

Abstract
Translocation of mRNA and tRNA through the ribosome is a crucial step in protein synthesis, whose mechanism is not yet understood. The crystal structures of three Thermus ribosome·tRNA·mRNA·EF-G complexes trapped with GDPNP or fusidic acid reveal conformational changes occurring during intermediate states of translocation, including large-scale rotation of the 30S subunit head and body. In all complexes, the tRNA acceptor ends occupy the 50S subunit E site, while their anticodon stem-loops move with the head of the 30S subunit to positions between the P and E sites, forming chimeric intermediate states. Two universally conserved bases of 16S rRNA that intercalate between bases of the mRNA may act as “pawls” of a translocational ratchet. These findings provide new insights into the molecular mechanism of ribosomal translocation.
Introduction
One of the most critical and complex steps of protein synthesis is the coupled translocation of mRNA and tRNAs through the ribosome, catalyzed by the GTPase elongation factor EF-G. Although several of the main steps have been identified, the underlying molecular mechanisms of translocation are poorly understood. Immediately following peptide bond formation, the A (aminoacyl-tRNA) site is occupied by the newly extended peptidyl-tRNA and the P (peptidyl-tRNA) site by a deacylated tRNA. In the first step of translocation, the acceptor end of the deacylated tRNA moves into the 50S subunit E (exit) site, forming the P/E hybrid state, and the acceptor end of the peptidyl-tRNA moves into the 50S P site, forming the A/P hybrid state (1, 2). Hybrid-states formation can occur spontaneously and reversibly in vitro (1, 3, 4), and is correlated with intersubunit rotational movement (4–6). Although this step can proceed in the absence of EF-G or GTP, the observation that EF-G favors hybrid-states formation (5) suggests that it is catalyzed by EF-G in vivo. Translocation is completed when the anticodon stem-loops (ASLs) of the tRNAs move from the 30S A and P sites to the P and E sites, respectively, coupled to movement of their associated mRNA codons. The latter step is rate-limiting (7), and strongly dependent on EF-G. There is increasing evidence that it is coupled to a rotational movement of the head of the small subunit (8–11). Although several cryo-EM reconstructions of EF-G-ribosome complexes have been determined (8, 12–14), until now only a single crystal structure has been reported, for a post-translocation complex in which EF-G was trapped on the ribosome with fusidic acid in the non-rotated state (15). Here, we describe crystal structures for three ribosome complexes containing EF-G, mRNA and tRNA trapped in intermediate states in which the 30S subunit head undergoes large-scale (15°–18°) intrasubunit rotations and the 30S body smaller (3°–5°) intersubunit rotations. Our findings provide insights into the molecular mechanisms of EF-G-catalyzed translocation.
Results
EF-G-Dependent Structural Changes in the Ribosome
Ribosomes from Thermus thermophilus were co-crystallized with the 27-nucleotide mv27 mRNA, elongator tRNAMet and EF-G in the presence of GTP and the antibiotic fusidic acid, which allows GTP hydrolysis but prevents release of EF-G (16), or with EF-G and GDPNP, a non-hydrolyzable analogue of GTP. Crystals of the fusidic acid complex (Fus) diffracted to 3.6Å, and those of the two other complexes, GDPNP-I and GDPNP-II, which crystallized under slightly different conditions, to 3.5Å and 4.1Å, respectively (Table S1). The most notable structural change is a large-scale counterclockwise rotation of the 30S head domain by 15° (Fus and GDPNP-I) or 18° (GDPNP-II), relative to the classical-state ribosome (17), around an axis roughly parallel to the long axis of the subunit (Fig. 1C–E). This is accompanied by smaller 3° (Fus and GDPNP-I) or 5° (complex GDPNP-II) counterclockwise rotations of the 30S body relative to the 50S subunit. These rotational movements resemble those observed for an EF-G·ribosome cryo-EM reconstruction (8) and for an RF3·ribosome crystal structure (18). Additional large-scale changes include a 14Å inward movement of the 50S subunit L1 stalk to contact the elbow of the tRNA (Fig. S1), and a 12Å inward movement of the L11 stalk to avoid clash with its contact site on domain V of EF-G. Rotation of the 30S head results in 25–30Å displacements of the contacts forming intersubunit bridge B1a between 30S protein S13 and the A-site finger (helix H38 of 23S rRNA), resulting in formation of a new bridge with protein S19, and an 11Å upward movement of H38 in the Fus and GDPNP-I complexes; in the GDPNP-II complex, this increases to 14Å because of greater rotation of the 30S head.
Figure 1. Structures of trapped 70S ribosome·EF-G complexes.

(A,B) Overall views of the (A) non-rotated 70S·EF-G-post complex (15) and (B) Fus 70S·EF-G complex with tRNA bound in the pe*/E state. (C–E) Interface views showing 30S subunit body and head rotation in the (C) EF-G-post state (15); (D) GDPNP-I and Fus complex; and (E) GDPNP-II complex. (F–H) Close-up views of EF-G domain IV interactions with the 30S subunit head in the (F) EF-G-post complex (15); (G) GDPNP-I and Fus complex; and (H) GDPNP-II complex. 16S rRNA, cyan; 30S proteins, blue; 23S rRNA, grey; 50S proteins, magenta; mRNA, green; P/P tRNA, red; E/E tRNA, yellow; pe*/E tRNA, red; EF-G, orange.
