

Abstract
Heart failure is a leading cause of morbidity and mortality worldwide, currently affecting 5 million Americans. A syndrome defined on clinical terms, heart failure is the end-result of events occurring in multiple heart diseases, including hypertension, myocardial infarction, genetic mutations and diabetes, and metabolic dysregulation is a hallmark feature. Mounting evidence from clinical and preclinical studies suggests strongly that fatty acid uptake and oxidation are adversely affected, especially in end-stage heart failure. Moreover, metabolic flexibility, the heart’s ability to move freely among diverse energy substrates, is impaired in heart failure. Indeed, impairment of the heart’s ability to adapt to its metabolic milieu, and associated metabolic derangement, are important contributing factors in heart failure pathogenesis. Elucidation of molecular mechanisms governing metabolic control in heart failure will provide critical insights into disease initiation and progression, raising the prospect of advances with clinical relevance.
Introduction
Cardiovascular disease is the leading cause of mortality worldwide, posing an enormous burden to individuals and society.1 Heart failure, a syndrome defined by clinical criteria, is the point of convergence of numerous cardiovascular diseases – the final common pathway of an injured heart. Thanks to tremendous advances in modern diagnostic and therapeutic technologies, survival of acute myocardial infarction has increased markedly in recent years. This, combined with an aging population, the epidemic of obesity and diabetes, and successes in other realms of medicine, such as oncology, has culminated in an ever-rising prevalence of heart failure.
Heart failure affects approximately 5 million Americans and is especially prevalent among the elderly, where it is the leading cause of hospitalization.1 Heart failure prevalence – already rampant – is expected to increase 25% by 2030, with total annual costs slated to reach $70 billion. Despite new therapies and considerably improved survival in recent years, 50% of patients still die within 5 years of diagnosis. Current therapeutic strategies emphasize suppression of neurohumoral activation and normalization of hemodynamics. However, despite significant improvements in clinical symptoms and attenuated pace of clinical decline, the progression of heart failure remains all-too-often unremitting. Novel therapeutic approaches are urgently needed.
Over the past years, numerous studies have firmly established metabolic derangement as a cardinal feature of the pathophysiology of heart failure.2–4 Heart failure-related changes in uptake and utilization of metabolic substrates, combined with alterations in cardiomyocyte energetics, have been described in numerous preclinical and clinical studies.5, 6 Availability of new technologies to detect alterations in metabolic events, coupled with major advances in our understanding of metabolic regulation, has raised the prospect of targeting cardiac metabolism to treat heart failure. Here, we provide an overview of normal cardiac metabolism and a description of the metabolic derangements in heart failure, followed by discussion of potential therapies targeting cardiac metabolism. Due to the complexity of these events, all aspects of cardiac metabolism are not included. Several recent reviews are therefore recommended for further reading.7–9
Myocardial metabolism in normal heart
The heart is the most avid ATP-consuming organ in the body. The adult human heart weighs approximately 300 grams, yet it utilizes 6 kg of ATP to pump 10 tons of blood daily. Cardiac ATP reserve is quite low, representing enough stored energy for fewer than 10 contractions.2 Thus, to maintain uninterrupted contraction and relaxation, the myocardium requires constant and robust ATP synthesis. To accomplish this, the heart functions as an omnivore, extracting energy from a wide range of metabolic substances, including free fatty acids (FFAs), glucose, lactate, and ketones. At generally prevailing concentrations of circulating substrates, the heart derives 50–70% of its newly generated ATP from oxidation of FFA.
Triglyceride metabolism
Plasma triglycerides (TG) are largely associated with lipoproteins and chylomicrons. Triacylglycerol-enriched lipoproteins are catabolized by lipoprotein lipase, whose localization at the luminal surface of coronary blood vessels may serve to increase local levels of FFA.10
Cardiomyocyte uptake of FFA is driven mainly by a transmembrane concentration gradient and mediated by either passive diffusion or transporter-facilitated transport (Figure 1). CD36 is a fatty acid translocase which is abundant in the cardiomyocyte. Deficiency of CD36 in cardiomyocytes leads to reduced FFA oxidation and triglyceride storage.11 Conversely, over-expression of CD36 in muscle enhances FFA oxidation in myocytes and triglyceride clearance from the circulation.12 Together, these data highlight the fact that CD36 is the major translocase facilitating FFA uptake in heart.
Figure 1. Glucose and fatty acid metabolic pathways in the cardiomyocyte.

Glucose is transported into the cardiomyocyte by glucose transporters (GLUT). Following phosphorylation by hexokinase (HK), glucose-6-phosphate (G6P) is fed into the glycogen synthetic pathway, glycolysis, or the pentose phosphate pathway (PPP). Phosphofructokinase 1 (PFK 1) is the first commitment enzyme of glycolysis. The glycolytic product pyruvate is transported into mitochondria or converted to lactate by lactate dehydrogenase (LDH). Pyruvate dehydrogenase (PDH) is a pivotal enzyme to catabolize pyruvate to acetyl CoA, which ultimately enters the citrate acid cycle. PDH enzymatic activity is inhibited by PDK4-dependent phosphorylation and stimulated by dephosphorylation via PDH phosphatase. Free fatty acids (FFAs) are transported into cardiomyocytes by diffusion or via a fatty acid translocase, such as CD36. Once inside the cell, FFAs are esterified to fatty acyl CoA by fatty acyl CoA synthase (FACS). The carnitine shuttle, composed of CPT-I, CPT-II, and CAT, is responsible for the transport of FFA from the cytosol to mitochondria for beta-oxidation, which generates acetyl CoA for the citric acid cycle. Glucose and FFA metabolic pathways are subject to complex reciprocal regulation. Citrate can be transported to the cytosol, where it undergoes lysis to acetyl CoA. Cytosolic acetyl CoA is converted to malonyl CoA by acetyl CoA carboxylase (ACC). Malonyl CoA is a potent inhibitor of CPT-1 and FFA metabolism.
Once inside the cardiomyocyte, FFA is esterified by fatty acyl CoA synthase (FACS). The resulting fatty acyl CoA is then either re-esterified to triglyceride for storage or transported to mitochondria for beta-oxidation. In addition, intramyocyte TG may be synthesized de novo from glycerol-3-phosphate. Myocardial TG is a dynamic FFA pool, subject to stringent hormonal regulation, and recent studies suggest that metabolism and turnover of myocardial TG play critical roles in cardiac physiology.13 Banke et al found the turnover of palmitoyl units of endogenous TG is 3.75-fold faster than oxidation of palmitate in normal heart, suggesting preferred oxidation of TG-derived FFA.14 Over-expression of PPARα in cardiomyocytes further enhances TG turnover, which is associated with up-regulation of several key enzymes involved in TG synthesis and lipolysis.
Derangements in TG dynamics contribute to the pathophysiology of various heart diseases. Indeed, myocardial steatosis has been implicated in the initiation and progression of diabetic cardiomyopathy, a prominent disorder associated with metabolic syndrome.15 Intramyocyte accumulation of TG is also associated with pressure overload-induced cardiac hypertrophy and myopathy.16 Collectively, intra-myocyte TG serves as a dynamic FFA reserve, and its metabolism participates in various aspects of cardiac health and disease.
FFA metabolism
Circulating FFA levels are subject to stringent metabolic control. During starvation, declines in insulin levels lead to enhanced lipolysis in adipocytes via activation of hormone-sensitive lipase, which liberates FFAs from triglyceride stores, releasing them into the circulation. Lipolysis is also enhanced under various pathological conditions, such as acute myocardial infarction, obesity, and diabetes.
TG-derived FFA and BSA-bound FFA from lipolysis are activated by FACS to fatty acyl CoA, which are readily fueled into the mitochondrial beta-oxidation pathway. In addition, FFA may be released from the intramyocyte pool of TG and channeled into catabolism. While short and medium chain FFAs diffuse passively into mitochondria where they are catabolized, long chain fatty acyl CoA requires the carnitine shuttle for transport. Long chain fatty acyl CoA is first converted to long chain acylcarnitine in the outer mitochondrial membrane by carnitine palmitoyltransferase I (CPT-I), followed by transport across the inner mitochondrial membrane mediated by carnitine translocase. Finally, long chain fatty acylcarnitine is reverted to long chain fatty acyl CoA by CPT-II, and carnitine is recycled back to the cytosol for the next round of fatty acid shuttling.
In this process, CPT-I is the rate-limiting enzyme and subject itself to metabolic regulation. Malonyl CoA is a potent allosteric inhibitor of CPT-I and hence, fatty acid metabolism. Malonyl CoA is synthesized by acetyl CoA carboxylase (ACC), a direct target of AMPK.17 Phosphorylation of ACC by AMPK strongly suppresses ACC enzymatic activity and inhibits production of malonyl CoA, a process deemed the most important mechanism of AMPK-stimulated fatty acid catabolism and ATP generation.18 CPT-I is also an established pharmacological target to suppress fatty acid utilization (see below).
