirus type 18 variant lineages in United States populations characterized by sequence analysis of LCR-E6, E2, and L1 regions. Virology. 2005;338:22–34. doi: 10.1016/j.virol.2005.04.022. [DOI] [PubMed] [Google Scholar]
- Barzon L, Militello V, Lavezzo E, Franchin E, Peta E, Squarzon L, Trevisan M, Pagni S, Dal Bello F, Toppo S, Palu G. Human papillomavirus genotyping by 454 next generation sequencing technology. J Clin Virol. 2011;52:93–97. doi: 10.1016/j.jcv.2011.07.006. [DOI] [PubMed] [Google Scholar]
- Bennett MD, Reiss A, Stevens H, Heylen E, Van Ranst M, Wayne A, Slaven M, Mills JN, Warren KS, O'Hara AJ, Nicholls PK. The first complete papillomavirus genome characterized from a marsupial host: a novel isolate from Bettongia penicillata. J Virol. 2010;84:5448–5453. doi: 10.1128/JVI.02635-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard HU. Coevolution of papillomaviruses with human populations. Trends Microbiol. 1994;2:140–143. doi: 10.1016/0966-842x(94)90602-5. [DOI] [PubMed] [Google Scholar]
- Bernard HU. Taxonomy and phylogeny of papillomaviruses: An overview and recent developments. Infect Genet Evol. 2013;18:357–361. doi: 10.1016/j.meegid.2013.03.011. [DOI] [PubMed] [Google Scholar]
- Bernard HU, Burk RD, Chen Z, van Doorslaer K, Hausen H, de Villiers EM. Classification of papillomaviruses (PVs) based on 189 PV types and proposal of taxonomic amendments. Virology. 2010;401:70–79. doi: 10.1016/j.virol.2010.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard HU, Calleja-Macias IE, Dunn ST. Genome variation of human papillomavirus types: phylogenetic and medical implications. Int J Cancer. 2006;118:1071–1076. doi: 10.1002/ijc.21655. [DOI] [PubMed] [Google Scholar]
- Bernard HU, Chan SY, Delius H. Evolution of papillomaviruses. Curr Top Microbiol Immunol. 1994;186:33–54. doi: 10.1007/978-3-642-78487-3_3. [DOI] [PubMed] [Google Scholar]
- Berumen J, Ordonez RM, Lazcano E, Salmeron J, Galvan SC, Estrada RA, Yunes E, Garcia-Carranca A, Gonzalez-Lira G, Madrigal-de la Campa A. Asian–American variants of human papillomavirus 16 and risk for cervical cancer: a case-control study. J Natl Cancer Inst. 2001;93:1325–1330. doi: 10.1093/jnci/93.17.1325. [DOI] [PubMed] [Google Scholar]
- Bottalico D, Chen Z, Dunne A, Ostoloza J, McKinney S, Sun C, Schlecht NF, Fatahzadeh M, Herrero R, Schiffman M, Burk RD. The oral cavity contains abundant known and novel human papillomaviruses from the Betapapillomavirus and Gammapapillomavirus genera. J Infect Dis. 2011;204:787–792. doi: 10.1093/infdis/jir383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouvard V, Baan R, Straif K, Grosse Y, Secretan B, El Ghissassi F, Benbrahim-Tallaa L, Guha N, Freeman C, Galichet L, Cogliano V. A review of human carcinogens–Part B: biological agents. Lancet Oncol. 2009;10:321–322. doi: 10.1016/s1470-2045(09)70096-8. [DOI] [PubMed] [Google Scholar]
- Burk RD. Pernicious papillomavirus infection. N Engl J Med. 1999;341:1687–1688. doi: 10.1056/NEJM199911253412209. [DOI] [PubMed] [Google Scholar]
- Burk RD, Chen Z, Harari A, Smith BC, Kocjan BJ, Maver PJ, Poljak M. Classification and nomenclature system for human Alphapapillomavirus variants: general features, nucleotide landmarks and assignment of HPV6 and HPV11 isolates to variant lineages. Acta Dermatovenerologica Alp Panonica Adriat. 2011;20:113–123. [PMC free article] [PubMed] [Google Scholar]
