

Abstract
SIRT6 is a histone deacetylase that has been proposed as a potential therapeutic target for metabolic disorders and the prevention of age-associated diseases. We have previously reported on the identification of quercetin and vitexin as SIRT6 inhibitors, and studied structurally related flavonoids including luteolin, kaempferol, apigenin and naringenin. It was determined that the SIRT6 protein remained active after immobilization and that a single frontal displacement could correctly predict the functional activity of the immobilized enzyme. The previous study generated a preliminary pharmacophore for the quercetin binding site on SIRT6, containing 3 hydrogen bond donors and one hydrogen bond acceptor. In this study, we have generated a refined pharmacophore with an additional twelve quercetin analogs. The resulting model had a positive linear behavior between the experimental elution time verses the fit values obtained from the model with a correlation coefficient of 0.8456.
Keywords: SIRT6, Pharmacophore Modeling, Frontal Displacement Chromatography, HAT, HDAC
1. Introduction
Yeast Silent Information Regulator 2 (SIR2) is the founding member of the sirtuin family of NAD+-dependent histone deacetylases (HDACs) that function as regulators of many important cellular processes [1, 2]. In humans, there are seven sirtuins, SIRT1-7, which have been implicated in pathways controlling aging, cancers and metabolic diseases [1]. The catalytic activity of sirtuins is closely associated with deacetylation of histones, however, they have also been shown to deacetylate non-histone proteins [3], possess mono-ADP-ribosyltransferase activity [4], and in the case of SIRT6 carry out deacylation of long-chain fatty acyl groups from lysines [5]. Every year, an increasing number of important targets of each of the sirtuins are reported, demonstrating their role as central regulators and illustrating the complexity of their function.
SIRT6 gained increasing attention when SIRT6-deficient mice exhibited shortened lifespan with a premature aging phenotype, similar to the SIR2 mutants in yeast, worms, and flies [6]. Interestingly these mice developed several acute degenerative processes by three weeks of age, including decreased serum glucose and insulin-like growth factor 1 (IGF-1) levels [2]. Importantly, activation of SIRT6 also reduces glycolysis activity by regulating the expression of multiple glycolytic genes [7, 8]. In addition, SIRT6 activity suppresses gluconeogenesis and normalizes glycemia in animal models [9]. Taken together, these findings suggest that SIRT6 is a master regulator of glucose homeostasis. Thus it has been proposed that SIRT6 might be a suitable therapeutic target in the context of age-associated diseases, as well as metabolic disorders including obesity and insulin resistant diabetes [8–11]. Therefore the identification of compounds that can modulate SIRT6 activity is likely to be of therapeutic relevance.
We had previously used a SIRT6 open tubular column (SIRT6-OT) to characterize the quercetin binding site of the SIRT6 protein [12]. A preliminary pharmacophore model was generated with a small subset of compounds (luteolin, apigenin, vitexin, quercetin, kaempferol and genistein) to identify key functional features of the quercetin binding site of the SIRT6 protein [12]. The top ranked pharmacophore model that we had reported previously contained three hydrogen bond donor (HBD) features and one hydrogen bond acceptor (HBA) which correlated with the experimentally determined Kd’s. However, as this was only a preliminary model based on a small number of compounds, a more robust pharmacophore model was generated with a subset of 14 training compounds and a test set of 6 compounds. Of the twenty compounds used, six were from the initial study, naringenin and daidzein were purchased and the remaining twelve quercetin analogs were synthesized and screened against SIRT6. Herein we report and discuss the refinement of this model.
2. Materials and Methods
2.1 Materials
1-ethyl-3-(3-methylaminopropyl) carbodiimide (EDC), gluteraldehyde, hydroxylamine hydrochloride, nicotinamide adenine dinucleotide (NAD+), Dithiothreitol (DTT) potassium phosphate dibasic, sodium cyanoborohydride, and sodium phosphate monobasic, quercetin, vitexin, naringenin, kaempferol, apigenin were obtained from Sigma-Aldrich Chemical Co. (Milwaukee, WI). Solutions were prepared using purified water from a Millipore MilliQ system (Millipore Corporation, Bedford, MA).
2.2 General procedure
Solvents and reagents were obtained from commercial suppliers and were used without further purification. NMR experiments were run on a Bruker Avance 500 equipped with a BBI probe and Z-gradients. Spectra were acquired at 300 K, using deuterated dimethyl sulfoxide (DMSO-d6) or deuterated chloroform (CDCl3) as the solvent. Chemical shifts for 1H and 13C spectra were recorded in parts per million using the residual nondeuterated solvent as the internal standard (for DMSO-d6, 2.50 ppm 1H, and for CDCl3, 7.26 ppm 1H). Data are reported as follows: chemical shift (ppm), multiplicity (indicated as br, broad signal, s, singlet, d, doublet, t, triplet, q, quartet, m, multiplet, and combinations thereof), coupling constant (J, Hz), and integrated intensity. All final compounds displayed ≥95% purity as determined by NMR analysis.
The target Quercetin derivatives were synthesized using the Frederique’s method [13].
Synthesis of the Chalcones (step 1)
Substituted 2′-hydroxyacetophenone (10 mmol) and the substituted aldehyde (10 mmol) were dissolved in ethanol (100 ml) and potassium hydroxide (1.12g, 20 mmol) was added and the mixture was stirred at 50 °C for 15 hours. After this time the pH was adjusted to 1–2 by the addition of HCl(c) and the precipitate formed was filtered under vacuum to obtain the target chalcone as a yellow solid.
Synthesis of the Flavanols (step 2)
The Chalcone was dissolved in 100 ml of methanol and 5 ml of 30% hydrogen peroxide and sodium hydroxide (1.2g 30 mmol) were added and the mixture was stirred at room temperature for 15 hours. HCl(c) was added until pH 1 and precipitate formed was filtered under vacuum to obtain the title compound as a solid.
QRmCH3 (76%)
1H-NMR (500 MHz; CDCl3): δ 8.31 (dd, J = 8.0, 1.5 Hz, 1H), 8.11 (d, J = 6.8 Hz, 2H), 7.75 (td, J = 7.8, 1.5 Hz, 1H), 7.65 (d, J = 8.5 Hz, 1H), 7.49-7.45 (m, 2H), 7.34 (d, J = 7.5 Hz, 1H), 7.11 (s, 1H), 2.52 (s, 3H).