Within the mitochondrial lumen, fatty acyl CoA is broken down by beta-oxidation to acetyl CoA. There are four enzymes involved in beta-oxidation, each regulated at distinct levels. While acetyl CoA is fed into the citric acid cycle, the other products of beta-oxidation, NADH and FADH2, are utilized by the electron transfer chain to generate a transmembrane proton gradient and synthesize ATP.
Glucose metabolism
Glucose uptake into cells is mediated by glucose transporters (GLUT) (Figure 1). In cardiomyocytes, two GLUT isoforms are abundant, GLUT1 and GLUT4.19 The translocation of GLUT4 from intracellular compartments to the sarcolemmal membrane is stimulated by insulin.20 AMPK can similarly trigger GLUT4 translocation to enhance glucose uptake.18 Once inside the cell, glucose is immediately phosphorylated by hexokinase (HK), generating glucose-6-phosphate (G6P). G6P, in turn, serves as the starting point for various metabolic processes, such as glycolysis, glycogen synthesis, and the pentose phosphate pathway.
Phosphofructokinase (PFK)1 is a critical enzyme in glycolysis, catalyzing the first commitment step from fructose 6-phosphate to fructose 1,6-biphosphate. PFK1 is subject to complex allosteric regulation.2 PFK1 enzymatic activity is inhibited by ATP and citrate and stimulated by AMP and fructose 2,6-biphosphate. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) catalyzes the first ATP-generating step in glycolysis. The product of GAPDH, NADH, functions as a potent inhibitor of GAPDH. Under anaerobic conditions, glycolytic flux is facilitated by removal of NADH through lactate dehydrogenase and generation of lactate. Finally, pyruvate kinase stimulates the second ATP-generating step in glycolysis, conversion of phosphoenoylpyruvate to pyruvate. There are two pyruvate kinase isoforms in muscle, PKM1 and PKM2, deriving from a single gene that undergoes alternative splicing21. Recent studies suggest that levels and activities of PKM2 are critical for cellular coordination of ATP generation and growth.22 However, the role of these pyruvate kinases in heart failure remains largely unknown. Pyruvate is translocated to mitochondria by recently identified pyruvate carriers.23, 24
Pyruvate dehydrogenase (PDH) complex is a pivotal multiprotein regulatory nexus in glucose metabolism. PDH is subject to elegant regulation by allosteric metabolites and post-translational modifications. The products of PDH, acetyl CoA and NADH, inhibit PDH enzymatic activity, while NAD+ and CoA stimulate it. PDH kinase (PDK) and phosphatase are associated within the PDH complex. Phosphorylation by PDK (mainly PDK4 in heart) suppresses PDH enzymatic activity, and dephosphorylation enhances it.25 Thus, PDK may be another important target for pharmacological intervention to stimulate glucose metabolism.
Glycogen metabolism
One branch of glucose metabolism results in synthesis of glycogen. G6P is first converted to G1P by phosphoglucomutase. G1P is then activated to UDP-glucose, which can be readily incorporated into the growing chain of glycogen by glycogen synthase. Glycogen is an important energy repository in heart, which can be quickly mobilized when needed.26 During energy shortage or increased workload, the rise of AMP activates PKA, which in turn phosphorylates and activates phosphorylase kinase, leading to glycogen breakdown and glycogenolysis.
Glycogen occupies as much as 2% of cardiomyocyte cell volume, and this component is significantly greater during fetal stages, suggesting an important role of glycogen homeostasis during development.26 Indeed, absence of cardiac glycogen by disrupting glycogen synthase 1 leads to early defects in cardiac development and increases in mortality.27 This result is consistent with the finding that glycogen synthase 1 is the major synthase isoform in heart.28
Glycogen metabolism contributes significantly to aerobic myocardial glucose use. Early studies showed that glycogen oxidation may provide >40% of total ATP from glucose.29 Importantly, more than 50% of glycogen-derived glucose is oxidized instead of generating lactate. Contemporaneously, Taegtmeyer and colleagues employed dual labeling techniques to simultaneously measure glucose and glycogen metabolism.30 They found that epinephrine stimulation leads to almost complete oxidation of glycogen, while about 50% of exogenous glucose passes through a non-oxidative pathway. Further, glycogenolysis responds rapidly to epinephrine, during the lag phase seen in glucose utilization. Glycogen is therefore a critical reservoir to accommodate rapid changes in cardiac demand, but not at the expense of creating more lactate or inefficient utilization of substrates.31
Preferential oxidation of glycogen-derived glucose, as opposed to lactate generation, has also been observed under disease conditions. Allard et al found that myocardial hypertrophy is associated with increases in glycolysis.32 However, a greater proportion of glycogen is oxidized as compared to exogenous glucose, suggesting hypertrophied heart maintains the ability to efficiently metabolize glycogen.
In ischemic conditions, however, the metabolism and functional role of glycogen is more complicated. Cross et al found that high glycogen content protects heart from brief ischemia and consequent contracture, probably due to enhanced nutrient reserves. However, prolonged ischemia provoked exacerbated injury in the presence of higher levels of glycogen, which may result from lower ischemic pH and increased sodium-proton exchange during reperfusion.33
Interplay between FFA and glucose metabolism
Fifty years ago, Philip Randle and colleagues first described a glucose/fatty acid cycle to explain the reciprocal relationship between these two substances in metabolism.34 Acetyl CoA generated from FFA beta-oxidation stimulates PDK, which in turn, phosphorylates PDH and suppresses PDH activity (Figure 1). On the other hand, acetyl CoA derived from glycolysis and PDH imposes direct inhibition on beta-oxidation reactions. Moreover, citrate from the citric acid cycle can be transported back to the cytosol, where it is lysed to acetyl CoA.35 Acetyl CoA in the cytosol, then, is subject to carboxylation to malonyl CoA by ACC. Malonyl CoA strongly inhibits CPT-I and FFA uptake. This “Randle hypothesis” appears to hold true under controlled experimental conditions. However, accumulating findings point to more complex metabolic interactions with insulin signaling in chronic situations, such as insulin resistance.36
As a consequence of these interlacing events involving enzymatic substrates and products, gene expression, and post-translational protein modifications, metabolism of both FFA and glucose is maintained in careful balance.37 Indeed, the metabolic flexibility of the myocardium – the ability to move rapidly among energy sources and coordinate a wide range of intracellular metabolic processes – is pivotal to mechanical contraction and relaxation in the setting of cardiac stress.38 Disruption of this flexibility is held to be a significant contributor to cardiomyocyte dysfunction and heart failure.
Amino acid metabolism
Amino acids are also important substrates for energy production in heart. Through transamination and deamination reactions, various amino acids generate metabolic intermediates and feed into the citric acid cycle. Early studies have shown that alanine is effectively secreted from the myocardium during pacing stress, while glutamate manifests net uptake.39 These differences are significantly larger in patients with ischemic heart disease, suggesting pathological implications. Moreover, the increase in alanine production is due largely to de novo synthesis, which occurs proportionate to intracellular pyruvate levels.40 The ischemic heart may also adapt to the hypoxic situation by selectively reducing lactate concentrations through conversion of pyruvate to alanine. Since lactate accumulation during ischemia severely inhibits anaerobic metabolism, secretion of pyruvate through transamination may relieve this inhibition and maximize energy production. In addition, glutamate uptake and metabolism under ischemic conditions may lead to significant increases in succinate, which can contribute to substrate phosphorylation and energy production.41
Recently, catabolism of branched chain amino acids has garnered attention due to connections to cardiac hypertrophy and heart failure.42 Through a series of reactions, branched chain amino acids can be converted to metabolic intermediates for the citric acid cycle metabolism. The rate-limiting enzyme is the branched chain α-keto acids dehydrogenase complex, whose activity is inhibited by phosphorylation. The mitochondrial phosphatase PP2C is capable of dephosphorylating and activating the dehydrogenase complex. PP2C expression is significantly reduced in failing heart, which raises the possibility that accumulation of branched chain amino acids may participate in the pathogenesis of heart failure.43 Moreover, silencing of the gene coding for the enzyme catalyzing the first step in peripheral branched-chain amino acid metabolism leads to remarkable improvements in glucose tolerance and insulin sensitivity, which involves an active futile cycle of increased protein synthesis and degradation.44
Nucleotide metabolism
Degradation of nucleic acids may be another important substrate for energy production in heart. Riboses derived from nucleotide catabolism may feed into glycolysis and the citric acid cycle through the non-oxidative phase of the pentose phosphate pathway. Recent studies identified a specific form of autophagy, ribophagy, targeting mature ribosomes under nutrient deprivation in yeast.45 Considering the rich content of nucleic acids in ribosomes, it is tempting to speculate that the pentose phosphate pathway is involved. Purine catabolism in cardiomyocytes, however, involves xanthine oxidase, which is also an important source of reactive oxygen species.46 Indeed, accumulating evidence suggests inhibition of xanthine oxidase leads to improvements in cardiac function in heart failure patients. Future work is therefore warranted to examine the role of ribophagy and nucleotide metabolism in cardiac energy homeostasis and function.