- Burk RD, Chen Z, Van Doorslaer K. Human papillomaviruses: genetic basis of carcinogenicity. Public Health Genomics. 2009;12:281–290. doi: 10.1159/000214919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burk RD, Terai M, Gravitt PE, Brinton LA, Kurman RJ, Barnes WA, Greenberg MD, Hadjimichael OC, Fu L, McGowan L, Mortel R, Schwartz PE, Hildesheim A. Distribution of human papillomavirus types 16 and 18 variants in squamous cell carcinomas and adenocarcinomas of the cervix. Cancer Res. 2003;63:7215–7220. [PubMed] [Google Scholar]
- Calleja-Macias IE, Kalantari M, Huh J, Ortiz-Lopez R, Rojas-Martinez A, Gonzalez-Guerrero JF, Williamson AL, Hagmar B, Wiley DJ, Villarreal L, Bernard HU, Barrera-Saldana HA. Genomic diversity of human papillomavirus-16, 18, 31, and 35 isolates in a Mexican population and relationship to European, African, and Native American variants. Virology. 2004;319:315–323. doi: 10.1016/j.virol.2003.11.009. [DOI] [PubMed] [Google Scholar]
- Calleja-Macias IE, Villa LL, Prado JC, Kalantari M, Allan B, Williamson AL, Chung LP, Collins RJ, Zuna RE, Dunn ST, Chu TY, Cubie HA, Cuschieri K, von Knebel-Doeberitz M, Martins CR, Sanchez GI, Bosch FX, Munoz N, Bernard HU. Worldwide genomic diversity of the high-risk human papillomavirus types 31, 35, 52, and 58, four close relatives of human papillomavirus type 16. J Virol. 2005;79:13630–13640. doi: 10.1128/JVI.79.21.13630-13640.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan PK, Zhang C, Park JS, Smith-McCune KK, Palefsky JM, Giovannelli L, Coutlee F, Hibbitts S, Konno R, Settheetham-Ishida W, Chu TY, Ferrera A, Alejandra Picconi M, De Marco F, Woo YL, Raiol T, Pina-Sanchez P, Bae JH, Wong MC, Chirenje MZ, Magure T, Moscicki AB, Fiander AN, Capra G, Young KiE, Tan Y, Chen Z, Burk RD, Chan MC, Cheung TH, Pim D, Banks L. Geographical distribution and oncogenic risk association of human papillomavirus type 58 E6 and E7 sequence variations. Int J Cancer. 2013;132:2528–2536. doi: 10.1002/ijc.27932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan SY, Chew SH, Egawa K, Grussendorf-Conen EI, Honda Y, Rubben A, Tan KC, Bernard HU. Phylogenetic analysis of the human papillomavirus type 2 (HPV-2), HPV-27, and HPV-57 group, which is associated with common warts. Virology. 1997;239:296–302. doi: 10.1006/viro.1997.8896. [DOI] [PubMed] [Google Scholar]
- Chang YJ, Chen HC, Lee BH, You SL, Lin CY, Pan MH, Chou YC, Hsieh CY, Chen YM, Cheng YJ, Chen CJ, Group CHS. Unique variants of human papillomavirus genotypes 52 and 58 and risk of cervical neoplasia. Int J Cancer. 2011;129:965–973. doi: 10.1002/ijc.25724. [DOI] [PubMed] [Google Scholar]
- Chen Z, DeSalle R, Schiffman M, Herrero R, Burk RD. Evolutionary dynamics of variant genomes of human papillomavirus types 18, 45, and 97. J Virol. 2009;83:1443–1455. doi: 10.1128/JVI.02068-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Schiffman M, Herrero R, Desalle R, Anastos K, Segondy M, Sahasrabuddhe VV, Gravitt PE, Hsing AW, Burk RD. Evolution and taxonomic classification of human papillomavirus 16 (HPV16)-related variant genomes: HPV31, HPV33, HPV35, HPV52, HPV58 and HPV67. PLoS One. 2011;6:e20183. doi: 10.1371/journal.pone.0020183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Schiffman M, Herrero R, Desalle R, Anastos K, Segondy M, Sahasrabuddhe V, Gravitt PE, Hsing A, Burk RD. Evolution and taxonomic classification of Alphapapillomavirus 7 complete genomes: HPV18, HPV39, HPV45, HPV59, HPV68 and HPV70. PLoS One. 2013;8(8):e72565. doi: 10.1371/journal.pone.0072565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Terai M, Fu L, Herrero R, DeSalle R, Burk RD. Diversifying selection in human papillomavirus type 16 lineages based on complete genome analyses. J Virol. 2005;79:7014–7023. doi: 10.1128/JVI.79.11.7014-7023.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conway C, Chalkley R, High A, Maclennan K, Berri S, Chengot P, Alsop M, Egan P, Morgan J, Taylor GR, Chester J, Sen M, Rabbitts P, Wood HM. Next-generation sequencing for simultaneous determination of human papillomavirus load, subtype, and associated genomic copy number changes in tumors. J Mol Diagnostics. 2012;14:104–111. doi: 10.1016/j.jmoldx.2011.10.003. [DOI] [PubMed] [Google Scholar]