QRpOH (34%)
1H-NMR (500 MHz; DMSO-d6): δ 10.10 (s, 1H), 9.35 (s, 1H), 8.14-8.10 (m, 3H), 7.78 (td, J = 7.7, 1.5 Hz, 1H), 7.74 (d, J = 8.2 Hz, 1H), 7.46 (t, J = 7.4 Hz, 1H), 6.96 (d, J = 8.8 Hz, 2H).
QRpCF3 (67%)
1H-NMR (500 MHz; CDCl3): δ 8.38 (d, J = 8.3 Hz, 2H), 8.26 (dd, J = 8.0, 1.1 Hz, 1H), 7.78 (d, J = 8.4 Hz, 2H), 7.76-7.72 (m, 1H), 7.61 (d, J = 8.5 Hz, 1H), 7.44 (t, J = 7.5 Hz, 1H), 7.23 (s, 1H).
QRmOH (43%)
1H-NMR (500 MHz; DMSO-d6): δ 9.71 (s, 1H), 9.56 (s, 1H), 8.12 (d, J = 7.9 Hz, 1H), 7.81 (ddd, J = 8.5, 7.0, 1.4 Hz, 1H), 7.74 (d, J = 8.4 Hz, 1H), 7.69 (s, 1H), 7.66 (d, J = 7.9 Hz, 1H), 7.47 (t, J = 7.5 Hz, 1H), 7.36 (t, J = 8.0 Hz, 1H), 6.90 (d, J = 7.9 Hz, 1H).
QRmNH2 (98%)
1H-NMR (500 MHz; DMSO-d6): δ 9.38 (s, 1H), 8.11 (d, J = 7.6 Hz, 1H), 7.79 (t, J = 7.5 Hz, 1H), 7.69 (d, J = 8.2 Hz, 1H), 7.46 (t, J = 7.4 Hz, 1H), 7.43 (s, 1H), 7.37 (d, J = 7.4 Hz, 1H), 7.19 (t, J = 7.7 Hz, 1H), 6.70 (d, J = 7.3 Hz, 1H), 5.31 (s, 2H).
QRmNO2 (59%)
1H-NMR (500 MHz; CDCl3): δ 9.09 (s, 1H), 8.63 (d, J = 7.9 Hz, 1H), 8.30 (d, J = 8.0 Hz, 1H), 8.26 (d, J = 7.9 Hz, 1H), 7.76 (t, J = 7.7 Hz, 1H), 7.72 (t, J = 8.1 Hz, 1H), 7.65 (d, J = 8.5 Hz, 1H), 7.45 (t, J = 7.5 Hz, 1H), 7.33 (s, 1H).
QRmBr (87%)
1H-NMR (500 MHz; CDCl3): δ 8.38 (t, J = 1.7 Hz, 1H), 8.25-8.20 (m, 2H), 7.72 (ddd, J = 8.6, 6.9, 1.7 Hz, 1H), 7.60-7.57 (m, 2H), 7.43-7.37 (m, 2H), 7.19 (s, 1H).
QRoOH (32%)
1H-NMR (500 MHz; DMSO-d6): δ 9.78 (s, 1H), 8.99 (s, 1H), 8.14 (dd, J = 8.0, 1.3 Hz, 1H), 7.76 (ddd, J = 8.4, 7.1, 1.4 Hz, 1H), 7.62 (d, J = 8.4 Hz, 1H), 7.48-7.44 (m, 2H), 7.35 (td, J = 7.8, 1.3 Hz, 1H), 6.99 (d, J = 8.3 Hz, 1H), 6.93 (t, J = 7.5 Hz, 1H).
QRpOMe (64%)
1H-NMR (500 MHz; CDCl3): δ 8.26-8.24 (m, 3H), 7.70 (ddd, J = 8.7, 6.9, 1.7 Hz, 1H), 7.59 (d, J = 8.4 Hz, 1H), 7.41 (t, J = 7.5 Hz, 1H), 7.06 (d, J = 9.0 Hz, 2H), 6.95 (s, 1H), 3.90 (s, 3H).
6OHQR (23%)
1H-NMR (500 MHz; DMSO-d6): δ 9.97 (s, 1H), 9.41 (s, 1H), 8.19 (d, J = 7.5 Hz, 2H), 7.62 (d, J = 9.0 Hz, 1H), 7.55 (t, J = 7.5 Hz, 2H), 7.48 (t, J = 7.5 Hz, 1H), 7.37 (d, J = 2.9 Hz, 1H), 7.26 (dd, J = 9.0, 2.9 Hz, 1H).
6MeOQR (55%)
1H-NMR (500 MHz; CDCl3): δ 8.25 (d, J = 7.4 Hz, 2H), 7.57 (d, J = 3.0 Hz, 1H), 7.55-7.51 (m, 3H), 7.47 (t, J = 7.3 Hz, 1H), 7.31 (dd, J = 9.2, 3.1 Hz, 1H), 7.03 (s, 1H), 3.92 (s, 3H).
6MeQR (71%)
1H-NMR (500 MHz; CDCl3): δ 8.25 (d, J = 7.5 Hz, 2H), 8.03 (s, 1H), 7.55-7.45 (m, 5H), 7.05 (s, 1H), 2.48 (s, 3H).
2.3. Frontal Displacement Chromatography
The SIRT6 (CT)-OT column was prepared as previously described [12]. The column was attached to the chromatographic system Series 1100 Liquid Chromatography/Mass Selective Detector (Agilent Technologies, Palo Alto, CA, USA) equipped with a vacuum de-gasser (G 1322 A), a binary pump (1312 A), an autosampler (G1313 A) with a 20 μL injection loop, a mass selective detector (G1946 B) supplied with atmospheric pressure ionization electrospray and an on-line nitrogen generation system (Whatman, Haverhill, MA, USA). The chromatographic system was interfaced to a 250 MHz Kayak XA computer (Hewlett-Packard, Palo Alto, CA, USA) running ChemStation software (Rev B.10.00, Hewlett-Packard). In the chromatographic studies, the mobile phase consisted of ammonium acetate [10 mM, pH 7.4]: methanol (90:10v/v) containing 0.2 mM NAD+ and 5 μM quercetin delivered at 0.05 mL min-1 at room temperature. Then 10 μM concentration of each of the polyphenols was placed in the mobile phase and the change in retention volume was obtained to rank the compounds in order of affinity based on the displacement of quercetin. Quercetin was monitored in the negative ion mode using single ion monitoring at m/z = 301.00 [MW - H]-ion for quercetin, with the capillary voltage at 3000 V, the nebulizer pressure at 35 psi, and the drying gas flow at 11 L/min at a temperature of 350°C.