Trapping of tRNA in the Chimeric pe*/E Intermediate State of Translocation
Binding of EF-G to the ribosome with either GTP and fusidic acid or as a GDPNP complex, under the conditions of complex formation and crystallization, results in movement of the tRNA from its initial binding state in the classical P site (P/P state) into new binding states. During rotation of the head of the 30S subunit, the P-site contacts between the ASL and the head (19, 20) are maintained, while those between the ASL and the 30S body and platform are disrupted. In the classical-state ribosome, a 14Å constriction between nucleotides 790 in the platform and 1340 in the head blocks movement of the ASL between the 30S P and E sites (21). Rotation of the 30S head widens this gap to 22Å in the Fus and GDPNP-I complexes, and to 24Å in GDPNP-II, allowing sufficient room for the ASL to move between the P and E sites (Fig. 2). The ASL precisely follows the rotational movement of the 30S subunit head, placing it midway between the positions of the classical P and E sites (Fig. 2). The positions of the ASLs in the Fus and GDPNP-I complexes superimpose closely, while the ASL in GDPNP-II is displaced further toward the E site. Interactions between tRNA and the 50S subunit are similar to the E-site interactions observed for tRNA bound in the classical E/E and hybrid P/E states (Fig. S1) (19, 20). Because the ASL is simultaneously bound to P-site elements of the 30S head and to features that lie between the P and E sites of the 30S body and platform, we refer to this new state as a chimeric pe*/E hybrid state. Repositioning of the tRNA is promoted not only by structural changes in the ribosome, but also by conformational changes in the tRNA itself. We imagine that the pe*/E state represents an intermediate between the P/E and E/E states.
Fig. 2. Movement of tRNA from the P/P to the pe*/E state.

(A) Electron density map (2Fo-Fc) contoured at 1.5σ for pe*/E tRNA from the Fus complex. (B) Superimposition of tRNA positions for the P/P (blue), E/E (grey) and pe*/E states from the Fus, (red); and GDPNP-II (pink) complexes, aligned on the 23S rRNAs from each complex. The pe*/E tRNAs move from the P site to positions midway between the P and E sites. (C) Positions of the ASLs of tRNA in the Fus (red) and GDPNP-II (pink) pe*/E states compared with those of the classical P/P (blue) and E/E (grey) (17) and hybrid-state P/E (yellow)(6) tRNAs, aligned on the 30S subunit body. (D) Interactions of (left) a P/P classical-state ASL (17) and (right) the pe*/E ASL from the 70S·EF-G Fus complex with the 30S subunit. The 15° rotation of the 30S subunit head in the EF-G opens the constriction blocking passage of the tRNA from the P site to the E site from 14Å to 22 Å, allowing translocation. (E) Side views of the complexes shown in (D). Rotation of the head moves the tRNA ASL away from its contacts with C1400 and A790 in the body and platform as it translocates into the pe*/E state.
Aligning the structures of tRNAs bound in these three states on their ASLs (Fig. 3) shows that the body of the E/E tRNA bends downward and sideways toward the P site, relative to the P/E tRNA (Fig. 3A,D). The pe*/E tRNA closely matches the downward bend of the E/E tRNA (Fig. 3C,F), but with a sideways bend half the magnitude of that of the E/E tRNA, consistent with its intermediate state of translocation. The main position of flexing of the tRNAs is localized to the junction between their D and anticodon stems at the non-canonical 26–44 base pair, as has been seen for kinking of the A/T-state tRNA in the EF-Tu ternary complex during tRNA selection (22, 23). These findings provide further evidence that tRNA does not transit the ribosome as a rigid body during protein synthesis, but exhibits considerable flexibility.
Fig. 3. Flexing of tRNA during translocation.
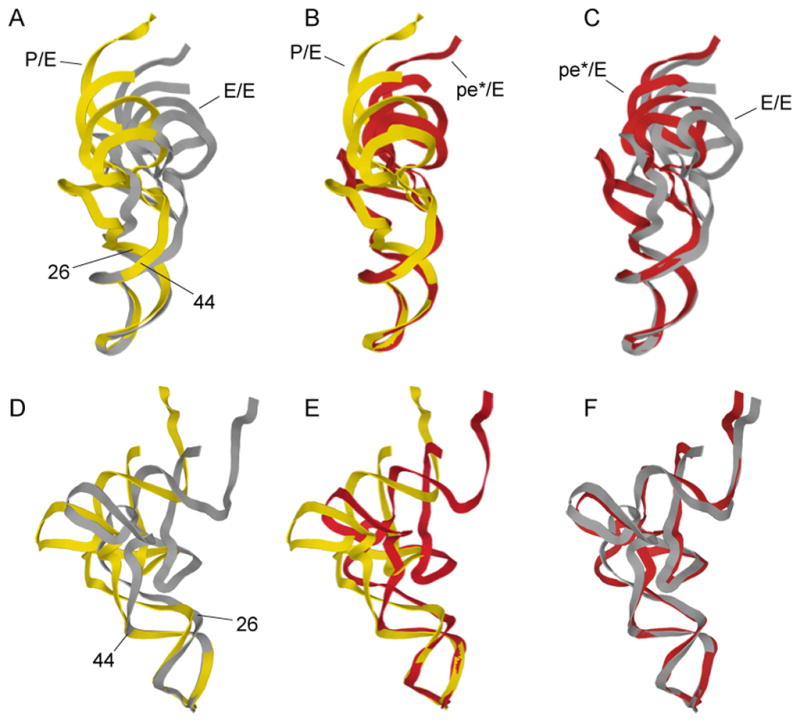
Comparison of the conformations of tRNA in the (A,D) hybrid P/E and classical E/E states; (B,E) chimeric pe*/E and P/E states; (C,F) pe*/E and E/E states, aligned on their respective ASLs. The main site of flexing is localized to the non-canonical base pair at positions 26–42, between the D and anticodon stems. The P/E and E/E structures are from (6, 15).