- Cornet I, Gheit T, Franceschi S, Vignat J, Burk RD, Sylla BS, Tommasino M, Clifford GM, Group IHVS. Human papillomavirus type 16 genetic variants: phylogeny and classification based on E6 and LCR. J Virol. 2012;86:6855–6861. doi: 10.1128/JVI.00483-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornet I, Gheit T, Clifford GM, Combes JD, Dalstein V, Franceschi S, Tommasino M, Clavel C. Human papillomavirus type 16 E6 variants in France and risk of viral persistence. Infect Agents Cancer. 2013a;8:4. doi: 10.1186/1750-9378-8-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornet I, Gheit T, Iannacone MR, Vignat J, Sylla BS, Del Mistro A, Franceschi S, Tommasino M, Clifford GM. HPV16 genetic variation and the development of cervical cancer worldwide. Br J Cancer. 2013b;108:240–244. doi: 10.1038/bjc.2012.508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Araujo Souza PS, Sichero L, Maciag PC. HPV variants and HLA polymorphisms: the role of variability on the risk of cervical cancer. Future Oncol. 2009;5:359–370. doi: 10.2217/fon.09.8. [DOI] [PubMed] [Google Scholar]
- De Boer MA, Peters LA, Aziz MF, Siregar B, Cornain S, Vrede MA, Jordanova ES, Fleuren GJ. Human papillomavirus type 18 variants: histopathology and E6/E7 polymorphisms in three countries. Int J Cancer. 2005;114:422–425. doi: 10.1002/ijc.20727. [DOI] [PubMed] [Google Scholar]
- de Villiers EM, Fauquet C, Broker TR, Bernard HU, zur Hausen H. Classification of papillomaviruses. Virology. 2004;324:17–27. doi: 10.1016/j.virol.2004.03.033. [DOI] [PubMed] [Google Scholar]
- de Villiers EM, Gissmann L, zur Hausen H. Molecular cloning of viral DNA from human genital warts. J Virol. 1981;40:932–935. doi: 10.1128/jvi.40.3.932-935.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deau MC, Favre M, Jablonska S, Rueda LA, Orth G. Genetic heterogeneity of oncogenic human papillomavirus type 5 (HPV5) and phylogeny of HPV5 variants associated with epidermodysplasia verruciformis. J Clin Microbiol. 1993;31:2918–2926. doi: 10.1128/jcm.31.11.2918-2926.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deau MC, Favre M, Orth G. Genetic heterogeneity among human papillomaviruses (HPV) associated with epidermodysplasia verruciformis: evidence for multiple allelic forms of HPV5 and HPV8 E6 genes. Virology. 1991;184:492–503. doi: 10.1016/0042-6822(91)90419-c. [DOI] [PubMed] [Google Scholar]
- Edgar RC. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics. 2004;5:113. doi: 10.1186/1471-2105-5-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekstrom J, Bzhalava D, Svenback D, Forslund O, Dillner J. High throughput sequencing reveals diversity of Human Papillomaviruses in cutaneous lesions. Int J Cancer. 2011;129:2643–2650. doi: 10.1002/ijc.26204. [DOI] [PubMed] [Google Scholar]
- Formentin A, Archambault J, Koushik A, Richardson H, Brassard P, Franco EL, Coutlee F. Human papillomavirus type 52 polymorphism and high-grade lesions of the uterine cervix. Int J Cancer. 2013;132:1821–1830. doi: 10.1002/ijc.27874. [DOI] [PubMed] [Google Scholar]
- Gheit T, Cornet I, Clifford GM, Iftner T, Munk C, Tommasino M, Kjaer SK. Risks for persistence and progression by human papillomavirus type 16 variant lineages among a population-based sample of Danish women. Cancer Epidemiol Biomarkers Prev. 2011;20:1315–1321. doi: 10.1158/1055-9965.EPI-10-1187. [DOI] [PubMed] [Google Scholar]