2.4. Molecular Modeling
Molecular structures used in modeling were either downloaded from the PubChem database or modified from their close analogs using Discovery Studio (version 3.5; Accelrys, Inc., San Diego, CA, USA). Each molecule was checked for correct bond-order and chemical structure, and hydrogen atoms were added using Discovery Studio. Using the experimental elution times and our previously published pharmacophore model as guidance [12], we created a training set that consisted of 14 flavonoid molecules (see Table 1, Fig. 1 and Supplementary Table S3) and a test set (see Table 2, Fig. 2 and Supplementary Table S4) of six compounds making up a total of 20 compounds.
Table 1.
Training set compounds:
| Compounds | PubChem ID | IUPAC NAME |
|---|---|---|
| Luteolin | 5280445 | 3′,4′,5,7-Tetrahydroxyflavone |
| Kaempferol | 5280863 | 3,5,7-trihydroxy-2-(4-hydroxyphenyl)chromen-4-one |
| QRMCH3 | 44457131 | 3-hydroxy-2-(3-methylphenyl)chromen-4-one |
| QRPOH | 688715 | 3-Hydroxy-2-(4-hydroxy-phenyl)-chromen-4-one |
| QRMCF3 | 15316112 | 3-hydroxy-2-[4-(trifluoromethyl)phenyl]chromen-4-one |
| QRMOH | 676295 | 3-hydroxy-2-(3-hydroxyphenyl)chromen-4-one |
| Quercetin | 5280343 | 2-(3,4-dihydroxyphenyl)-3,5,7-trihydroxychromen-4-one |
| Apigenin | 5280443 | 4′,5,7-Trihydroxyflavone |
| 6OHQR | 688659 | 3,6-dihydroxy-2-phenylchromen-4-one |
| 6MEOQR | 688676 | 3-hydroxy-6-methoxy-2-phenyl-4h-chromen-4-on |
| 6MEQR | 227445 | 6-Methylflavonol |
| QRPOME | 97141 | 4′-Methoxyflavonol |
| Daidzein | 5281708 | 7-hydroxy-3-(4-hydroxyphenyl)chromen-4-one |
| Genistein | 5280961 | 5,7-dihydroxy-3-(4 hydroxyphenyl) chromen-4-one |
Fig 1.
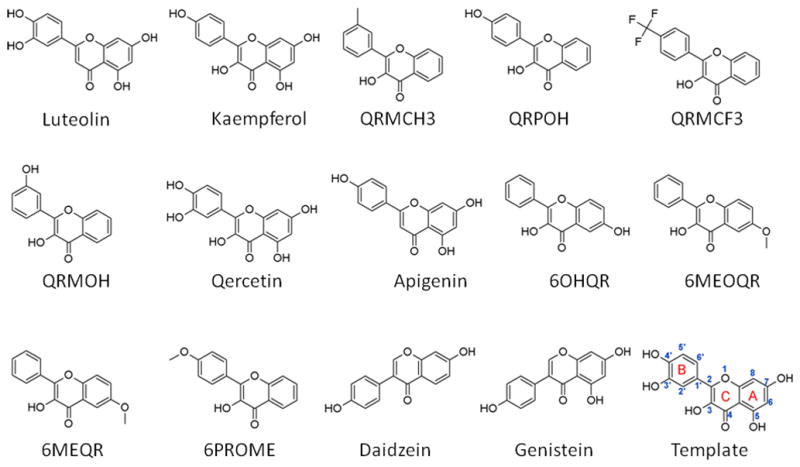
Chemical structures and the names of the training set compounds. Please refer to Table 1 for the IUPAC name and Pubchem ID. For convenience, the atom/ring numbering that is referred to in this study is shown using quercetin as a template (last compound).
Table 2.
Test Set compounds
| Compounds | PubChem ID | IUPAC NAME |
|---|---|---|
| QRMNH2 | NA | 3-hydroxy-6-methyl-2-(3-nitrophenyl)chromen-4-one |
| QRMNO2 | 1659460 | 3-hydroxy-6-methyl-2-(3-nitrophenyl)chromen-4-one |
| QRMBR | 466298 | 3-Hydroxy-3′-bromoflavanone |
| Naringenin | 932 | 5,7-dihydroxy-2-(4-hydroxyphenyl)-2,3-dihydrochromen-4-one |
| Vitexin | 5280441 | Apigenin 8-C-glucoside |
| QROOH | 578729 | 2′-methoxyflavonol |
Fig 2.
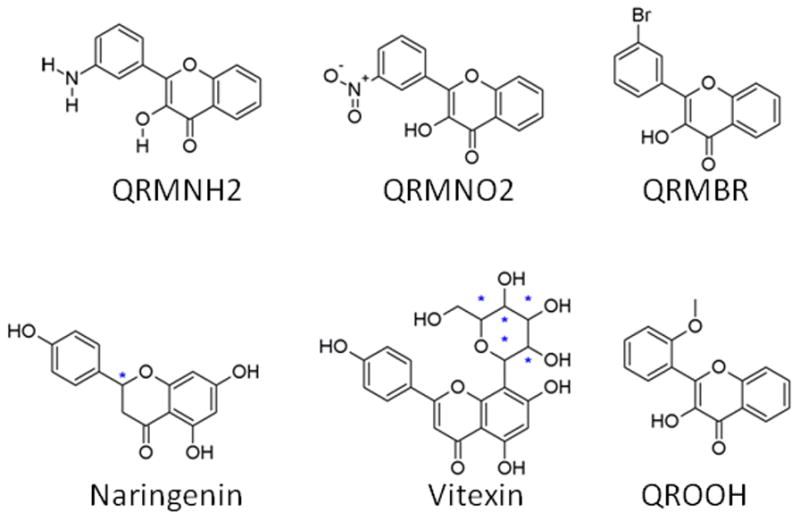
Chemical structures of the test set compounds. Please refer to Table 2 for the IUPAC name and Pubchem ID. The chiral center for Naringenin is marked with a star (*).