Considering the massive amounts of energy required by the heart to maintan unremitting mechanical contraction, it is not surprising that modulation of cardiac metabolism may influence whole body energy homeostasis. A recent study by Grueter et al reported that cardiomyocyte-specific over-expression of MED13, a subunit of the Mediator complex, protects mice from diet-induced obesity and improves glucose tolerance.47 Mechanistically, MED13 induction in heart increases whole body energy expenditure by selectively inhibiting nuclear receptor-responsive genes and enhancing energy homeostasis.
Metabolic derangements in the progression of heart failure
As noted, heart failure is a complex and highly prevalent clinical syndrome that results from a wide range of diseases, including hypertension, ischemic disease, diabetes, and many more.48 However, despite the diverse etiologies that trigger heart failure, altered cardiac metabolism is a common theme.4, 49 Generally, it is agreed that in mild-to-moderate heart failure, phosphocreatine and total creatine stores begin to decline; simultaneously, there is a shift in metabolic substrate utilization from FFA oxidation to glucose oxidation. [Recently, however, an essential role for creatine in heart has been challenged.50] In end-stage heart failure, however, FFA uptake and oxidation are impaired, and the heart becomes highly reliant on glycolysis for energy production (Figure 2).
Figure 2. Heart failure progression and metabolic derangements.
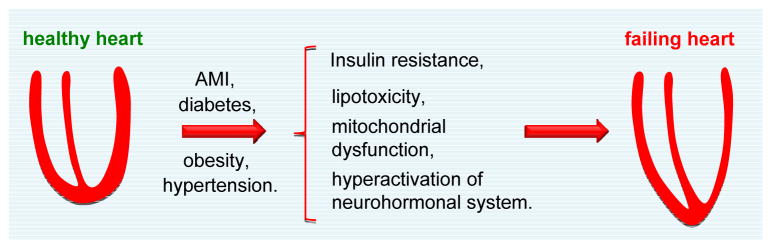
The healthy heart is a metabolic omnivore, capable of utilizing virtually any available nutrient. Progression of heart failure correlates with progressive metabolic derangements, which include insulin resistance, elevations in intracellular FFA levels, and mitochondrial dysfunction. Moreover, hyperactivation of the neurohumonal system leads to alterations in substrate availability in heart. In the early stages of heart failure, FFA uptake and oxidation remain either unchanged or moderately increased, while glucose oxidation is increased. When faced with persistent stress, advanced-stage heart failure emerges, and oxidation of both glucose and FFA are severely diminished. AMI, acute myocardial infarction.
Early-stage heart failure
Early stages of heart failure are marked by a wide range of compensatory processes, occurring at both molecular and functional levels. Clinical studies have revealed variable findings. For example, measurements of arterio-coronary sinus metabolite concentrations or fractional extractions among numerous substrates were not altered in a study of patients with moderate failure (NYHA II–III)51. Further, absolute FFA uptake was similar in heart failure patients and controls. On the other hand, significant increases in FFA oxidation were detected in similar patients (NYHA II–III) relative to healthy controls.52
Experimental studies have not revealed major alterations in substrate metabolism in early-stage heart failure. Using isotope tracer technology, Stanley and colleagues did not detect abnormalities in metabolic substrate utilization in a dog model of moderate heart failure.53 Total PDH activity was not altered. However, malonyl CoA levels were significantly reduced in the heart failure group compared to controls.53 Similarly, relatively normal rates of FFA and glucose metabolism have been described during the early stages of pacing-induced heart failure.54 In a comprehensive study using Dahl salt-sensitive rats, Kato et al showed that FFA uptake was not changed during the compensated stage of heart failure.55 Overall, these clinical and animal model studies support the notion that FFA utilization and global substrate metabolism are not negatively affected at early stages of heart failure.
Advanced heart failure
In contrast to the subtle metabolic changes occurring in early-stage heart failure, substrate extraction and oxidation are significantly altered at advanced stages of the syndrome. Metabolic labeling in patients of idiopathic dilated cardiomyopathy (DCM) uncovered correlations between reductions in FFA uptake and heart failure severity.56 Similarly, FFA uptake and oxidation were significantly impaired in patients with DCM; however, during pacing stress, the increase in glucose uptake seen in controls was severely diminished in DCM patients.57 In aggregate, these data highlight that loss of metabolic flexibility in response to stress is a consistent feature of heart failure.
These clinical observations have been validated and extended in animal studies. FFA uptake is severely reduced and glucose uptake significantly increased in end-stage heart failure (left ventricular end diastolic pressure ≥ 25 mmHg).54 Likewise, in a rapid pacing canine model, glucose uptake was significantly reduced in advanced heart failure, under both basal and insulin-stimulated conditions.58 Prolonged exposure to a high-salt diet in Dahl salt-sensitive rats led to a gradual increase in glucose uptake and sudden declines in FFA extraction at the onset of severe dysfunction.55 Thus, there is wide consensus that FFA uptake and utilization are significantly diminished in advanced heart failure. The discrepancies observed in glucose uptake may derive from differences in experimental model and/or experimental approaches employed. Thus, alterations in cardiac metabolism in heart failure derive from a combination of alterations in coronary flow and nutrient availability plus shifts in intrinsic myocardial substrate metabolism.
Metabolic derangements in non-ischemic heart failure
Metabolic alterations are considered among the most important contributors to the pathophysiology of non-ischemic heart failure. Various mechanisms have been proposed to participate in syndrome initiation and progression.
Lipotoxicity
Fat accumulation in heart, or fatty heart, has long been recognized by clinicians.59 Numerous experimental and clinical studies have established the correlation between TG accumulation and cardiomyopathy. Lipotoxicity is therefore held to be an important mechanism linking obesity and cardiac dysfunction.
Intramyocyte TG pools are maintained within a dynamic balance, determined by the rate of FFA uptake, TG synthesis, and TG catabolism. In Zucker diabetic rats, cardiac dilation and reduced contractility are associated with TG accumulation in cardiomyocytes as measured chemically and morphologically.60 The aberrant deposition of TG within the myocyte is due to reduced expression of fatty acid oxidative enzymes. More importantly, treatment of Zucker rats with troglitazone significantly lowered myocardial TG levels and improved cardiac function.
On the other hand, cardiomyocyte-restricted over-expression of enzymes involved in TG synthesis leads to TG accumulation and cardiac dysfunction. In two studies, Chiu et al over-expressed long chain FACS and fatty acid transport protein 1 in mouse heart and observed similar phenotypes of lipid accumulation and lipotoxicity.61, 62 Likewise, cardiomyocyte-specific over-expression of LPL or PPARα provoke increases in lipid uptake and cardiac dysfunction.63, 64 Collectively, these results indicate that breakdown of the finely-tuned balance of TG homeostasis in cardiomyocytes leads to abnormal accumulation of lipid and cardiomyopathy.