mRNA Interactions
Well-resolved electron density allows fitting of 23 of the 27 nucleotides of the mv27 mRNA in the GDPNP-I and Fus complexes (Figs. 4, S3). The path of the mRNA and its contacts with the 30S body and platform (which serve as static reference points for mRNA movement) in the GDPNP-I complex can be compared with those of a mRNA from a previously determined structure of a complex containing tRNAs bound in classical states (Fig. 4)(17). In the classical-state complex, protein S11 contacts the mRNA upstream from the E site at positions −6 and −7, (where position +1 is defined as the 5′ nucleotide of the P-site codon in the initially formed complex); in the EF-G Fus complex, the corresponding contacts are made with positions −3 and −4, respectively, indicating a displacement of the mRNA by 3 nucleotides, or one codon. Using another landmark, 16S nucleotide U1498 packs against riboses +1 and +2 of the mRNA in the classical complex; in the Fus and GDPNP-I complexes, U1498 packs against ribose +4 and phosphate +5, corresponding again to translocation of nearly 3 nucleotides, or one codon. However, calibration against G926 gives a smaller displacement; in the classical complex, G926 H-bonds with phosphate +1, whereas in the EF-G Fus complex, it interacts with phosphate +3. This net displacement of the mRNA by about 2 nucleotides in the EF-G·GDP·fusidic acid complex extends through the downstream region to position +11, while upstream of the P site, the mRNA translocates by about 3 nucleotides, or one full codon. This difference may be due to flexibility in the downstream part of the mRNA chain caused by the sparse downstream contacts resulting from a vacant A site. Codon-anticodon base pairing is disrupted in the Fus and GDPNP-I complexes; however, pairing with the first two codon bases is retained in the GDPNP-II complex (Fig. S4).
Fig. 4. Interactions of mRNA with the 30S subunit.

(A) mRNA bound to a classical-state 70S ribosome (17). (B) mRNA bound to the Fus complex with EF-G and pe*/E tRNA. The positions of proteins S3, S11 and S18 are shown as blue transparent molecular surfaces; also shown are the positions of EF-G; the anticodon stem-loops of EF-G, P-tRNA, E-tRNA and pe*/E tRNA; the Shine-Dalgarno helix (S/D). Elements of 16S rRNA are shown in cyan. The A-, P- and E-site codons for the mRNAs during complex formation are shown in yellow, orange and red, respectively. The mRNAs are numbered with +1 corresponding to the 5′ nucleotide of the P-site codon. (C,D) The conformations of the tertiary hairpin-like structures containing the intercalating bases C1397 and A1503 (shown in red) in (C) the classical-state ribosome and (D) the 70S·EF-G Fus complex. The structures of these features in the GDPNP I and II complexes are similar to those of the Fus complex. The universally conserved C1399-G1504 base pair is shown in dark blue.
An important question concerning the mechanics of translocation is how the translational reading frame is maintained. Slippage of the reading frame will occur if the movement of mRNA and tRNAs are not precisely synchronized, resulting in translation of a stream of incorrect codons and most likely, an out-of-frame termination codon that risks creating a dominant, toxic product. Of potential relevance to this question is the intercalation of two bases from 16S rRNA between bases at two positions of the mRNA (Figs. 4, S5). In the Fus and GDPNP-I complexes, C1397 stacks on A+10 (Fig. 4), partially intercalating between bases +9 and +10; it is not clear whether this interaction occurs in the GDPNP-II complex, because of disorder in this region of its mRNA. Intercalation of C1397 was also observed in a termination complex formed with release factor RF2 (24). Previously unreported is the intercalation of A1503 between positions -1 and -2 (Fig. 4), which is observed in all three complexes. These interactions would block mRNA movement in either the forward or reverse direction. We propose that they act as “pawls” of a translocational ratchet to prevent reversal of the movement of the mRNA chain during translocation. In the classical-state complex (17), where both the extended structure of the mRNA and bases 1397 and 1503 are well resolved, both bases are retracted from their intercalated conformations, a state that would permit mRNA movement; in the state observed for our EF-G-containing complexes, their intercalation would lock the register of the mRNA, preventing slippage of the translational reading frame. At some point prior to the next round of translocation, the bases would need to retract from their intercalated state to allow mRNA movement. The universal conservation of bases 1397 and 1503, from bacteria to archaea to eukarya, is consistent with such a critical role. Both bases project from the tips of conserved, compact, tertiary hairpin-like structures that seem optimized to facilitate intercalation (Fig. 4C,D). Moreover, the two hairpins are connected by a conserved, tertiary Watson-Crick base pair between C1399 and G1504, suggesting that their movements could be coordinated during translocation.