2.5. Pharmacophore Modeling
Currently, four crystal structures for the deacetylase sirtuin-type domain of human SIRT6 are available in the 3D structure database, RCSB PDB (http://www.rcsb.org; PDB IDs: 3K35, 3PKI and 3PKJ [14] and 3ZG6 [5]). However, recent studies [5, 14] showed that SIRT6 function is markedly different from other known SIRT types especially in how the cofactor (NAD+) binding lacks the necessity for an acetyl lysine substrate and possibly a different role for the key residue, H131, along with the possibility of a conformational modification during ligand binding. Keeping these critical properties of SIRT6 in mind, we have decided to model the inhibitory role of flavanoids using a pharmacophore modeling approach. Our initial modeling efforts [12] using a smaller set of compounds identified common features that showed strong correlation to the experimental data. With the availability of additional compounds, in this study we have refined the model.
2.6. Common features modeling
The pharmacophore models in this study are built by exploiting the common features present among the strongly inhibiting flavanoid set and the ones that are consistently absent from the weak inhibitors. Details of pharmacophore modeling have been explained in many reviews and articles [15, 16], here we provide only a brief overview of the method.
A pharmacophore model represents a collection of chemical features (e.g.. hydrogen bond donor, hydrogen bond acceptor, hydrophobic etc.) that can be identified to be present at specific 3D positions in the most-active compounds and consistently absent from the inactive compounds thus explaining the biological data. We have carried out pharmacophore modeling using the HipHop algorithm as implemented in Discovery Studio (ver 3.5, Accelrys Inc. San Diegeo, CA). Pharmacophore modeling details are elaborated in [15], and consists of 5 key steps: 1) Selection of an initial set with activity information 2) Conformation generation for the initial set to account for ligand flexibility 3) Extraction of common features from the set; and, 4) Combination of extracted features to create meaningful models. Excluded volumes can also be added at this step based on inactive molecules, and finally, the user-defined number of models are scored and ranked based on how each molecule maps (fit value) to the model. Chemical features in Discovery Studio are represented by either spheres or vectors. The spheres are used for non-directional features (single sphere) such as the hydrophobic property and vectors for directional properties such as hydrogen bond donors or acceptors. The end points of the vector are used to mark the location of hydrogen bond forming partners (e.g. the heavy atom and the projecting point location). Note that each end of the vector also contains a larger meshed sphere marking the tolerance volume where the mapped feature from the molecules must be located. Any deviation will be penalized in the fit value.
2.7. Model
Pharmacophore models were built in Discovery Studio using the training set (Fig. 1) and tested using the compounds shown in Fig. 2. For building pharmacophore models using the common features approach, two key parameters are needed: “Principal” and “MaxOmitFeat”. Principal is the molecular property that mimics the binding affinity or activity of compounds. Hence, each molecule should be assigned to one of the following values: Principal=2 (active), 1 (moderately active) or 0 (inactive). The active compounds that are assigned Principal=2 will be considered as a reference set and all the chemical features in this group will be considered during modeling, whereas the inactive ones will be ignored except when excluded volumes are modeled. The MaxOmitFeat parameter indicates how many chemical features (HBA, HBD etc.) a molecule is allowed to miss when mapped to a model. This parameter is set to “0” for all the compounds in our study. Table 3 lists the key modeling parameters used in this study and a more elaborate list is presented in Supplemental pages. The conformational flexibility of compounds is included in the model by generating conformers (generation algorithm: BEST, maximum conformers was set to 200) with an energy threshold of 20 kcal/mol and the conformers were used during model building. The detailed list of parameters used in pharmacophore modeling is provided in the Supplementary pages. The following chemical features were used as input for the modeling: Hydrogen Bond Acceptor/Donar, Hydrophobic, Hydrophobic Aromatic and Ring Aromatic. These features were selected based on a preliminary Feature Mapping analysis carried out using Discovery Studio (ver 3.5, Accelrys Inc. San Diegeo, CA) and also using our previous modeling efforts [12].
Table 3.
Table shows the key pharmacophore modeling parameters. Principal reflects whether the compound is active (Principal=2), moderately active (Principle = 1) or Inactive (Principle=0). Detailed list of other parameters used in the modeling are shown in Supplementary pages.
| Compounds | Principal |
|---|---|
| Kaempferol | 2 |
| Luteolin | 2 |
| QRMCH3 | 2 |
| QRPOH | 2 |
| QRMCF3 | 2 |
| QRMOH | 2 |
| Quercetin | 2 |
| Apigenin | 1 |
| 6OHQR | 1 |
| 6MEOQR | 1 |
| 6MEQR | 1 |
| QRPOME | 1 |
| Daidzein | 0 |
| Genistein | 0 |
Several pharmacophore modeling runs using Discovery Studio were carried out with default parameters, with the exception of Minimum Interface Distance (MID) and Maximum Excluded Volumes (MEV) (see Supplementary pages S1 and S2 for details) to identify the best model that was able to explain the experimental data. The conformation generation using the BEST algorithm was used as the default as this method is designed to explore and provide the elaborate coverage of conformational space. The MID is known to be influenced by the size of the molecule and is defined as the minimum spacing between the chemical features. Based on the size of the compounds in our set, we explored a range of values from 0.5 to 2.97Å. The MEV defines the space occupied by the inactive molecules but not by the active ones and the HipHopRefine algorithm, implemented in Discovery Studio 3.5, was used for adding excluded volumes to the model(s). For MEV, we used a range of values starting from as small as 10 to a high value of 500. Table 4 summarizes the results of our pharmacophore modeling.
Table 4.
Summary of Common Features Pharmacophore modeling. Input ligands used in the modeling are listed in Table 1. Pharmacophore feature components for each model are shown in Features column.
| No | Features | Rank | Direct Hit | Partial Hit | Max Fit |
|---|---|---|---|---|---|
| 01 | ZDA | 80.118 | 111111111111 | 000000000000 | 3 |
| 02 | ZAA | 79.957 | 111111111111 | 000000000000 | 3 |
| 03 | RHA | 78.087 | 111111111111 | 000000000000 | 3 |
| 04 | RHA | 78.087 | 111111111111 | 000000000000 | 3 |
| 05 | ZAA | 77.684 | 111111111111 | 000000000000 | 3 |
| 06 | RAA | 77.084 | 111111111111 | 000000000000 | 3 |
| 07 | RAA | 77.084 | 111111111111 | 000000000000 | 3 |
| 08 | RAA | 76.518 | 111111111111 | 000000000000 | 3 |
| 09 | RAA | 76.379 | 111111111111 | 000000000000 | 3 |
| 10 | ZAA | 74.430 | 111111111111 | 000000000000 | 3 |
Note the following symbols are used to denote the pharmacophore features: D: Hydrogen Bond Donar, A: Hydrogen Bond Acceptor, H: Hydrophobic, Z: Hydrophobic Aromatic and R: Ring-Aromatic. Max fit for the models are 3 because of 3 features (a combination of Z, R, D, A). If a compound maps to all features in the model then it will be identified by a mask value of 1 otherwise by a 0. Note that all the models are direct hit. Rank indicates how well the ligands map to the computed model.