Glucolipotoxicity
While it is generally accepted that long-term consumption of high caloric foods modifies cardiac metabolism and impairs heart performance, the interplay between glucose and FFA in the progression of heart disease may be complicated in diabetes. The levels of both glucose and FFA are elevated in diabetic conditions. Excessive uptake and metabolism of FFA may provoke accumulation of intermediate metabolites, such as ceramides, which can promote cell death.60 Moreover, when oxidation of FFA falls below uptake, FFA may be esterified to intramyocyte TG and this lipid deposition may deteriorate cardiac contractile function. On the other hand, increases in glucose levels in diabetes cause insulin resistance in cardiomyocytes, which may be mediated by up-regulation of the hexosamine biosynthetic pathway.65 Further, chronic hyperglycemia is also accompanied by increases in ROS, which may interfere with calcium handling, mitochondrial function, and cardiac contractility.9, 66 In addition, elevations in glucose metabolism also inhibit FFA oxidation, contributing to the severity of lipid accumulation in cardiomyocytes. Collectively, the co-existence of hyperlipidemia and hyperglycemia in diabetes may result in excessive lipid deposition within cardiomyocytes, insulin resistance, and ROS overproduction, a combination events which has been termed glucolipotoxicity.67
Metabolic hypertrophy
Recent clinical studies have suggested that defects in glycogen metabolism may predispose to hypertrophic cardiomyopathy. Arad et al identified mutations in LAMP2 and PRAKG2 in patients with hypertrophic cardiomyopathy and electrophysiological abnormalities.68 Indeed, gain-of-function mutations of PRKAG2 trigger enhanced glycogen synthesis and enlargement of cardiomyocytes harboring glycogen-laden vacuoles.69
Mitochondrial dysfunction
Mitochondria are the energy powerhouses of the cardiomyocyte, and loss of normal mitochondrial function has long been implicated in the pathogenesis of heart failure.70, 71 Early studies using guinea pigs and cats, however, revealed that mitochondrial oxidative phosphorylation, oxygen consumption, and ATPase activity do not differ between normal and diseased hearts.72 It was therefore proposed that the decrease in myocardial contractility in heart failure may stem from metabolic control instead of intrinsic mitochondrial defects. On the other hand, studies in a canine chronic heart failure model identified severe abnormalities of mitochondrial size and integrity.73 These morphological alterations are translated into deterioration of mitochondrial respiratory activity after addition of substrates.74 For example, stage 3 respiratory rate is reduced 50% in chronic heart failure. Additionally, a detailed time course analysis showed that mitochondrial dysfunction develops in parallel with the onset of systolic dysfunction.75 Inconsistencies among these studies may be due to different subpopulations of cardiomyocyte mitochondria evaluated. Indeed, recent studies suggest the two mitochondrial subpopulations in cardiomyocytes, subsarcolemmal and interfibrillar, exhibit distinct properties in terms of respiration capacity, protein synthesis, and sensitivity to increased calcium.76
Accumulating evidence supports a model in which decreases in FFA oxidation and mitochondrial biogenesis may account for reductions in mitochondrial activity.77 Enzymes of the first and third steps of FFA oxidation are severely reduced in heart failure patients, findings which have been confirmed in preclinical models.78 In another study based on canine heart failure models, Osorio et al found the activity of two key enzymes of FFA oxidation, CPT-I and medium chain acyl-CoA dehydrogenase, were significantly reduced, which may be due to diminished expression of retinoid X receptor α.79 At a molecular level, an extensive literature points to significant reductions in master regulators of FFA oxidation and mitochondrial biogenesis, including PPARα and PGC1α.77
In addition, defects in mitochondrial respiration during heart failure may lead to over-production of ROS, which in turn fosters further deteriorations in mitochondrial function, disruptions in mitochondrial membrane integrity, and cell death. Significant accumulations of cytochrome c have been described in the cardiomyocyte cytosol of myocardial samples from patients with cardiomyopathy, which were associated with activation of caspase 3 and cell death.80
Interplay between systemic hemodynamics and cardiac metabolism
Clinical heart failure is, of course, associated with profound alterations in systemic hemodynamics. As a compensatory mechanism to counteract the insufficiency of circulatory supply, a complex set of neurohormonal reflexes is activated.81 Whereas the acute response may be adaptive in the short term, chronic overdrive of the sympathetic system promotes deterioration of cardiac performance. Indeed, countering these reflexes is bedrock therapy in heart failure management currently. Now, accumulating evidence suggests that metabolic derangements elicited by hyperactivation of the sympathetic system play important roles in disease initiation and progression.
Substrate availability
Activation of neurohormonal systems serves to maintain cardiac output to support systemic demand. Part of this response, however, is accompanied by increases in circulating norepinephrine (NE), elevations in sympathetic outflow, and NE spillover. The augmentation of plasma NE levels activates beta 3-adrenergic receptors in adipose tissues and enhances TG lipolysis,82 triggering increases in FFA levels which provide substrate necessary for maintenance of cardiac performance. With time, however, elevations in plasma FFA can adversely impact cardiac metabolism.
Increases in plasma FFA levels promote FFA uptake and metabolism in cardiomyocytes, which may interfere with glucose utilization. Excessive reliance on FFA metabolism can also promote ROS formation, which may further impair mitochondrial function and cardiac function. Indeed, elevated plasma FFA levels are an independent risk factor of sudden cardiac death in patients referred for coronary angiography.83 Conversely, clinical therapy for neurohormonal hyperactivation is associated with significant declines in circulating FFA levels in healthy men due to suppression of lipolysis in adipose tissues.84 Further, blockage of beta adrenoreceptors using carvedilol leads to 57% decreases in FFA utilization in the myocardium in heart failure patients, which is associated with significant improvements in cardiac performance.85
Overdrive of the sympathetic nervous system also participates in the pathogenesis of whole body insulin resistance. Elevations in circulating FFA levels can cause profound insulin desensitization in various tissues, such as liver, skeletal muscle and heart. Underlying mechanisms may include ROS over-production, interference with the insulin signaling pathway, or lipid accumulation, which can further impair left ventricular function. A 20-year follow-up study identified insulin resistance and associated factors as independent risk factors for systolic dysfunction.86 Genetic modulation to enhance cardiac insulin sensitivity leads to dramatic improvements in cardiac function, highlighting the fact that insulin resistance plays an important role in the pathogenesis of heart failure.87
Ischemia in advanced heart failure
Myocardial metabolic situations in advanced heart failure are distinct relative to earlier phases of the disorder. As discussed above, there is a consistent reduction in FFA oxidation in late stages of the syndrome. Although some discrepancies exist in the literature regarding glucose utilization, it is generally agreed that glucose oxidation is impaired, promoting reductions in metabolic substrates and ATP production. Diastolic ventricular pressure is elevated and diastolic wall tension is increased. As a consequence, coronary perfusion can be compromised, which creates a circumstance of myocardial ischemia in the absence of obstructive coronary artery disease. This is consistent with the findings that glucose uptake and glycolysis are enhanced while glucose oxidation is suppressed in advanced heart failure, which phenocopies the metabolic presentation of ischemic heart disease.88 Collectively, heart failure is associated with prominent metabolic dysregulation, which may likely emerge as a major, underlying mechanism of cardiomyopathy.
Metabolic therapy for the failing heart
The failing heart has been likened to “an engine out of fuel”4, and targeting cardiac metabolism has been proposed as a novel means of enhancing cardiac energetics and function. However, caution is warranted in efforts to translate preclinical observations to patients. First, responses to metabolic manipulations may vary significantly depending on metabolic state and cardiac insulin sensitivity. Second, there are well established differences in cardiac metabolism across species, such that observations made in animals may not pertain, at least not completely so, to people. Similarly, drugs that manifest beneficial effects in small clinical trials may later prove neutral or even harmful in large well-controlled clinical trials. Despite these concerns and issues, accumulating evidence from animal studies and small clinical trials strongly suggests that cardiac metabolism is a promising target for novel heart failure therapy.
Of the potential therapeutic strategies targeting cardiac metabolism, modulating substrate utilization has garnered considerable attention. Glucose oxidation generates less ATP per molecule than does FFA oxidation. However, for each oxygen molecule consumed, glucose oxidation provides 12% more ATP, a difference which may be significantly greater in reality due to adverse effects of FFA metabolism on ATP generation. Glucose is therefore an energetically more efficient substrate.49 Further, the shift from FFA oxidation to glucose utilization can protect cardiomyocytes from ROS accumulation and damage, which may result from buildup of FFA metabolic intermediates.89 Thus, the preferential use of glucose as metabolic substrate could potentially ease the burden of the failing heart. In concert with this, a number of studies in animals and humans suggest that boosting glucose oxidation improves cardiac function in heart failure.26
Promoting glucose utilization
Glucose-Insulin-Potassium infusion
Glucose-Insulin-Potassium (GIK) was first proposed as a therapy for acute myocardial infarction, seeking to increase glucose utilization and suppress FFA oxidation.90 A meta-analysis of clinical trials was suggestive of potential benefit in reducing in-hospital mortality.91 However, two more recent, large, randomized clinical trials have shown that GIK infusion does not affect mortality or development of heart failure after acute MI.92, 93
Clinical studies of GIK as a potential treatment for chronic heart failure are less robust, but have revealed some beneficial effects. In one study of patients with ischemic cardiomyopathy, a single dose of GIK infusion elicited improvements in ventricular wall motion and ejection fraction.94 Further, intermittent and long-term GIK infusion in patients with systolic heart failure led to improvements in cardiac function at 1 week and 1 month.95 It is not clear, however, whether the beneficial effects are due to enhancement of glucose utilization or suppression of FFA availability, or both. Additional large-scale, randomized trials are therefore warranted to test for efficacy and assess potential side effects, such as hyperkalemia, hyper/hypoglycemia, etc.