Structuring of Switch Loop I of EF-G
EF-G is bound in the subunit interface of the ribosome, where its domain I binds to the sarcin-ricin loop (SRL) (Fig. S8) and protein L6 of the 50S subunit, domains II and III with 16S rRNA helices h5 and h15 and protein S12 in the 30S subunit, domain IV with the mRNA and helix h34 in the 30S subunit and domain V to the L11 stalk of the 50S subunit. In all previous structures of EF-G, the switch loop I region (25), was disordered. This functionally important feature of EF-G (residues 40–67 in T. thermophilus EF-G) is now well resolved in the GDPNP-I complex, revealing its contacts with other regions of EF-G, with the ribosome and with GDPNP (Fig. 6). Structuring of the conserved core of switch loop I (positions 59–67 in T. thermophilus EF-G) completes the cage that encloses GDPNP and fixes its position with a network of H-bonded and Van der Waals interactions (Fig. 6B). Arg61 in switch loop I interacts with the phosphate of G2663 in the SRL, while Thr64 contacts the β- and γ-phosphates of GDPNP via a coordinated magnesium ion. The NKXD motif (residues 137–140) interacts with the guanosine end and the phosphate binding loop (residues 20–27) wraps around the triphosphate end of the nucleotide cofactor (Fig. 6B). The position of the conserved His87 in the switch loop II does not allow it to act as a general base as in a previously proposed universal mechanism for the GTPase reaction (26)(Fig. S9). Structuring of switch loop I appears to fix the relative geometry of domains I, III and V through contacts with domain III. Comparison with the crystal structure of EF-G·GDP in its free form (27), whose switch loop I is unstructured, shows rearrangement of domains III and V relative to domain I (Fig. 6E,F). In the GDP form, contacts between domain III and the 30S subunit and domain V and the L11 stalk are disrupted, likely causing release of EF-G from the ribosome. The position of fusidic acid in the Fus complex overlaps that of residues 63–66 in the conserved core of switch loop I, mimicking the stabilizing contacts between domains I and III made by the structured switch loop (Fig. 6C,D). This finding explains how fusidic acid stabilizes a conformation virtually identical to that of the GDPNP-I complex, allowing EF-G to bind stably to the ribosome in the presence of GDP.
Fig. 6. Structuring of switch loop I.

(A) EF-G GDPNP-I showing the path of switch loop I (residues 40–67) which was disordered in all previous EF-G structures. The conserved core (residues 59–67) is shown in blue, and the rest of the switch loop I (residues 40–58) in magenta. (B) Structure of the GDPNP binding pocket, showing interactions with switch loop I (light blue), the guanosine recognition motif (cyan) and P loop (orange). A magnesium ion coordinating the β and γ phosphates of GDPNP is shown as a green sphere. (C) The switch I region in the GDPNP-I complex and (D) The fusidic acid binding site in the Fus complex, in the same view as for (C). In (C) and (D), showing the conserved core of switch loop I (blue) and the rest of switch loop I (magenta), guanosine recognition motif (cyan), phosphate binding loop (orange) and switch loop 2 (green) and domain III contacts (yellow); the components are shown with transparent molecular surface representations. (E) EF-G from the GDPNP-I structure containing a structured switch loop I (blue, magenta) compared with (F) the structure of free EFG·GDP containing a disordered switch loop I (27) showing movement of domains III (yellow), IV and V (cyan).
Comparison of the folds of the crystal structures of the four different translational G proteins in which switch loop I is ordered shows that while the conserved core of switch loop I (residues 59–67) is positioned similarly in all four cases, the folds of the N-terminal of the switch loop (residues 40–58) diverge into two classes (Fig. S10). In one class, which includes EF-G and EF-Tu (26), the N-terminal region of the loop continues in the direction of the α1 helix, then loops back toward the beta-phosphate of GDPNP where it joins the conserved core. The other class comprises EF-G-2 (28) and RF3 (18), in which the loop runs parallel to the α1 helix and then forms a complex fold that packs against GDPNP before joining the conserved core. The crystal structure of free EF-G-2·GTP was fitted to the electron density for EF-G from a cryo-EM reconstruction of an EF-G·ribosome complex (28). However, the resulting interdomain geometry for EF-G-2 differs from that of the EF-G structures, particularly for domain III, which is shifted by as much as 9Å relative to that seen in GDPNP-I. Details of the interactions between the C-terminal domain (CTD) of protein L12 and the G′ domain of EF-G, which are believed to be important for recruiting EF-G to the ribosome and for stimulating GTP hydrolysis (29, 30) can be visualized in both the Fus and GDPNP-I complexes (Figs. 5A–B, S6). These involve residues A67, K71, T82, K87 and K90 of the CTD with K215, E218, D222, Y231 and V232 of the G′ domain, the majority of which are universally conserved.