3. Results and Discussion
In our previous studies, we immobilized the SIRT6 protein onto the surface of an open tubular (OT) capillary generating the SIRT6-OT column [12]. It was determined that the SIRT6 protein remained active after immobilization and that a single frontal displacement could correctly predict the functional activity of the immobilized enzyme. A frontal chromatogram can be seen in Fig. 3, where an initial flat portion representing specific binding to the column is followed by a breakthrough curve and plateau representing saturation of the column. It was established that the change in retention volume from the displacement of 5 μM quercetin with a series of polyphenols (10 μM) correlated linearly with the level of inhibition of the deacetylation activity of SIRT6 with an r2 of 0.9363 (y=−0.7584x + 71.17) [12]. Using this data, we developed a preliminary pharmacophore model [12] containing three hydrogen bond donors and one hydrogen bond acceptor, with a small subset of polyphenols. An additional, fourteen training compounds were screened against SIRT6 activity, resulting in a more refined pharmacophore.
Fig 3.

Frontal displacement elution profiles of 5 μM quercetin (A) with 10 μM of QRMBr (B), 10 μM QRMNH2 (C), 10 μM quercitin (D) and 10 μM QRMCH3 (E) on the SIRT6-OT column on the agilent LC-MSD. Running buffer consisted of ammonium acetate [10 mM, pH 7.4]: methanol (90:10v/v) containing 0.2 mM NAD+ and the flow rate was 50 μl/min
Each modeling run was set to produce 10 pharmacophore models (hypothesis; see Table 4.). The run that produced the maximum number of models that correlated most closely with the activity observed on the SIRT6-OT column was chosen for further analysis. Values of 2.97 and 200 for MID and MEV, respectively, produced reasonable models, and was chosen as the default values for this study. The rest of the parameters were kept at default levels and detailed in the Supplementary pages. The models and the ranking scores of the fitting of the molecules into the model are also provided in Table 4. The Table lists two mask strings, Direct and Partial hit signature masks, which characterize how each compound in the training set was able to map to the models. A value of one or zero indicates whether the molecule is able to map to every feature (direct hit) or not (partial hit) in the generated pharmacophore model. In this model set, all the molecules were able to map to all the ten models (Table 4). Note that the two lowest binding compounds, daidzein and genistein, were assigned principal = 0 (see Table 3). Their conformations will not be used during the pharmacophore model building stage but will be used during the excluded volume placement stage and are therefore not shown as part of the mask values (Table 4). Analyzing how the molecules were able to map to the models and also taking into account the comparison of the relative order of how the compounds fit to the model (fit values) versus experimental elution times, the pharmacophore number 06 (table 4 and shown in red font, Tables 5 and 6) was identified as the best model for the quercetin binding site of SIRT6. This pharmacophore model (see Fig. 4, Table 4 and S5) consists of a ring aromatic feature (orange spheres) and two hydrogen-bond acceptors (identified with feature A in Table 4 and shown as green spheres in Fig. 4). The excluded volume spheres are shown as grey spheres and the distance constraints between the features are also labeled in the model. This model is very similar to our preliminary model [12] and will be identified as the default model for the rest of the manuscript. Scatter plot of experimental elution time verses the fit values obtained from the model is shown in Fig. 5A. The data highlights the positive linear behavior with a correlation coefficient of 0.65, calculated using the Pearson method (R software, version 2.15.2) (more details in Supplementary information S6). Of particular interest is apigenin. This molecule has consistently been an outlier from our ranking studies, specifically, when comparing the magnitude of displacement of quercetin with its Kd, indicating that its binding site may be separate from the quercetin binding pocket. For this reason, we decided to analyze our training set data with and without apigenin. The scatter plot of the subset without apigenin is shown in Fig. 5B, and the correlation coefficient using the Pearson method is 0.8456. The model was further tested using the test set (Fig. 2) and the fit values for the set are shown in Table 6. The scatter plot (Fig. 5c) for the test set, clearly demonstrates fit values closely follow the elution order with a correlation coefficient of 0.7486 using the Pearson method.
Table 5.
Comparison of experimental elution times for the training set compounds with the pharmacophore model (see Fig. 4 and Table 4) fit values.
| Compounds | Experimental Elution Times | Pharmacophore Fit Values |
|---|---|---|
| Luteolin | 1.2731 | 1.2476 |
| Kampferol | 1.1152 | 1.0371 |
| QRMCH3 | 1.0866 | 0.8686 |
| QRPOH | 1.0750 | 0.7660 |
| QRMCF3 | 1.0266 | 0.9329 |
| QRMOH | 1.0093 | 0.7642 |
| Quercetin | 1.0000 | 1.1051 |
| Apigenin | 0.7145 | 1.5893 |
| 6OHQR | 0.6882 | 0.9382 |
| 6MeOQR | 0.6571 | 0.8159 |
| 6MeQR | 0.6228 | 0.7242 |
| QRPOMe | 0.5984 | 0.7754 |
| Daidzein | 0.4000 | 0.1639 |
| Genistein | 0.0106 | 0.1617 |
Table 6.
Comparison of experimental elution times for the test set compounds with the pharmacophore model (see Fig. 3 and Table 4) fit values.
| Compounds | Experimental Elution Times | Pharmacophore Fit Values |
|---|---|---|
| QRMBR | 1.1152 | 0.9014 |
| QRMNH2 | 1.2731 | 0.8446 |
| Naringenin | 1.0866 | 0.6670* |
| QROOH | 1.0750 | 0.6603 |
| QMNO2 | 1.0266 | 0.6454 |
| Vitexin | 1.0093 | 0.5000 |
The fit values correspond to S-Naringenin, while experimental elution time is reported for racemic.