Glucagon-Like Peptide-1 (GLP-1)
Clinical studies have revealed robust correlation between heart failure and insulin resistance.96 That said, glucose intolerance is exceedingly common in heart failure patients. A positive correlation has been reported between blood glucose levels and the severity of heart failure.97 Mechanistically, insulin resistance leads to impaired insulin signaling in cardiomyocytes and reduced glucose utilization. Further, systemic insulin resistance provokes dyslipidemia and increases in circulating FFA levels, which worsen cardiomyocyte metabolic balance. Therefore, improving insulin sensitivity represents a potential means to enhance glucose utilization and inhibit lipotoxicity.49
GLP-1, an incretin peptide released from intestinal cells, potently stimulates insulin production from pancreatic islet β cells. Recombinant GLP-1 infusion has been shown to improve cardiac function in multiple preclinical models, including a canine heart failure model induced by right ventricular pacing,98 hypertensive heart failure-prone rats,99 and a post-ischemic rat model.100 However, only a few clinical trials, with limited numbers of patients, have been performed to evaluate the effects of GLP-1 in human heart failure.101 In NYHA class III-IV patients, both short-term (3 days)102, 103 and long-term (5 weeks)104 GLP-1 infusions improved cardiac function and exercise tolerance, independent of diabetes mellitus. In contrast, the GLP-1 effect in patients with NYHA class II/III heart failure in other studies was either not observed or without statistical significance.105 This raises the intriguing question of whether GLP-1 efficacy exists only in end-stage heart failure, when energy production is severely perturbed and insulin sensitivity is greatly suppressed. Overall, only modest increases in ejection fraction (4.4%) in GLP-1-treated patients have been noted. Additional studies with larger numbers of patients with diverse etiologies and heart failure stages are required to conclusively assess the clinical value of GLP-1 in heart failure. Recently, however, two large clinical trials evaluating saxagliptin and alogliptin, inhibiting DPP4 and increasing GLP-1 levels, failed to uncover beneficial cardiovascular effects in patients with type 2 diabetes.106, 107
At a mechanistic level, GLP-1 is generally considered to enhance insulin sensitivity and therefore promote glucose utilization. Infusion of recombinant GLP-1 in dogs with dilated cardiomyopathy leads to significant increases in insulin sensitivity measured by hyperinsulinemic-euglycemic clamp.98 Both basal and insulin-stimulated glucose uptake in myocardium are augmented by GLP-1 infusion, which is associated with up-regulation of oxygen consumption and coronary blood flow.
Other findings suggest that GLP-1 may improve cardiac function beyond the incretin effect and target-cell insulin sensitization. GLP-1 induces dose-dependent relaxation of preconstricted arteries.108 This endothelium-modulating effect was later extended in a clinical study, where infusion of recombinant GLP-1 elicited relative changes in brachial artery diameter from baseline without affecting plasma levels of glucose or insulin.109 Moreover, Elmquist and colleagues showed that both central and peripheral administration of a GLP-1 receptor agonist leads to increases in blood pressure and heart rate, highlighting a central regulatory role of GLP-1.110 Silencing of the GLP-1 receptor in mice causes profound cardiac phenotypes, including increased left ventricle thickness, reduced resting heart rate, and impaired contractility after insulin administration.111 Collectively, these findings suggest that the cardiac actions of GLP-1 stem from hemodynamic effects beyond incretin action.
Inhibiting FFA oxidation
As noted, metabolic alterations in heart failure are complex, partially due to the various disease etiologies of the syndrome.112 In addition, the stage and severity of heart failure also play important roles in determining the oxidative capacity of the heart. Overdrive of the sympathetic nervous system and insulin resistance contribute significantly to rises in circulating FFA levels and consequent increases in FFA oxidation. As a result, glucose uptake and utilization may be suppressed. Numerous studies have examined the approach of inhibiting FFA use in heart failure with some promising insights emerging.71 However, efficacy may vary, depending on the stage of syndrome progression. In advanced stage heart failure, mitochondrial oxidative capacity is already depressed. At that point, inhibition of FFA oxidation by pharmacological means may prove deleterious, as FFA is an important substrate of ATP production. Caution is warranted as this biology is moved forward toward the clinic.
Long chain 3-ketoacyl coenzyme A thiolase inhibitors
Trimetazidine suppresses free fatty acid oxidation by inhibiting long chain 3-ketoacyl coenzyme A thiolase, a key enzyme in the beta-oxidation pathway.113 Accumulating evidence suggests that trimetazidine confers protection in heart failure, especially of ischemic etiology. For example, in an ex vivo working heart model, trimetazidine infusion significantly improved functional recovery after 30 min of ischemia, which was associated with stimulation of glucose oxidation and decreases in lipid utilization.114 Additionally, long-term treatment of heart failure patients with trimetazidine improved left ventricular function and increased the ratio of phosphocreatine to ATP.115 Other work exploring different models suggests that additional mechanisms may be involved in trimetazidine-mediated cardioprotection, including suppression of fibrosis116 and enhancement of anti-oxidant capacity.117
More recently, an international multicenter retrospective cohort study of 669 heart failure patients from 2002 to 2010 reported that addition of trimetazidine on top of optimized standard therapy improved clinical outcomes and cardiovascular mortality.118 These results are promising, especially as the duration of observation was relatively long compared to other studies (38–40 months). However, this was not a prospective clinical trial and therefore causal relationships cannot be established. Additionally, two meta-analyses summarizing multiple randomized clinical trials of trimetazidine noted that most trials reported benefit.119, 120 Also, whereas trimetazidine is generally well tolerated, significant Parkinsonism-like side effects have been reported in multiple cases.121
CPT-I inhibitors
CPT-I is the rate-limiting enzyme in mitochondrial uptake and oxidation of long chain FFAs. It has been hypothesized that pharmacological suppression of CPT-I could suppress FFA uptake and oxidation, decrease futile oxygen use, reduce lipotoxicity, and potentially promote more efficient glucose oxidation.
Originally developed in the 1970’s as an anti-anginal agent, perhexiline has emerged with promise in the treatment of heart failure. Mechanistically, perhexiline suppresses CPT-I enzymatic activity, and therefore inhibits FFA oxidation and enhances glucose utilization and myocardial efficiency.122 A randomized, double blind clinical trial was carried out in two medical centers evaluating perhexiline’s effect in 56 heart failure patients.123 At 3 months, the perhexiline-treated group manifested improvements in symptoms, peak exercise oxygen consumption, left ventricular ejection fraction, and resting and peak stress myocardial function.123
Etoxomir was originally developed to treat diabetes by suppressing FFA uptake and utilization. However, long-term administration (12 weeks) was reported to improve cardiac function in a rat model of pressure overload.124 Additionally, in FFA-perfused rat heart, etoxomir elicited improvements in functional recovery associated with enhancement of glucose utilization.125 The first clinical trial of etoxomir in heart failure patients was conducted in 2000 with 10 patients (NYHA II–III). Three-month treatment with etoxomir led to significant benefits in cardiac stroke volume and maximum cardiac output during exercise.126 However, a larger clinical trial (ERGO, Etoxomir for the Recovery of Glucose Oxidation) was terminated due to detection of a liver injury signal.127
Oxfenicine, another CPT-I inhibitor, has been shown to prevent cardiac remodeling and delay decompensation in a canine model of pacing-induced heart failure.128 However, evidence of toxicity in heart, liver, and kidney, as well as inhibition of membrane ion channels, highlights the need for thorough evaluation before consideration of clinical use.129
PPAR agonists
Peroxisome proliferator-activated receptors (PPARs) are a class of nuclear receptors that regulate lipid metabolism by governing expression of multiple enzymes in the pathway. Of the three PPAR family members, PPAR-α is a major regulator of genes responsible for fatty acid uptake and oxidation. Cardiomyocyte-restricted over-expression of PPAR-α leads to up-regulation of FFA uptake and oxidation, which mimics the metabolic phenotype of diabetic cardiomyopathy.64 Also, PPAR-α levels are significantly repressed in cardiac hypertrophy elicited by pressure overload. A study by Young et al suggests that re-activation of PPAR-α may actually deteriorate cardiac contractile performance in hypertrophied rat heart.130 That said, systemic activation of PPAR-α pharmacologically triggers robust induction of mitochondrial FFA oxidation in liver, which may contribute to beneficial effects in heart.131 Increased hepatic extraction of circulating FFA can effectively reduce myocardial FFA uptake and utilization, especially under the conditions of heart failure. Previous studies showed that activation of PPAR-α protects heart from ischemia/reperfusion injury by metabolic modulation and suppression of inflammation.132 However, preclinical studies of the PPAR-α agonist fenofibrate in heart failure yielded variable results, ranging from beneficial to neutral, possibly due to differences in severity of disease, treatment duration, or dosage.133, 134 To date, no human studies on PPAR-α agonists in heart failure have been conducted.
PPAR-γ is the target of the thiazolidinedione (TZD) class of anti-diabetic drugs. PPAR-γ activation enhances lipid uptake and storage in adipose tissue. In so doing, circulating levels of FFA and TG can be significantly reduced, which may benefit cardiac function in heart failure. TZD administration in Zucker fatty rats can protect from ischemic injury through enhancing insulin sensitivity and glucose uptake.135 However, whether the improvement in cardiac metabolism contributes to the protection by TZD remains to be defined using loss-of-function approaches. A similar beneficial effect in cardiac function was reported in an ischemia/reperfusion model.136 Moreover, additional animal studies have demonstrated that PPAR-γ agonist administration improves left ventricular function and remodeling in ischemia-induced heart failure.137 More recently, TZD administration was reported to increase GLUT4 expression, improve insulin sensitivity, and enhance cardiac function in dogs.138 However, clinical use of PPAR-γ agonists to treat heart failure, cardiac ischemia/reperfusion, and even diabetes has been hindered due to potential cardiac toxicity manifested as fluid retention.139
Beta-adrenoreceptor antagonists
Patients with heart failure and ischemic heart disease are usually treated with medications targeting beta-adrenoreceptors. These pharmacological approaches improve cardiac function by decreasing workload and oxygen demand. Accumulating evidence, however, indicates that beta-receptor antagonists elicit profound metabolic changes in heart and body. Metoprolol treatment for 12 weeks led to significant reductions in CPT-I activity in a canine model of heart failure.140 Furthermore, 6-month treatment with beta-adrenoreceptor antagonists reduces resting energy production in moderate heart failure patients, which is accompanied by reductions in lipid oxidation and enhancement of glucose oxidation.141 These results suggest that beta-adrenoreceptor antagonism modulates cardiac metabolism, which may contribute to their pharmacological efficacy in presently unknown ways.