Fig. 5. EF-G interactions and dynamics.

(A) Overall view of the position of EF-G on the 50S subunit. (B) Detailed view of the contact surface between the G′-domain of EF-G and the C-terminal domain (CTD) of one of the four copies of protein L12. 23S rRNA (grey), 50S proteins (magenta), EF-G (orange), L12 CTD (magenta in A, blue in B). (C) Superimposition of EF-G from the fusidic acid post complex (grey) (15) with EF-G from the GDPNP-I complex (orange) by alignment on their domains I shows rearrangements in the orientation of domains III and IV. (D) Alignment of the same two EF-G structures in panel C on their respective domains IV shows local rearrangement of the loop containing the conserved Gly502-Gly503 residues (cf. Fig. 1F–H).
Interdomain Movement in EF-G
Comparison of the different structures of EF-G in its ribosome-bound state shows that the largest conformational changes occur in domains III and IV, between the EF-G-post complex (15) and the Fus and GDPNP-I complexes. Domain IV moves upward toward the head of the 30S subunit by ~6Å and domain III by ~3Å (Figs. 5C, S7) (Fig. 1G). At the tip of domain IV, the loop containing the conserved Gly502 moves by ~5Å between the post and GDPNP-I and Fus structures, as its contact with the mRNA backbone switches from position +2 to +4 (Fig. 1F,G). Although the largest displacement of domain IV in the ribosome is seen in the GDPNP-II complex, this is a mainly a result of the overall rotational movement of EF-G relative to the ribosome, rather than internal movement of the EF-G molecule. In the GDPNP-II complex, 23S rRNA features that contact EF-G, including the SRL, the L11 stalk and H89, move by 2–4 Å in concert with movement of EF-G, which follows the increased intersubunit rotation of the 30S subunit; on the 30S side, this results in disruption of contacts between domain III of EF-G and protein S12. Rotation of the 30S subunit head and body in the GDPNP-II complex brings the tip of domain IV into contact with helix 34 of 16S rRNA in the GDPNP-II complex, as observed previously by cryo-EM (8), where the backbone oxygens of Val530 and the universally conserved Gly531 form H-bonds with phosphate 1210 and ribose 1209 of 16S rRNA, respectively (Fig. 1H). This interaction appears to participate in fixing the position of the head of the 30S subunit in the rotational state observed for the GDPNP-II complex.
Discussion
In the absence of continuous rotary motion (31) (which is unlikely for an asymmetric structure like the ribosome), translocation of mRNA and tRNA through the ribosome must be based on some kind of ratchet mechanism, as was realized many decades ago (32). Intersubunit rotational movement, which is coupled to hybrid-states formation, has been termed “ratcheting” (4, 6, 12, 33–35); however, ribosomes containing bound tRNAs have been observed to undergo spontaneous, reversible, back-and-forth intersubunit rotation in the absence of EF-G or GTP (3), unlike the behavior of a true ratchet. Moreover, the step of translocation that is critically dependent on catalysis by EF-G is the movement of mRNA, coupled to that of the tRNA ASLs, on the 30S subunit (1, 7). In a cryo-EM reconstruction of fusidic acid-trapped EF-G complexes, Spahn and co-workers recently resolved a previously unobserved sub-population of particles in which rotation of the 30S subunit head was accompanied by movement of the ASL of a P-site tRNA into a novel intermediate state (8). Because the tRNA maintained contact with the P site on the 30S head and simultaneously established interaction with the E site on the 30S platform, they termed this the ‘pe/E’ hybrid state. In our crystal structures, head rotation in the three different trapped EF-G complexes results in movement of the ASLs to positions midway between the 30S P and E sites (Fig. 2); accordingly, we term these states chimeric pe*/E hybrid states. Close comparison with the cryo-EM structure (8) shows that the ASL in their pe/E state is positioned 5Å further toward the E site from the pe*/E ASL in the GDPNP-I and Fus structures, and 2.7Å further in the GDPNP-II structure. The pe/E and pe*/E states appear to represent intermediate states between the hybrid P/E and classical E/E states, near the completion of the EF-G-catalyzed translocation of a P-site tRNA. The larger body rotations observed for some ribosome complexes (6, 18, 36), together with the large head rotation observed in the GDPNP-II complex, may be sufficient to complete the movement of tRNA fully into the eventual E/E state, as predicted from the rotational values observed for a ribosome·RF3 complex (18). Finally, our structures suggest that C1397 and A1503 act as molecular pawls to fix the position of the mRNA ratchet; we do not know if there are separate pawls to prevent back-slippage of tRNA during reverse rotation of the 30S head, or indeed, whether the movements of mRNA and tRNA are coupled simply through their codon-anticodon interactions, or instead are translocated by separate structural features of the translation apparatus, albeit in a coordinated fashion.