Fig 4.
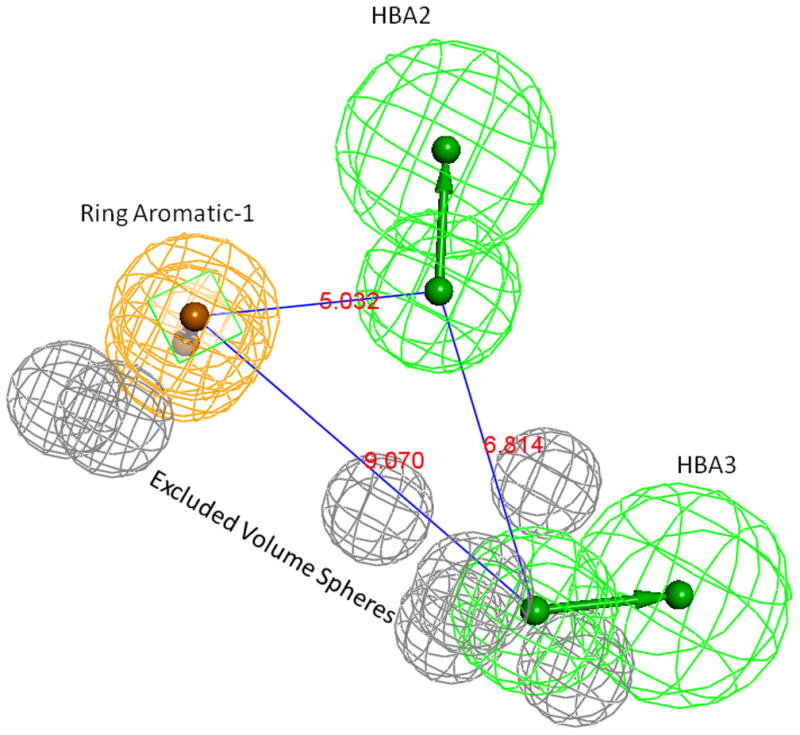
Pharmacophore model-06. Grey spheres indicate the excluded volumes, Green spheres are HBA and the Ring-Aromatic feature is shown in orange color spheres. Supplementary pages show the model with exclusion spheres.
Fig 5.
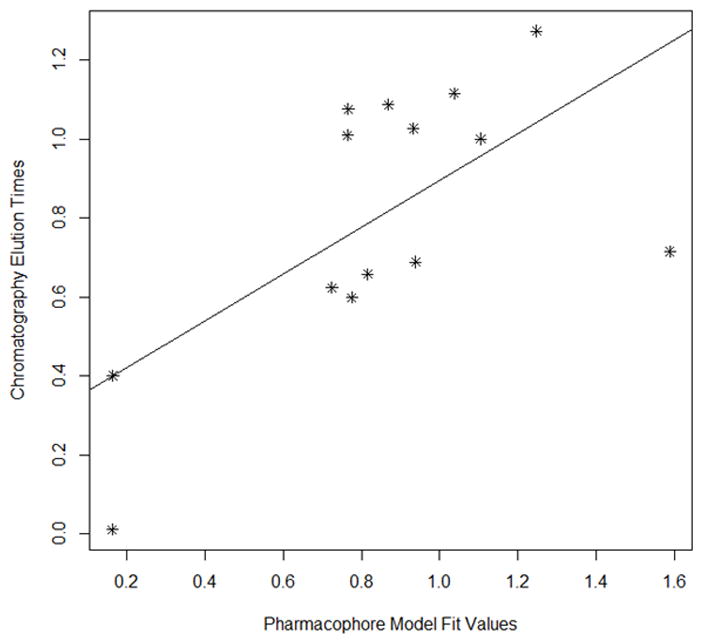
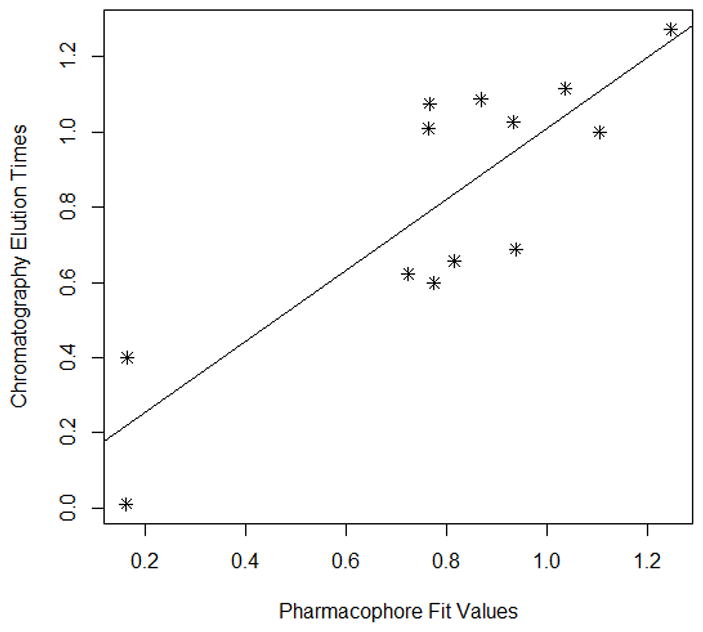
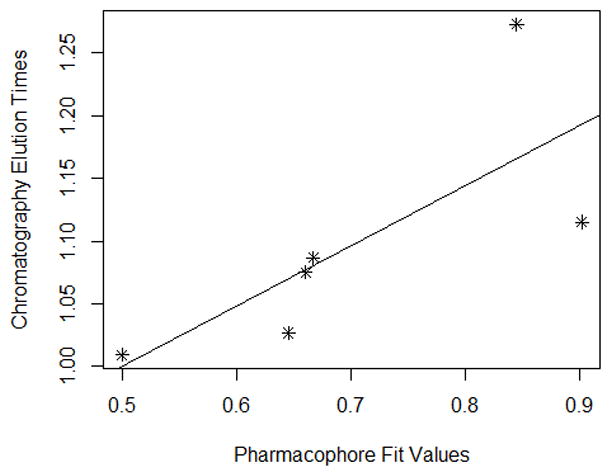
Fig 5A Scatter plot of chromatographic elution times and pharmacophore model (see Table 5). The best fit line for the data is also provided.