Collectively, multiple potential targets governing FFA and glucose utilization have been evaluated for therapeutic efficacy. CPT-I inhibitor-dependent suppression of FFA oxidation, and GLP-1-dependent enhancement of glucose oxidation hold the greatest promise to stimulate myocardial efficiency and enhance cardiac recovery in heart failure. However, a number of questions must be answered in comprehensive clinical trials prior to consideration of clinical use. At what stage of heart failure should patients be treated? Do different etiologies of heart failure affect drug efficacy and usage? Can metabolic drugs improve heart failure with preserved ejection fraction, a syndrome presently without effective therapy?
Conclusion and perspectives
Despite the longstanding, pervasive, and rapidly expanding prevalence of heart failure, optimal therapy has evolved little in recent years beyond the widespread use of mechanical devices. Novel targets and therapeutic strategies are therefore urgently needed. Among these, disease-associated metabolic derangements have long been the focus of great interest. Yet, it is only recently that appreciation of the importance of metabolic events, and metabolic flexibility, in cardiac stress has emerged. Concomitantly, several drugs targeting substrate metabolism have been developed.
Due to the intrinsic complexities of cardiac metabolism and the profound interplay among various substrates during disease progression, additional work is warranted to advance our understanding of fuel utilization, signal transduction, mitochondrial functionality, and cardiac contractility. Together, these events will converge on a rapidly evolving biology, where promise is heightened for novel means of targeting the devastating clinical syndrome of heart failure.
Acknowledgments
We thank the Hill lab for valuable discussions. This work was supported by grants from the NIH (HL-080144; HL-0980842; HL-100401), Cancer Prevention and Research Institute of Texas CPRIT (CPRIT, RP110486P3), the AHA DeHaan Foundation (0970518N), and the Fondation Leducq (11CVD04). Zhao V. Wang was supported by a postdoctoral fellowship from the AHA (10POST4320009).
Footnotes
Conflicts of interest
The authors declare no conflicts of interest.
References
- 1.Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Borden WB, Bravata DM, Dai S, Ford ES, Fox CS, Franco S, Fullerton HJ, Gillespie C, Hailpern SM, Heit JA, Howard VJ, Huffman MD, Kissela BM, Kittner SJ, Lackland DT, Lichtman JH, Lisabeth LD, Magid D, Marcus GM, Marelli A, Matchar DB, McGuire DK, Mohler ER, Moy CS, Mussolino ME, Nichol G, Paynter NP, Schreiner PJ, Sorlie PD, Stein J, Turan TN, Virani SS, Wong ND, Woo D, Turner MB. Heart disease and stroke statistics--2013 update: A report from the american heart association. Circulation. 2013;127:e6–e245. doi: 10.1161/CIR.0b013e31828124ad. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stanley WC, Recchia FA, Lopaschuk GD. Myocardial substrate metabolism in the normal and failing heart. Physiol Rev. 2005;85:1093–1129. doi: 10.1152/physrev.00006.2004. [DOI] [PubMed] [Google Scholar]
- 3.Lopaschuk GD, Ussher JR, Folmes CD, Jaswal JS, Stanley WC. Myocardial fatty acid metabolism in health and disease. Physiol Rev. 2010;90:207–258. doi: 10.1152/physrev.00015.2009. [DOI] [PubMed] [Google Scholar]
- 4.Neubauer S. The failing heart--an engine out of fuel. N Engl J Med. 2007;356:1140–1151. doi: 10.1056/NEJMra063052. [DOI] [PubMed] [Google Scholar]
- 5.Tuunanen H, Knuuti J. Metabolic remodelling in human heart failure. Cardiovascular research. 2011;90:251–257. doi: 10.1093/cvr/cvr052. [DOI] [PubMed] [Google Scholar]
- 6.van Bilsen M, van Nieuwenhoven FA, van der Vusse GJ. Metabolic remodelling of the failing heart: Beneficial or detrimental? Cardiovascular research. 2009;81:420–428. doi: 10.1093/cvr/cvn282. [DOI] [PubMed] [Google Scholar]
- 7.Des Rosiers C, Labarthe F, Lloyd SG, Chatham JC. Cardiac anaplerosis in health and disease: Food for thought. Cardiovascular research. 2011;90:210–219. doi: 10.1093/cvr/cvr055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cotter DG, Schugar RC, Crawford PA. Ketone body metabolism and cardiovascular disease. Am J Physiol Heart Circ Physiol. 2013;304:H1060–1076. doi: 10.1152/ajpheart.00646.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tsutsui H, Kinugawa S, Matsushima S. Oxidative stress and heart failure. Am J Physiol Heart Circ Physiol. 2011;301:H2181–2190. doi: 10.1152/ajpheart.00554.2011. [DOI] [PubMed] [Google Scholar]
- 10.Yu YH, Ginsberg HN. Adipocyte signaling and lipid homeostasis: Sequelae of insulin-resistant adipose tissue. Circ Res. 2005;96:1042–1052. doi: 10.1161/01.RES.0000165803.47776.38. [DOI] [PubMed] [Google Scholar]
- 11.Irie H, Krukenkamp IB, Brinkmann JF, Gaudette GR, Saltman AE, Jou W, Glatz JF, Abumrad NA, Ibrahimi A. Myocardial recovery from ischemia is impaired in cd36-null mice and restored by myocyte cd36 expression or medium-chain fatty acids. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:6819–6824. doi: 10.1073/pnas.1132094100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ibrahimi A, Bonen A, Blinn WD, Hajri T, Li X, Zhong K, Cameron R, Abumrad NA. Muscle-specific overexpression of fat/cd36 enhances fatty acid oxidation by contracting muscle, reduces plasma triglycerides and fatty acids, and increases plasma glucose and insulin. The Journal of biological chemistry. 1999;274:26761–26766. doi: 10.1074/jbc.274.38.26761. [DOI] [PubMed] [Google Scholar]
- 13.Kienesberger PC, Pulinilkunnil T, Nagendran J, Dyck JR. Myocardial triacylglycerol metabolism. Journal of molecular and cellular cardiology. 2013;55:101–110. doi: 10.1016/j.yjmcc.2012.06.018. [DOI] [PubMed] [Google Scholar]
- 14.Banke NH, Wende AR, Leone TC, O’Donnell JM, Abel ED, Kelly DP, Lewandowski ED. Preferential oxidation of triacylglyceride-derived fatty acids in heart is augmented by the nuclear receptor pparalpha. Circ Res. 2010;107:233–241. doi: 10.1161/CIRCRESAHA.110.221713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Turer AT, Hill JA, Elmquist JK, Scherer PE. Adipose tissue biology and cardiomyopathy: Translational implications. Circ Res. 2012;111:1565–1577. doi: 10.1161/CIRCRESAHA.111.262493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Krishnan J, Suter M, Windak R, Krebs T, Felley A, Montessuit C, Tokarska-Schlattner M, Aasum E, Bogdanova A, Perriard E, Perriard JC, Larsen T, Pedrazzini T, Krek W. Activation of a hif1alpha-ppargamma axis underlies the integration of glycolytic and lipid anabolic pathways in pathologic cardiac hypertrophy. Cell metabolism. 2009;9:512–524. doi: 10.1016/j.cmet.2009.05.005. [DOI] [PubMed] [Google Scholar]
- 17.Ruderman NB, Saha AK, Kraegen EW. Minireview: Malonyl coa, amp-activated protein kinase, and adiposity. Endocrinology. 2003;144:5166–5171. doi: 10.1210/en.2003-0849. [DOI] [PubMed] [Google Scholar]
- 18.Beauloye C, Bertrand L, Horman S, Hue L. Ampk activation, a preventive therapeutic target in the transition from cardiac injury to heart failure. Cardiovascular research. 2011;90:224–233. doi: 10.1093/cvr/cvr034. [DOI] [PubMed] [Google Scholar]
- 19.Abel ED. Glucose transport in the heart. Frontiers in bioscience : a journal and virtual library. 2004;9:201–215. doi: 10.2741/1216. [DOI] [PubMed] [Google Scholar]
- 20.Abel ED. Insulin signaling in heart muscle: Lessons from genetically engineered mouse models. Current hypertension reports. 2004;6:416–423. doi: 10.1007/s11906-004-0034-4. [DOI] [PubMed] [Google Scholar]