Methods
Construction of Thermus thermophilus strain HB27_L9d
The L9-protein-coding region plus an additional 1000 nucleotides upstream and 2200 nucleotides downstream was PCR-amplified from Thermus thermophilus HB27 genomic DNA with primers 5′TTGAATTCTAGACGCCATTTTGGACCTGACCCTCGCCGGTCAGG and 5′TTTCCATTTAAGCTTGGCTTCGCAACATGGGAGACCCTGGCTAGCCC, which add Xba 1 and Hind III sites to facilitate cloning. Site-directed mutagenesis (37) with oligo 5′GGAGGGGCCTAAGGGGTTTGCCCCTGCAGCGTTCACCCCCTACTTCCGCAC C was used to delete the entire L9-protein-coding region and replace it with a Pst 1 restriction site, generating plasmid pL9d. The KAT cassette (coding for thermostable kanamycin adenyl transferase downstream of a Thermus thermophilus promoter) was cut from plasmid pKT1 (38) and ligated into the Pst 1 site of pL9d, to generate plasmid pL9d_KAT. For unknown reasons, pL9d_KAT made E. coli (DH5alpha) cells very sick, and plasmid prepped from the transformed E. coli strain contained a mixture of pL9d and pL9d_KAT. HB27 cells were transformed with pL9d/pL9d_KAT as described (38), plated on ATCC 697 medium plus 20 ug/ml kanamycin, and grown at 70°C for 24 hours. Kanamycin-resistant colonies were screened for deletion of L9 by PCR, using primers that flank the L9-coding region. Surprisingly, we did not detect colonies that contain the KAT-protein-coding sequence inserted into the genome at the L9 position, but instead obtained “clean deletions” of L9 in which the genomic copy of L9 was replaced with a Pst 1 restriction site. The resulting HB27 L9 deletion strain was only transiently kanamycin resistant, and we suspect it arose from double-transformation with pL9d and pL9d_KAT.
Purification of Thermus thermophilus EF-G
Thermus thermophilus EF-G, cloned into pET24b (Novagen), was expressed in E. coli strain BLR(DE3) by induction with IPTG (1 mM final concentration). Cells were lysed in 25 mM Tris-Cl (pH 7.5), 60 mM NH4Cl, 10 mM MgC2, and 5 mM βME, and ribosomes were removed (pelleted) by centrifugation in a Ti70 (Beckman) rotor at 55,000 rpm for 2 hours at 4°C. The supernatant was heated at 65°C for 20 minutes to denature E. coli proteins, which were removed (pelleted) by centrifugation in a JA20 rotor at 30,000 × g for 20 minutes at 4°C. EFG was purified by FPLC chromatography using a 6 ml Resource-Q anion-exchange column (Pharmacia) and eluted with a 200 ml salt gradient from 60 to 300 mM KCl in buffer A [25 mM Tris-Cl (pH 7.5), 60 mM NH4Cl, 5 mM βME]. EFG-containing fractions were concentrated to less than 0.4 ml using an Amicon Ultra 15 concentrator (Millipore; 10,000 molecular weight cutoff), before additional purification on a 24 ml Superdex 75 gel-filtration column (Pharmacia) preequilibrated in buffer A. Aliquots were flash-frozen in liquid nitrogen and stored at −80°C.
Ribosome preparation
Ribosomes were prepared from Thermus thermophilus strain HB27_L9d. Cells were resuspended in buffer B [25 mM Tris-Cl (pH 7.5), 100 mM NH4Cl, 52.5 mM MgCl2, 2.5 mM EDTA, and 5 mM βME], lysed, and pelleted through cushions containing 1.1 M sucrose and buffer B supplemented with NH4Cl to 1M. Ribosomes were purified over a Toyopearl Butyl-650S column, and eluted with a gradient of 0.7 M to 0.2 M AS. To remove endogenous tRNA, ribosomes were dissociated into subunits by dialysis against buffer C [55 mM Tris-Cl (pH 7.5), 100 mM NH4Cl, 1 mM MgCl2, and 5 mM βME], then pelleted through cushions containing 25% sucrose in buffer C. The pellets were resuspended in buffer D [20 mM KHepes (pH 7.5), 100 mM NH4Cl, 10 mM MgCl2, 1 mM spermine, and 5 mM βME] and ribosomes were re-associated by heating at 65°C for 5 minutes before loading onto gradients containing 10–35% sucrose in buffer D. The 70S ribosome peak was collected and the ribosomes were pelleted, resuspended in buffer D, flash-frozen in liquid nitrogen and stored at −80°C.
Complex formation
400 pmol ribosomes, 800 pmol tRNA-Met (MP Biomedicals), and 1000 pmol mRNA mv27 (GGCAAGGAGGUAAAAAUGGUAAAAAAA; IDT) were incubated at 37°C for 30 minutes in 10 mM KHepes (pH 7.5), 100 mM NH4Cl, 5 mM MgCl2, and 1 mM spermine. 3000 pmol of EF-G pre-incubated for 30 minutes at 37°C with either GTP plus fusidic acid, or GDPNP, in 10 mM KHepes (pH 7.5) and 100 mM NH4Cl, were added to the ribosome-tRNA complex, and incubated at 37°C for 15 minutes. The final conditions were 10 mM KHepes (pH 7.5), 100 mM NH4Cl, 1–1.5 mM MgCl2 (GDPNP-I and Fus complexes) or 0.5 mM MgCl2 (GDPNP-II complex), 0.5–0.8 mM spermine, 2 mM DTT, 0.25 mM fusidic acid and 0.5 mM GTP, or 1 mM GDPNP in a total volume of 100 μl. In complexes containing viomycin, it was added to a final concentration of 0.5–1 mM. Deoxy Big Chap (Hampton) was added to 0.5 mM right before crystallization.