- Giannoudis A, Herrington CS. Human papillomavirus variants and squamous neoplasia of the cervix. J Pathol. 2001;193:295–302. doi: 10.1002/1096-9896(2000)9999:9999<::AID-PATH809>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- Gottschling M, Stamatakis A, Nindl I, Stockfleth E, Alonso A, Bravo IG. Multiple evolutionary mechanisms drive papillomavirus diversification. Mol Biol Evol. 2007;24:1242–1258. doi: 10.1093/molbev/msm039. [DOI] [PubMed] [Google Scholar]
- Guan P, Howell-Jones R, Li N, Bruni L, de Sanjose S, Franceschi S, Clifford GM. Human papillomavirus types in 115,789 HPV-positive women: a meta-analysis from cervical infection to cancer. Int J Cancer. 2012;131:2349–2359. doi: 10.1002/ijc.27485. [DOI] [PubMed] [Google Scholar]
- Hecht JL, Kadish AS, Jiang G, Burk RD. Genetic characterization of the human papillomavirus (HPV) 18 E2 gene in clinical specimens suggests the presence of a subtype with decreased oncogenic potential. Int J Cancer. 1995;60:369–376. doi: 10.1002/ijc.2910600317. [DOI] [PubMed] [Google Scholar]
- Heinzel PA, Chan SY, Ho L, O'Connor M, Balaram P, Campo MS, Fujinaga K, Kiviat N, Kuypers J, Pfister H, et al. Variation of human papillomavirus type 6 (HPV-6) and HPV-11 genomes sampled throughout the world. J Clin Microbiol. 1995;33:1746–1754. doi: 10.1128/jcm.33.7.1746-1754.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbst LH, Lenz J, Van Doorslaer K, Chen Z, Stacy BA, Wellehan JF, Jr, Manire CA, Burk RD. Genomic characterization of two novel reptilian papillomaviruses, Chelonia mydas papillomavirus 1 and Caretta caretta papillomavirus 1. Virology. 2009;383:131–135. doi: 10.1016/j.virol.2008.09.022. [DOI] [PubMed] [Google Scholar]
- Hildesheim A, Schiffman M, Bromley C, Wacholder S, Herrero R, Rodriguez A, Bratti MC, Sherman ME, Scarpidis U, Lin QQ, Terai M, Bromley RL, Buetow K, Apple RJ, Burk RD. Human papillomavirus type 16 variants and risk of cervical cancer. J Natl Cancer Inst. 2001;93:315–318. doi: 10.1093/jnci/93.4.315. [DOI] [PubMed] [Google Scholar]
- Hildesheim A, Wang SS. Host and viral genetics and risk of cervical cancer: a review. Virus Res. 2002;89:229–240. doi: 10.1016/s0168-1702(02)00191-0. [DOI] [PubMed] [Google Scholar]
- Ho L, Chan SY, Burk RD, Das BC, Fujinaga K, Icenogle JP, Kahn T, Kiviat N, Lancaster W, Mavromara-Nazos P, Labropoulou V, Mitrani-Rosenbaum S, Norrild B, Pillai MR, Stoerker J, Syrjaenen K, Syrjaenen S, Tay SK, Villa LL, Wheeler CM, Williamson AL, Bernard HU. The genetic drift of human papillomavirus type 16 is a means of reconstructing prehistoric viral spread and the movement of ancient human populations. J Virol. 1993;67:6413–6423. doi: 10.1128/jvi.67.11.6413-6423.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho L, Chan SY, Chow V, Chong T, Tay SK, Villa LL, Bernard HU. Sequence variants of human papillomavirus type 16 in clinical samples permit verification and extension of epidemiological studies and construction of a phylogenetic tree. J Clin Microbiol. 1991;29:1765–1772. doi: 10.1128/jcm.29.9.1765-1772.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA: Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- Kim YW, Bae SM, Kim YW, Park DC, Lee KH, Liu HB, Kim IW, Jang CK, Ahn WS. Target-based molecular signature characteristics of cervical adenocarcinoma and squamous cell carcinoma. Int J Oncol. 2013;43:539–547. doi: 10.3892/ijo.2013.1961. [DOI] [PubMed] [Google Scholar]