Fig 5B. Scatter plot of training set (Fig 6A) without Apigenin vs the experimental elution times. The best fit line for the data is also provided. (Pearson (r) : 0.8456, Spearman(r) = 0.7088
Fig 5C. Scatter plot of chromatographic elution times and pharmacophore model (see Table 6). The best fit line for the data is also provided. (Pearson (r): 0.7486, Spearman(r) = 0.9429)
The aromatic ring feature in the model (orange sphere, Fig. 4) is where most of the compounds were able to place their B aromatic ring (see Fig. 1, template). The hydrogen bond acceptor, HBA2 (Fig. 4) is found mapped to the ring oxygen (1O; atom number followed by the type, Fig. 1). The second acceptor feature, HBA1, is mapped to either the carbonyl oxygen (4C=O) or the hydroxyl oxygen (5C-OH). The feature set that includes B-Ring, C-ring oxygen (O1) (Fig. 1), and either 4C=O or 5CO-H (atoms 4 and 5) appears to be one of the key SIRT6 binding site interacting atoms based on the mapping conformations of the most active members of the training set. Figs. 6 and 7 show the mapping of luteolin and kaempferol to the model. The active members that lack oxygen atom, A:5C-OH, can still bind using the carbonyl oxygen 4C=O, if the molecule is small enough to fit in the cavity. For example, Fig. 8 shows the possible binding mode of the compound 6OHQR to the model. Since compounds like 6OHQR lack the hydroxyl group at 5C-OH, they might be forced to use the carbonyl group and still satisfy the mapping of the other two features. Analyses of other compounds reveal the likelihood of this binding mode driving the activity of the set.
Fig 6.
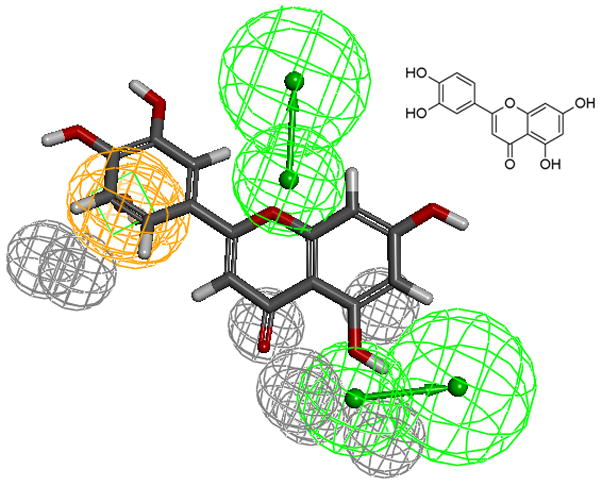
Luteolin (inset) mapped to the pharmacophore
Fig 7.
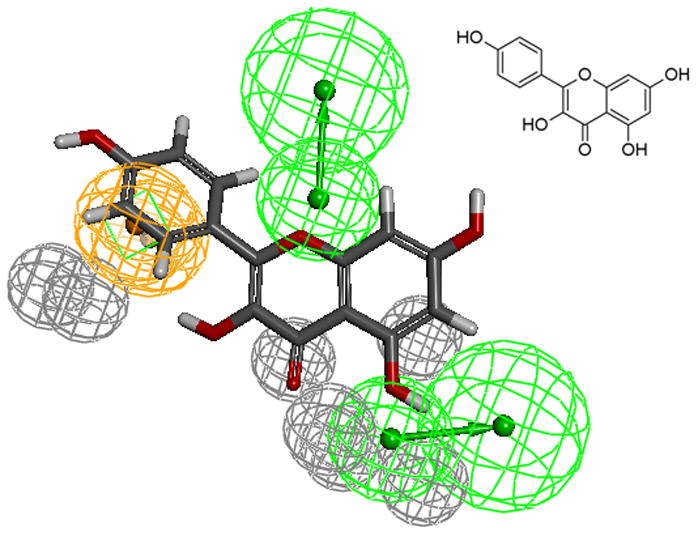
kaempferol mapped to the pharmacophore
Fig 8.
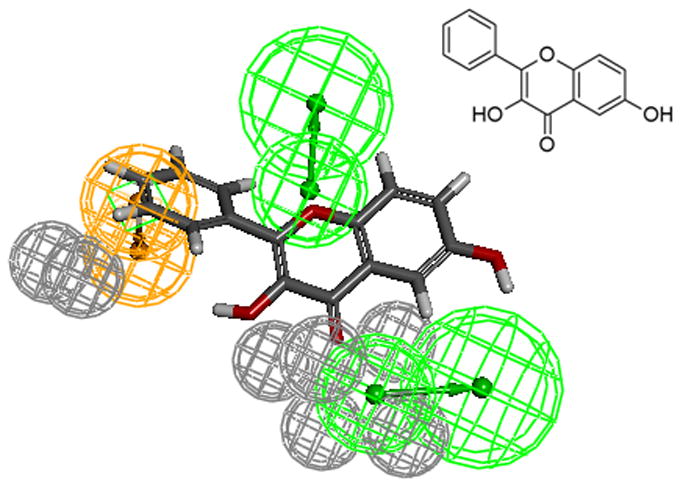
6OHQR mapped to the model.
The test set (Table 6) pharmacophore mapping and elution time data also agree with the model. Test set compounds seem to reinforce the necessity of the constraint locations of the aromatic ring (ring C) and the fused ring location along with the substituents. For instance, the sugar substituent for vitexin (Fig. 9) prevented the mapping preference observed for luteolin or kaempferol (see Figs. 6 and 7). The large sugar substituent adds strain in the mapping of HBA and decreases interactions with the HBD in the receptor, thus changing the binding mode of vitexin. This result in weak binding, as reflected in the lower fit/elution values (0.5/1.0) compared to luteolin (1.25/1.27). As naringenin is a chiral compound, R- and S- forms were included as separate compounds in our test set. The chiral center and the possibility of non-planar conformation of the C ring in naringenin makes S- form to fit more favorably to the model than its isomer. Hence, based on the model results, we would expect S- form to be a better binder compared to the R- form. Using a larger set of flavanoids, we have refined our previous model [12] for the quercetin binding site of SIRT6. This model was also subsequently verified using a test set of active and inactive compounds.
Fig 9.