Crystallization
Both the EF-G·GDPNP·ribosome·mRNA·tRNA and EFG·GDP·fusidic acid·ribosome·mRNA·tRNA complexes were grown via vapor diffusion in 96-well plates by adding 1–2 μL of reservoir solution [100 mM Tris (pH 7.0), 200 mM KSCN, 2.9% PEG20K, 2.8% PPG P400, 5.1% PEG550MME), to 1–2 μL of sample. Viomycin-containing complexes were crystallized under the same conditions. After 10 days at 22°C, crystals grew to dimensions of up to 1000×600×60 μM. After slowly adding PEG 400 to a final concentration of 25% (v/v) for the GDPNP-I and Fus complexes or 20% for the GDPNP-II complex, the crystals were harvested and flash-frozen in liquid nitrogen.
Crystal screening and data collection
Crystals were screened at beamline 12-2 at the Stanford Synchrotron Radiation Laboratory (SSRL) and at beamline 12-3-1 at the Advanced Light Source (ALS), Lawrence Berkeley National Laboratory. Data were collected at beamline 12-3-1 at the ALS, and at beamline 23ID-D at the Advanced Photon Source at Argonne National Laboratory using 0.3° oscillations. Diffraction data were indexed and integrated in XDS (39) and scaled in SCALA (40).
Molecular replacement, model building and structure refinement
The 50S and 30S models from previously published structures (24) were used as initial search models for molecular replacement using the program Phaser (41). Two 70S ribosomes were found per asymmetric unit in space group C2. Clear density was visible in unbiased Fo–Fc difference electron density maps for EF-G, mRNA and tRNA in all of the above complexes. Four-rigid-body refinements were performed initially in Phenix (42), and the refined models were subjected to 600-rigid-body refinement and TLS group, B group and B individual minimization refinements in Phenix and in CNS (43).
Models for tRNA, mRNA, EF-G domain IV and switch loop, GDPNP, GDP and fusidic acid were initially built manually in O (44) or Coot (45) against simulated-annealing Fo-Fc omit maps calculated in Phenix. The C-terminal domain of L12 was built manually based on a previous poly(Ala) model (15).
Inclusion of viomycin improved the resolution of EF-G complex crystals as found previously for crystals of RF3·ribosome complexes (18). Statistics for the fusidic acid complex crystallized in the absence of viomycin are presented in Table S1 for comparison. Structures of viomycin-free complexes were also solved at 4.1Å and 4.3Å, respectively, for the GDPNP-I and GDPNP-II complexes (data not shown); the sole structural differences detected between viomyin-containing or viomycin-free structures are that16S rRNA bases A1492 and A1493 and 23S rRNA base A1913 are flipped out in the viomycin complexes while retaining their original positions in the viomycin-free complexes.
Supplementary Material
Acknowledgments
We thank Hossein Amiri for critical reading of the manuscript, Srividya Mohan for helpful discussions and the synchtrotron beamline staffs at the ALS, APS and SSRL for expert help with data collection. This work was supported by grants (to H.F.N.) from the NIH. Coordinates and structure factors have been deposited with the following PDB accession codes: (GDPNP-I) 4KCY, 4KCZ, 4KD0, 4KD2; (GDPNP-2) 4KBT, 4KBU, 4KBV, 4KBW; (Fus-3.6) 4KD8, 4KD9, 4KDA, 4KDB; (Fus-4.2) 4KDG, 4KDH, 4KDJ, 4KDK.
References
- 1.Moazed D, Noller HF. Nature. 1989;342:142. doi: 10.1038/342142a0. [DOI] [PubMed] [Google Scholar]
- 2.Dorner S, Brunelle JL, Sharma D, Green R. Nat Struct Mol Biol. 2006;13:234. doi: 10.1038/nsmb1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cornish PV, Ermolenko DN, Noller HF, Ha T. Mol Cell. 2008;30:578. doi: 10.1016/j.molcel.2008.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Agirrezabala X, et al. Mol Cell. 2008;32:190. doi: 10.1016/j.molcel.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Spiegel PC, Ermolenko DN, Noller HF. RNA. 2007;13:1473. doi: 10.1261/rna.601507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dunkle JA, et al. Science. 2011;332:981. doi: 10.1126/science.1202692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rodnina MV, Wintermeyer W. Biochem Soc Trans. 2011;39:658. doi: 10.1042/BST0390658. [DOI] [PubMed] [Google Scholar]
- 8.Ratje AH, et al. Nature. 2010;468:713. doi: 10.1038/nature09547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guo Z, Noller HF. Proc Natl Acad Sci U S A. 2012;109:20391. doi: 10.1073/pnas.1218999109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Borovinskaya MA, Shoji S, Holton JM, Fredrick K, Cate JH. ACS Chem Biol. 2007;2:545. doi: 10.1021/cb700100n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Taylor DJ, et al. EMBO J. 2007;26:2421. doi: 10.1038/sj.emboj.7601677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Frank J, Agrawal RK. Nature. 2000;406:318. doi: 10.1038/35018597. [DOI] [PubMed] [Google Scholar]