- Kohler A, Gottschling M, Manning K, Lehmann MD, Schulz E, Kruger-Corcoran D, Stockfleth E, Nindl I. Genomic characterization of ten novel cutaneous human papillomaviruses from keratotic lesions of immunosuppressed patients. J Gen Virol. 2011;92:1585–1594. doi: 10.1099/vir.0.030593-0. [DOI] [PubMed] [Google Scholar]
- Li J, Cai H, Xu Z, Wang Q, Hang D, Shen N, Liu M, Zhang C, Abliz A, Ke Y. Nine complete genome sequences of cutaneous human papillomavirus genotypes isolated from healthy skin of individuals living in rural he nan province, China. J Virol. 2012;86:11936. doi: 10.1128/JVI.01988-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Franceschi S, Howell-Jones R, Snijders PJ, Clifford GM. Human papillomavirus type distribution in 30,848 invasive cervical cancers worldwide: Variation by geographical region, histological type and year of publication. Int J Cancer. 2011;128:927–935. doi: 10.1002/ijc.25396. [DOI] [PubMed] [Google Scholar]
- Lizano M, Berumen J, Garcia-Carranca A. HPV-related carcinogenesis: basic concepts, viral types and variants. Arch Med Res. 2009;40:428–434. doi: 10.1016/j.arcmed.2009.06.001. [DOI] [PubMed] [Google Scholar]
- Luhn P, Walker J, Schiffman M, Zuna RE, Dunn ST, Gold MA, Smith K, Mathews C, Allen RA, Zhang R, Wang S, Wentzensen N. The role of co-factors in the progression from human papillomavirus infection to cervical cancer. Gynecol Oncol. 2013;128:265–270. doi: 10.1016/j.ygyno.2012.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matos RP, Sichero L, Mansur IM, Bonfim CM, Bittar C, Nogueira RL, Kupper DS, Valera FC, Nogueira ML, Villa LL, Rahal P, Calmon MF. Nucleotide and phylogenetic analysis of human papillomavirus types 6 and 11 isolated from recurrent respiratory papillomatosis in Brazil. Infect Genet Evol. 2013;16:282–289. doi: 10.1016/j.meegid.2012.12.033. [DOI] [PubMed] [Google Scholar]
- Mokili JL, Dutilh BE, Lim YW, Schneider BS, Taylor T, Haynes MR, Metzgar D, Myers CA, Blair PJ, Nosrat B, Wolfe ND, Rohwer F. Identification of a novel human papillomavirus by metagenomic analysis of samples from patients with febrile respiratory illness. PLoS One. 2013;8:e58404. doi: 10.1371/journal.pone.0058404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mounts P, Shah KV, Kashima H. Viral etiology of juvenile- and adult-onset squamous papilloma of the larynx. Proc Natl Acad Sci USA. 1982;79:5425–5429. doi: 10.1073/pnas.79.17.5425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz N, Bosch FX, de Sanjose S, Herrero R, Castellsague X, Shah KV, Snijders PJ, Meijer CJ. Epidemiologic classification of human papillomavirus types associated with cervical cancer. N Engl J Med. 2003;348:518–527. doi: 10.1056/NEJMoa021641. [DOI] [PubMed] [Google Scholar]
- Ong CK, Chan SY, Campo MS, Fujinaga K, Mavromara-Nazos P, Labropoulou V, Pfister H, Tay SK, ter Meulen J, Villa LL, Bernard HU. Evolution of human papillomavirus type 18: an ancient phylogenetic root in Africa and intratype diversity reflect coevolution with human ethnic groups. J Virol. 1993;67:6424–6431. doi: 10.1128/jvi.67.11.6424-6431.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prado JC, Calleja-Macias IE, Bernard HU, Kalantari M, Macay SA, Allan B, Williamson AL, Chung LP, Collins RJ, Zuna RE, Dunn ST, Ortiz-Lopez R, Barrera-Saldana HA, Cubie HA, Cuschieri K, von Knebel-Doeberitz M, Sanchez GI, Bosch FX, Villa LL. Worldwide genomic diversity of the human papillomaviruses-53, 56, and 66, a group of high-risk HPVs unrelated to HPV-16 and HPV-18. Virology. 2005;340:95–104. doi: 10.1016/j.virol.2005.06.024. [DOI] [PubMed] [Google Scholar]