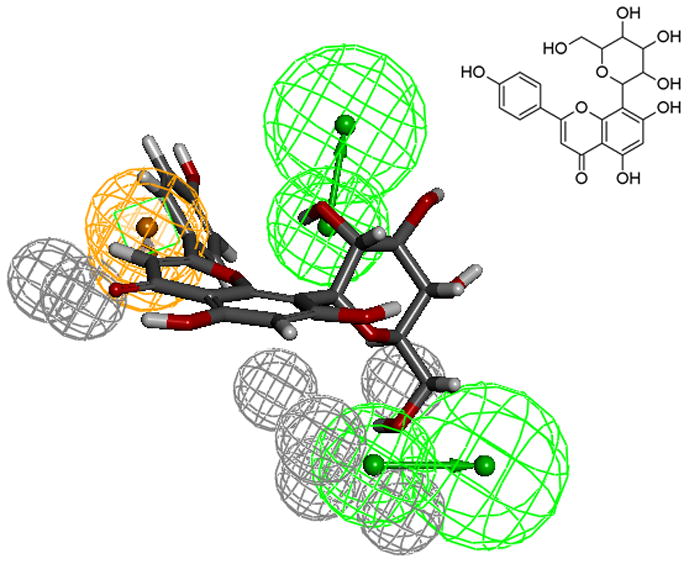
Compound Vitexin mapped to the pharmacophore
Supplementary Material
Highlights.
Ranking of quercetin analogs using Frontal Displacement Chromatography
Refined model of the quercetin binding site of SIRT6 protein
Synthesis of quercetin analogs
Acknowledgments
This work was supported in part by funds from the NIA Intramural Research Program (RM), and from the National Cancer Institute/National Institutes of Health contract No. HHSN261200800001E.
Footnotes
The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. government.
There are no financial conflicts
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Haigis MC, Sinclair DA. Mammalian sirtuins: biological insights and disease relevance. Annual review of pathology. 2010;5:253–95. doi: 10.1146/annurev.pathol.4.110807.092250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mostoslavsky R, Chua KF, Lombard DB, Pang WW, Fischer MR, Gellon L, et al. Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell. 2006;124:315–29. doi: 10.1016/j.cell.2005.11.044. [DOI] [PubMed] [Google Scholar]
- 3.Cheng HL, Mostoslavsky R, Saito S, Manis JP, Gu Y, Patel P, et al. Developmental defects and p53 hyperacetylation in Sir2 homolog (SIRT1)-deficient mice. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:10794–9. doi: 10.1073/pnas.1934713100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Borra MT, O’Neill FJ, Jackson MD, Marshall B, Verdin E, Foltz KR, et al. Conserved enzymatic production and biological effect of O-acetyl-ADP-ribose by silent information regulator 2-like NAD+-dependent deacetylases. The Journal of biological chemistry. 2002;277:12632–41. doi: 10.1074/jbc.M111830200. [DOI] [PubMed] [Google Scholar]
- 5.Jiang H, Khan S, Wang Y, Charron G, He B, Sebastian C, et al. SIRT6 regulates TNF-alpha secretion through hydrolysis of long-chain fatty acyl lysine. Nature. 2013;496:110–3. doi: 10.1038/nature12038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim HS, Xiao C, Wang RH, Lahusen T, Xu X, Vassilopoulos A, et al. Hepatic-specific disruption of SIRT6 in mice results in fatty liver formation due to enhanced glycolysis and triglyceride synthesis. Cell metabolism. 2010;12:224–36. doi: 10.1016/j.cmet.2010.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu TF, Vachharajani VT, Yoza BK, McCall CE. NAD+-dependent sirtuin 1 and 6 proteins coordinate a switch from glucose to fatty acid oxidation during the acute inflammatory response. The Journal of biological chemistry. 2012;287:25758–69. doi: 10.1074/jbc.M112.362343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhong L, D’Urso A, Toiber D, Sebastian C, Henry RE, Vadysirisack DD, et al. The histone deacetylase Sirt6 regulates glucose homeostasis via Hif1alpha. Cell. 2010;140:280–93. doi: 10.1016/j.cell.2009.12.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dominy JE, Jr, Lee Y, Jedrychowski MP, Chim H, Jurczak MJ, Camporez JP, et al. The deacetylase Sirt6 activates the acetyltransferase GCN5 and suppresses hepatic gluconeogenesis. Molecular cell. 2012;48:900–13. doi: 10.1016/j.molcel.2012.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rodgers JT, Puigserver P. Certainly can’t live without this: SIRT6. Cell metabolism. 2006;3:77–8. doi: 10.1016/j.cmet.2006.01.009. [DOI] [PubMed] [Google Scholar]
- 11.Kanfi Y, Naiman S, Amir G, Peshti V, Zinman G, Nahum L, et al. The sirtuin SIRT6 regulates lifespan in male mice. Nature. 2012;483:218–21. doi: 10.1038/nature10815. [DOI] [PubMed] [Google Scholar]
- 12.Singh N, Ravichandran S, Norton DD, Fugmann SD, Moaddel R. Synthesis and characterization of a SIRT6 open tubular column: Predicting deacetylation activity using frontal chromatography. Analytical biochemistry. 2013;436:78–83. doi: 10.1016/j.ab.2013.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.van Acker FAA, Hageman JA, Haenen GRMM, van der Vijgh WJF, Bast A, Menge WMPB. Synthesis of novel 3,7-substituted-2-(3′,4′-dihydroxyphenyl)flavones with improved antioxidant activity. J Med Chem. 2000;43:3752–60. doi: 10.1021/jm000951n. [DOI] [PubMed] [Google Scholar]
- 14.Pan PW, Feldman JL, Devries MK, Dong A, Edwards AM, Denu JM. Structure and biochemical functions of SIRT6. The Journal of biological chemistry. 2011;286:14575–87. doi: 10.1074/jbc.M111.218990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Clement OO, Mehl AT. HipHop: Pharmacophores Based on Multiple Common-Feature Alignments. La Jolla, CA, USA: Intl Univ Line; [Google Scholar]
- 16.Vaidyanathan J, Vaidyanathan TK, Ravichandran S. Computer simulated screening of dentin bonding primer monomers through analysis of their chemical functions and their spatial 3D alignment. Journal of biomedical materials research Part B, Applied biomaterials. 2009;88:447–57. doi: 10.1002/jbm.b.31134. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.