- 21.Mazurek S, Boschek CB, Hugo F, Eigenbrodt E. Pyruvate kinase type m2 and its role in tumor growth and spreading. Seminars in cancer biology. 2005;15:300–308. doi: 10.1016/j.semcancer.2005.04.009. [DOI] [PubMed] [Google Scholar]
- 22.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the warburg effect: The metabolic requirements of cell proliferation. Science (New York, NY ) 2009;324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Herzig S, Raemy E, Montessuit S, Veuthey JL, Zamboni N, Westermann B, Kunji ER, Martinou JC. Identification and functional expression of the mitochondrial pyruvate carrier. Science (New York, NY ) 2012;337:93–96. doi: 10.1126/science.1218530. [DOI] [PubMed] [Google Scholar]
- 24.Bricker DK, Taylor EB, Schell JC, Orsak T, Boutron A, Chen YC, Cox JE, Cardon CM, Van Vranken JG, Dephoure N, Redin C, Boudina S, Gygi SP, Brivet M, Thummel CS, Rutter J. A mitochondrial pyruvate carrier required for pyruvate uptake in yeast, drosophila, and humans. Science (New York, NY ) 2012;337:96–100. doi: 10.1126/science.1218099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wu P, Sato J, Zhao Y, Jaskiewicz J, Popov KM, Harris RA. Starvation and diabetes increase the amount of pyruvate dehydrogenase kinase isoenzyme 4 in rat heart. The Biochemical journal. 1998;329 (Pt 1):197–201. doi: 10.1042/bj3290197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Depre C, Vanoverschelde JL, Taegtmeyer H. Glucose for the heart. Circulation. 1999;99:578–588. doi: 10.1161/01.cir.99.4.578. [DOI] [PubMed] [Google Scholar]
- 27.Pederson BA, Chen H, Schroeder JM, Shou W, DePaoli-Roach AA, Roach PJ. Abnormal cardiac development in the absence of heart glycogen. Mol Cell Biol. 2004;24:7179–7187. doi: 10.1128/MCB.24.16.7179-7187.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Laughlin MR, Petit WA, Jr, Dizon JM, Shulman RG, Barrett EJ. Nmr measurements of in vivo myocardial glycogen metabolism. The Journal of biological chemistry. 1988;263:2285–2291. [PubMed] [Google Scholar]
- 29.Henning SL, Wambolt RB, Schonekess BO, Lopaschuk GD, Allard MF. Contribution of glycogen to aerobic myocardial glucose utilization. Circulation. 1996;93:1549–1555. doi: 10.1161/01.cir.93.8.1549. [DOI] [PubMed] [Google Scholar]
- 30.Goodwin GW, Ahmad F, Taegtmeyer H. Preferential oxidation of glycogen in isolated working rat heart. The Journal of clinical investigation. 1996;97:1409–1416. doi: 10.1172/JCI118561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Taegtmeyer H, Wilson CR, Razeghi P, Sharma S. Metabolic energetics and genetics in the heart. Annals of the New York Academy of Sciences. 2005;1047:208–218. doi: 10.1196/annals.1341.019. [DOI] [PubMed] [Google Scholar]
- 32.Allard MF, Henning SL, Wambolt RB, Granleese SR, English DR, Lopaschuk GD. Glycogen metabolism in the aerobic hypertrophied rat heart. Circulation. 1997;96:676–682. doi: 10.1161/01.cir.96.2.676. [DOI] [PubMed] [Google Scholar]
- 33.Cross HR, Opie LH, Radda GK, Clarke K. Is a high glycogen content beneficial or detrimental to the ischemic rat heart? A controversy resolved. Circ Res. 1996;78:482–491. doi: 10.1161/01.res.78.3.482. [DOI] [PubMed] [Google Scholar]
- 34.Randle PJ, Garland PB, Hales CN, Newsholme EA. The glucose fatty-acid cycle. Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet. 1963;1:785–789. doi: 10.1016/s0140-6736(63)91500-9. [DOI] [PubMed] [Google Scholar]
- 35.Zaidi N, Swinnen JV, Smans K. Atp-citrate lyase: A key player in cancer metabolism. Cancer Res. 2012;72:3709–3714. doi: 10.1158/0008-5472.CAN-11-4112. [DOI] [PubMed] [Google Scholar]
- 36.Samuel VT, Shulman GI. Mechanisms for insulin resistance: Common threads and missing links. Cell. 2012;148:852–871. doi: 10.1016/j.cell.2012.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kelley DE, Mandarino LJ. Fuel selection in human skeletal muscle in insulin resistance: A reexamination. Diabetes. 2000;49:677–683. doi: 10.2337/diabetes.49.5.677. [DOI] [PubMed] [Google Scholar]
- 38.Taegtmeyer H, Golfman L, Sharma S, Razeghi P, van Arsdall M. Linking gene expression to function: Metabolic flexibility in the normal and diseased heart. Annals of the New York Academy of Sciences. 2004;1015:202–213. doi: 10.1196/annals.1302.017. [DOI] [PubMed] [Google Scholar]
- 39.Mudge GH, Jr, Mills RM, Jr, Taegtmeyer H, Gorlin R, Lesch M. Alterations of myocardial amino acid metabolism in chronic ischemic heart disease. The Journal of clinical investigation. 1976;58:1185–1192. doi: 10.1172/JCI108571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Taegtmeyer H, Peterson MB, Ragavan VV, Ferguson AG, Lesch M. De novo alanine synthesis in isolated oxygen-deprived rabbit myocardium. The Journal of biological chemistry. 1977;252:5010–5018. [PubMed] [Google Scholar]
- 41.Taegtmeyer H. Metabolic responses to cardiac hypoxia. Increased production of succinate by rabbit papillary muscles. Circ Res. 1978;43:808–815. doi: 10.1161/01.res.43.5.808. [DOI] [PubMed] [Google Scholar]
- 42.Huang Y, Zhou M, Sun H, Wang Y. Branched-chain amino acid metabolism in heart disease: An epiphenomenon or a real culprit? Cardiovascular research. 2011;90:220–223. doi: 10.1093/cvr/cvr070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lu G, Ren S, Korge P, Choi J, Dong Y, Weiss J, Koehler C, Chen JN, Wang Y. A novel mitochondrial matrix serine/threonine protein phosphatase regulates the mitochondria permeability transition pore and is essential for cellular survival and development. Genes & development. 2007;21:784–796. doi: 10.1101/gad.1499107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.She P, Reid TM, Bronson SK, Vary TC, Hajnal A, Lynch CJ, Hutson SM. Disruption of bcatm in mice leads to increased energy expenditure associated with the activation of a futile protein turnover cycle. Cell metabolism. 2007;6:181–194. doi: 10.1016/j.cmet.2007.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kraft C, Deplazes A, Sohrmann M, Peter M. Mature ribosomes are selectively degraded upon starvation by an autophagy pathway requiring the ubp3p/bre5p ubiquitin protease. Nature cell biology. 2008;10:602–610. doi: 10.1038/ncb1723. [DOI] [PubMed] [Google Scholar]
- 46.Hajjar RJ, Leopold JA. Xanthine oxidase inhibition and heart failure: Novel therapeutic strategy for ventricular dysfunction? Circ Res. 2006;98:169–171. doi: 10.1161/01.RES.0000204557.09610.01. [DOI] [PubMed] [Google Scholar]
- 47.Grueter CE, van Rooij E, Johnson BA, DeLeon SM, Sutherland LB, Qi X, Gautron L, Elmquist JK, Bassel-Duby R, Olson EN. A cardiac microrna governs systemic energy homeostasis by regulation of med13. Cell. 2012;149:671–683. doi: 10.1016/j.cell.2012.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Owens AT, Jessup M. The year in heart failure. Journal of the American College of Cardiology. 2012;60:359–368. doi: 10.1016/j.jacc.2012.01.064. [DOI] [PubMed] [Google Scholar]
- 49.Ashrafian H, Frenneaux MP, Opie LH. Metabolic mechanisms in heart failure. Circulation. 2007;116:434–448. doi: 10.1161/CIRCULATIONAHA.107.702795. [DOI] [PubMed] [Google Scholar]
- 50.Lygate CA, Aksentijevic D, Dawson D, ten Hove M, Phillips D, de Bono JP, Medway DJ, Sebag-Montefiore L, Hunyor I, Channon KM, Clarke K, Zervou S, Watkins H, Balaban RS, Neubauer S. Living without creatine: Unchanged exercise capacity and response to chronic myocardial infarction in creatine-deficient mice. Circ Res. 2013;112:945–955. doi: 10.1161/CIRCRESAHA.112.300725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Funada J, Betts TR, Hodson L, Humphreys SM, Timperley J, Frayn KN, Karpe F. Substrate utilization by the failing human heart by direct quantification using arterio-venous blood sampling. PloS one. 2009;4:e7533. doi: 10.1371/journal.pone.0007533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Paolisso G, Gambardella A, Galzerano D, D’Amore A, Rubino P, Verza M, Teasuro P, Varricchio M, D’Onofrio F. Total-body and myocardial substrate oxidation in congestive heart failure. Metabolism: clinical and experimental. 1994;43:174–179. doi: 10.1016/0026-0495(94)90241-0. [DOI] [PubMed] [Google Scholar]