- Quint KD, de Koning MN, van Doorn LJ, Quint WG, Pirog EC. HPV genotyping and HPV16 variant analysis in glandular and squamous neoplastic lesions of the uterine cervix. Gynecol Oncol. 2010;117:297–301. doi: 10.1016/j.ygyno.2010.02.003. [DOI] [PubMed] [Google Scholar]
- Rector A, Lemey P, Tachezy R, Mostmans S, Ghim SJ, Van Doorslaer K, Roelke M, Bush M, Montali RJ, Joslin J, Burk RD, Jenson AB, Sundberg JP, Shapiro B, Van Ranst M. Ancient papillomavirus-host co-speciation in Felidae. Genome Biol. 2007;8:R57. doi: 10.1186/gb-2007-8-4-r57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabol I, Matovina M, Si-Mohamed A, Grce M. Characterization and whole genome analysis of human papillomavirus type 16 e1-1374\widehat63nt variants. PLoS One. 2012;7:e41045. doi: 10.1371/journal.pone.0041045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiffman M, Castle PE, Jeronimo J, Rodriguez AC, Wacholder S. Human papillomavirus and cervical cancer. Lancet. 2007;370:890–907. doi: 10.1016/S0140-6736(07)61416-0. [DOI] [PubMed] [Google Scholar]
- Schiffman M, Herrero R, Desalle R, Hildesheim A, Wacholder S, Rodriguez AC, Bratti MC, Sherman ME, Morales J, Guillen D, Alfaro M, Hutchinson M, Wright TC, Solomon D, Chen Z, Schussler J, Castle PE, Burk RD. The carcinogenicity of human papillomavirus types reflects viral evolution. Virology. 2005;337:76–84. doi: 10.1016/j.virol.2005.04.002. [DOI] [PubMed] [Google Scholar]
- Schiffman M, Rodriguez AC, Chen Z, Wacholder S, Herrero R, Hildesheim A, Desalle R, Befano B, Yu K, Safaeian M, Sherman ME, Morales J, Guillen D, Alfaro M, Hutchinson M, Solomon D, Castle PE, Burk RD. A population-based prospective study of carcinogenic human papillomavirus variant lineages, viral persistence, and cervical neoplasia. Cancer Res. 2010;70:3159–3169. doi: 10.1158/0008-5472.CAN-09-4179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiffman M, Wentzensen N. Human papillomavirus infection and the multistage carcinogenesis of cervical cancer. Cancer Epidemiol Biomarkers Prev. 2013;22:553–560. doi: 10.1158/1055-9965.EPI-12-1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seedorf K, Krammer G, Durst M, Suhai S, Rowekamp WG. Human papillomavirus type 16 DNA sequence. Virology. 1985;145:181–185. doi: 10.1016/0042-6822(85)90214-4. [DOI] [PubMed] [Google Scholar]
- Sichero L, Ferreira S, Trottier H, Duarte-Franco E, Ferenczy A, Franco EL, Villa LL. High grade cervical lesions are caused preferentially by non-European variants of HPVs 16 and 18. Int J Cancer. 2007;120:1763–1768. doi: 10.1002/ijc.22481. [DOI] [PubMed] [Google Scholar]
- Sichero L, Villa LL. Epidemiological and functional implications of molecular variants of human papillomavirus. Braz J Med Biol Res. 2006;39:707–717. doi: 10.1590/s0100-879x2006000600002. [DOI] [PubMed] [Google Scholar]
- Smith B, Chen Z, Reimers L, van Doorslaer K, Schiffman M, Desalle R, Herrero R, Yu K, Wacholder S, Wang T, Burk RD. Sequence imputation of HPV16 genomes for genetic association studies. PLoS One. 2011;6:e21375. doi: 10.1371/journal.pone.0021375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JS, Lindsay L, Hoots B, Keys J, Franceschi S, Winer R, Clifford GM. Human papillomavirus type distribution in invasive cervical cancer and high-grade cervical lesions: a meta-analysis update. Int J Cancer. 2007;121:621–632. doi: 10.1002/ijc.22527. [DOI] [PubMed] [Google Scholar]
- Stamatakis A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 2006;22:2688–2690. doi: 10.1093/bioinformatics/btl446. [DOI] [PubMed] [Google Scholar]