- 13.Gao H, Valle M, Ehrenberg M, Frank J. J Struct Biol. 2004;147:283. doi: 10.1016/j.jsb.2004.02.008. [DOI] [PubMed] [Google Scholar]
- 14.Stark H, Rodnina MV, Wieden HJ, van Heel M, Wintermeyer W. Cell. 2000;100:301. doi: 10.1016/s0092-8674(00)80666-2. [DOI] [PubMed] [Google Scholar]
- 15.Gao YG, et al. Science. 2009;326:694. doi: 10.1126/science.1179709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bodley JW, Zieve FJ, Lin L, Zieve ST. Biochem Biophys Res Commun. 1969;37:437. doi: 10.1016/0006-291x(69)90934-6. [DOI] [PubMed] [Google Scholar]
- 17.Jenner LB, Demeshkina N, Yusupova G, Yusupov M. Nat Struct Mol Biol. 2010;17:555. doi: 10.1038/nsmb.1790. [DOI] [PubMed] [Google Scholar]
- 18.Zhou J, Lancaster L, Trakhanov S, Noller HF. RNA. 2011;18:230. doi: 10.1261/rna.031187.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Korostelev A, Trakhanov S, Laurberg M, Noller HF. Cell. 2006;126:1065. doi: 10.1016/j.cell.2006.08.032. [DOI] [PubMed] [Google Scholar]
- 20.Selmer M, et al. Science. 2006;313:1935. doi: 10.1126/science.1131127. [DOI] [PubMed] [Google Scholar]
- 21.Schuwirth BS, et al. Science. 2005;310:827. doi: 10.1126/science.1117230. [DOI] [PubMed] [Google Scholar]
- 22.Schmeing TM, et al. Science. 2009;326:688. doi: 10.1126/science.1179700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schuette JC, et al. EMBO J. 2009;28:755. doi: 10.1038/emboj.2009.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Korostelev A, et al. Proc Natl Acad Sci U S A. 2008;105:19684. doi: 10.1073/pnas.0810953105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bourne HR, Sanders DA, McCormick F. Nature. 1991;349:117. doi: 10.1038/349117a0. [DOI] [PubMed] [Google Scholar]
- 26.Voorhees RM, Schmeing TM, Kelley AC, Ramakrishnan V. Science. 2010;330:835. doi: 10.1126/science.1194460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Laurberg M, et al. J Mol Biol. 2000;303:593. doi: 10.1006/jmbi.2000.4168. [DOI] [PubMed] [Google Scholar]
- 28.Connell SR, et al. Mol Cell. 2007;25:751. doi: 10.1016/j.molcel.2007.01.027. [DOI] [PubMed] [Google Scholar]
- 29.Mohr D, Wintermeyer W, Rodnina MV. Biochemistry. 2002;41:12520. doi: 10.1021/bi026301y. [DOI] [PubMed] [Google Scholar]
- 30.Helgstrand M, et al. J Mol Biol. 2007;365:468. doi: 10.1016/j.jmb.2006.10.025. [DOI] [PubMed] [Google Scholar]
- 31.Noji H, Yasuda R, Yoshida M, Kinosita K., Jr Nature. 1997;386:299. doi: 10.1038/386299a0. [DOI] [PubMed] [Google Scholar]
- 32.Woese C. Nature. 1970;226:817. doi: 10.1038/226817a0. [DOI] [PubMed] [Google Scholar]
- 33.Frank J. Curr Opin Struct Biol. 2012;22:778. doi: 10.1016/j.sbi.2012.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Julian P, et al. Proc Natl Acad Sci U S A. 2008;105:16924. doi: 10.1073/pnas.0809587105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Korostelev A, Noller HF. J Mol Biol. 2007;373:1058. doi: 10.1016/j.jmb.2007.08.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jin H, Kelley AC, Ramakrishnan V. Proc Natl Acad Sci U S A. 2011;108:15798. doi: 10.1073/pnas.1112185108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kunkel TA, Bebenek K, McClary J. Methods Enzymol. 1991;204:125. doi: 10.1016/0076-6879(91)04008-c. [DOI] [PubMed] [Google Scholar]
- 38.Lasa I, Caston JR, Fernandez-Herrero LA, de Pedro MA, Berenguer J. Mol Microbiol. 1992;6:1555. doi: 10.1111/j.1365-2958.1992.tb00877.x. [DOI] [PubMed] [Google Scholar]
- 39.Kabsch W. J Appl Cryst. 1993;26:795. [Google Scholar]
- 40.Evans P. Acta Crystallogr D Biol Crystallogr. 2006;62:72. doi: 10.1107/S0907444905036693. [DOI] [PubMed] [Google Scholar]
- 41.McCoy AJ, et al. J Appl Crystallogr. 2007;40:658. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Adams PD, et al. Acta Crystallogr D Biol Crystallogr. 2010;66:213. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brunger AT, et al. Acta Crystallogr D Biol Crystallogr. 1998;54:905. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 44.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Acta Crystallogr A. 1991;47(Pt 2):110. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 45.Emsley P, Cowtan K. Acta Crystallogr D Biol Crystallogr. 2004;60:2126. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.