- Stewart AC, Eriksson AM, Manos MM, Munoz N, Bosch FX, Peto J, Wheeler CM. Intratype variation in 12 human papillomavirus types: a worldwide perspective. J Virol. 1996;70:3127–3136. doi: 10.1128/jvi.70.5.3127-3136.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun M, Gao L, Liu Y, Zhao Y, Wang X, Pan Y, Ning T, Cai H, Yang H, Zhai W, Ke Y. Whole genome sequencing and evolutionary analysis of human papillomavirus type 16 in central china. PloS One. 2012;7:e36577. doi: 10.1371/journal.pone.0036577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terai M, DeSalle R, Burk RD. Lack of canonical E6 and E7 open reading frames in bird papillomaviruses: Fringilla coelebs papillomavirus and Psittacus erithacus timneh papillomavirus. J Virol. 2002;76:10020–10023. doi: 10.1128/JVI.76.19.10020-10023.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villa LL, Sichero L, Rahal P, Caballero O, Ferenczy A, Rohan T, Franco EL. Molecular variants of human papillomavirus types 16 and 18 preferentially associated with cervical neoplasia. J Gen Virol. 2000;81:2959–2968. doi: 10.1099/0022-1317-81-12-2959. [DOI] [PubMed] [Google Scholar]
- Wheeler CM. Natural history of human papillomavirus infections, cytologic and histologic abnormalities, and cancer. Obstet Gynecol Clin North Am. 2008;35(vii):519–536. doi: 10.1016/j.ogc.2008.09.006. [DOI] [PubMed] [Google Scholar]
- Xi LF, Kiviat NB, Hildesheim A, Galloway DA, Wheeler CM, Ho J, Koutsky LA. Human papillomavirus type 16 and 18 variants: race-related distribution and persistence. J Natl Cancer Inst. 2006;98:1045–1052. doi: 10.1093/jnci/djj297. [DOI] [PubMed] [Google Scholar]
- Xi LF, Koutsky LA, Hildesheim A, Galloway DA, Wheeler CM, Winer RL, Ho J, Kiviat NB. Risk for high-grade cervical intraepithelial neoplasia associated with variants of human papillomavirus types 16 and 18. Cancer Epidemiol Biomarkers Prev. 2007;16:4–10. doi: 10.1158/1055-9965.EPI-06-0670. [DOI] [PubMed] [Google Scholar]
- Xi LF, Schiffman M, Koutsky LA, He Z, Winer RL, Hulbert A, Lee SK, Ke Y, Kiviat NB. Persistence of newly detected human papillomavirus type 31 infection, stratified by variant lineage. Int J Cancer. 2013;132:549–555. doi: 10.1002/ijc.27689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi LF, Schiffman M, Koutsky LA, Hulbert A, Lee SK, Defilippis V, Shen Z, Kiviat NB. Association of human papillomavirus type 31 variants with risk of cervical intraepithelial neoplasia grades 2–3. Int J Cancer. 2012;131:2300–2307. doi: 10.1002/ijc.27520. [DOI] [PubMed] [Google Scholar]
- Yamada T, Manos MM, Peto J, Greer CE, Munoz N, Bosch FX, Wheeler CM. Human papillomavirus type 16 sequence variation in cervical cancers: a worldwide perspective. J Virol. 1997;71:2463–2472. doi: 10.1128/jvi.71.3.2463-2472.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada T, Wheeler CM, Halpern AL, Stewart AC, Hildesheim A, Jenison SA. Human papillomavirus type 16 variant lineages in United States populations characterized by nucleotide sequence analysis of the E6, L2, and L1 coding segments. J Virol. 1995;69:7743–7753. doi: 10.1128/jvi.69.12.7743-7753.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuna RE, Moore WE, Shanesmith RP, Dunn ST, Wang SS, Schiffman M, Blakey GL, Teel T. Association of HPV16 E6 variants with diagnostic severity in cervical cytology samples of 354 women in a US population. Int J Cancer. 2009;125:2609–2613. doi: 10.1002/ijc.24706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuna RE, Tuller E, Wentzensen N, Mathews C, Allen RA, Shanesmith R, Dunn ST, Gold MA, Wang SS, Walker J, Schiffman M. HPV16 variant lineage, clinical stage, and survival in women with invasive cervical cancer. Infect Agents Cancer. 2011;6:19. doi: 10.1186/1750-9378-6-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.