- 53.Chandler MP, Kerner J, Huang H, Vazquez E, Reszko A, Martini WZ, Hoppel CL, Imai M, Rastogi S, Sabbah HN, Stanley WC. Moderate severity heart failure does not involve a downregulation of myocardial fatty acid oxidation. Am J Physiol Heart Circ Physiol. 2004;287:H1538–1543. doi: 10.1152/ajpheart.00281.2004. [DOI] [PubMed] [Google Scholar]
- 54.Recchia FA, McConnell PI, Bernstein RD, Vogel TR, Xu X, Hintze TH. Reduced nitric oxide production and altered myocardial metabolism during the decompensation of pacing-induced heart failure in the conscious dog. Circ Res. 1998;83:969–979. doi: 10.1161/01.res.83.10.969. [DOI] [PubMed] [Google Scholar]
- 55.Kato T, Niizuma S, Inuzuka Y, Kawashima T, Okuda J, Tamaki Y, Iwanaga Y, Narazaki M, Matsuda T, Soga T, Kita T, Kimura T, Shioi T. Analysis of metabolic remodeling in compensated left ventricular hypertrophy and heart failure. Circulation. Heart failure. 2010;3:420–430. doi: 10.1161/CIRCHEARTFAILURE.109.888479. [DOI] [PubMed] [Google Scholar]
- 56.Yazaki Y, Isobe M, Takahashi W, Kitabayashi H, Nishiyama O, Sekiguchi M, Takemura T. Assessment of myocardial fatty acid metabolic abnormalities in patients with idiopathic dilated cardiomyopathy using 123i bmipp spect: Correlation with clinicopathological findings and clinical course. Heart (British Cardiac Society) 1999;81:153–159. doi: 10.1136/hrt.81.2.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Neglia D, De Caterina A, Marraccini P, Natali A, Ciardetti M, Vecoli C, Gastaldelli A, Ciociaro D, Pellegrini P, Testa R, Menichetti L, L’Abbate A, Stanley WC, Recchia FA. Impaired myocardial metabolic reserve and substrate selection flexibility during stress in patients with idiopathic dilated cardiomyopathy. Am J Physiol Heart Circ Physiol. 2007;293:H3270–3278. doi: 10.1152/ajpheart.00887.2007. [DOI] [PubMed] [Google Scholar]
- 58.Nikolaidis LA, Sturzu A, Stolarski C, Elahi D, Shen YT, Shannon RP. The development of myocardial insulin resistance in conscious dogs with advanced dilated cardiomyopathy. Cardiovascular research. 2004;61:297–306. doi: 10.1016/j.cardiores.2003.11.027. [DOI] [PubMed] [Google Scholar]
- 59.Szczepaniak LS, Victor RG, Orci L, Unger RH. Forgotten but not gone: The rediscovery of fatty heart, the most common unrecognized disease in america. Circ Res. 2007;101:759–767. doi: 10.1161/CIRCRESAHA.107.160457. [DOI] [PubMed] [Google Scholar]
- 60.Zhou YT, Grayburn P, Karim A, Shimabukuro M, Higa M, Baetens D, Orci L, Unger RH. Lipotoxic heart disease in obese rats: Implications for human obesity. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:1784–178. doi: 10.1073/pnas.97.4.1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chiu HC, Kovacs A, Blanton RM, Han X, Courtois M, Weinheimer CJ, Yamada KA, Brunet S, Xu H, Nerbonne JM, Welch MJ, Fettig NM, Sharp TL, Sambandam N, Olson KM, Ory DS, Schaffer JE. Transgenic expression of fatty acid transport protein 1 in the heart causes lipotoxic cardiomyopathy. Circ Res. 2005;96:225–233. doi: 10.1161/01.RES.0000154079.20681.B9. [DOI] [PubMed] [Google Scholar]
- 62.Chiu HC, Kovacs A, Ford DA, Hsu FF, Garcia R, Herrero P, Saffitz JE, Schaffer JE. A novel mouse model of lipotoxic cardiomyopathy. The Journal of clinical investigation. 2001;107:813–822. doi: 10.1172/JCI10947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yagyu H, Chen G, Yokoyama M, Hirata K, Augustus A, Kako Y, Seo T, Hu Y, Lutz EP, Merkel M, Bensadoun A, Homma S, Goldberg IJ. Lipoprotein lipase (lpl) on the surface of cardiomyocytes increases lipid uptake and produces a cardiomyopathy. The Journal of clinical investigation. 2003;111:419–426. doi: 10.1172/JCI16751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Finck BN, Lehman JJ, Leone TC, Welch MJ, Bennett MJ, Kovacs A, Han X, Gross RW, Kozak R, Lopaschuk GD, Kelly DP. The cardiac phenotype induced by pparalpha overexpression mimics that caused by diabetes mellitus. The Journal of clinical investigation. 2002;109:121–130. doi: 10.1172/JCI14080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hanover JA, Krause MW, Love DC. The hexosamine signaling pathway: O-glcnac cycling in feast or famine. Biochimica et biophysica acta. 2010;1800:80–95. doi: 10.1016/j.bbagen.2009.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414:813–820. doi: 10.1038/414813a. [DOI] [PubMed] [Google Scholar]
- 67.Young ME, McNulty P, Taegtmeyer H. Adaptation and maladaptation of the heart in diabetes: Part ii: Potential mechanisms. Circulation. 2002;105:1861–1870. doi: 10.1161/01.cir.0000012467.61045.87. [DOI] [PubMed] [Google Scholar]
- 68.Arad M, Maron BJ, Gorham JM, Johnson WH, Jr, Saul JP, Perez-Atayde AR, Spirito P, Wright GB, Kanter RJ, Seidman CE, Seidman JG. Glycogen storage diseases presenting as hypertrophic cardiomyopathy. N Engl J Med. 2005;352:362–372. doi: 10.1056/NEJMoa033349. [DOI] [PubMed] [Google Scholar]
- 69.Arad M, Benson DW, Perez-Atayde AR, McKenna WJ, Sparks EA, Kanter RJ, McGarry K, Seidman JG, Seidman CE. Constitutively active amp kinase mutations cause glycogen storage disease mimicking hypertrophic cardiomyopathy. The Journal of clinical investigation. 2002;109:357–362. doi: 10.1172/JCI14571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Abel ED, Doenst T. Mitochondrial adaptations to physiological vs. Pathological cardiac hypertrophy. Cardiovascular research. 2011;90:234–242. doi: 10.1093/cvr/cvr015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lionetti V, Stanley WC, Recchia FA. Modulating fatty acid oxidation in heart failure. Cardiovascular research. 2011;90:202–209. doi: 10.1093/cvr/cvr038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.SOBEL BES, JF, Pool PE, Sonnenblick EH, Braunwald E. Normal oxidative phosphorylation in mitochondria from the failing heart. Circ Res. 1967;21:355–364. [Google Scholar]
- 73.Sabbah HN, Sharov V, Riddle JM, Kono T, Lesch M, Goldstein S. Mitochondrial abnormalities in myocardium of dogs with chronic heart failure. Journal of molecular and cellular cardiology. 1992;24:1333–1347. doi: 10.1016/0022-2828(92)93098-5. [DOI] [PubMed] [Google Scholar]
- 74.Sharov VG, Goussev A, Lesch M, Goldstein S, Sabbah HN. Abnormal mitochondrial function in myocardium of dogs with chronic heart failure. Journal of molecular and cellular cardiology. 1998;30:1757–1762. doi: 10.1006/jmcc.1998.0739. [DOI] [PubMed] [Google Scholar]
- 75.Doenst T, Pytel G, Schrepper A, Amorim P, Farber G, Shingu Y, Mohr FW, Schwarzer M. Decreased rates of substrate oxidation ex vivo predict the onset of heart failure and contractile dysfunction in rats with pressure overload. Cardiovascular research. 2010;86:461–470. doi: 10.1093/cvr/cvp414. [DOI] [PubMed] [Google Scholar]
- 76.Holmuhamedov EL, Oberlin A, Short K, Terzic A, Jahangir A. Cardiac subsarcolemmal and interfibrillar mitochondria display distinct responsiveness to protection by diazoxide. PloS one. 2012;7:e44667. doi: 10.1371/journal.pone.0044667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Huss JM, Kelly DP. Mitochondrial energy metabolism in heart failure: A question of balance. The Journal of clinical investigation. 2005;115:547–555. doi: 10.1172/JCI200524405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sack MN, Rader TA, Park S, Bastin J, McCune SA, Kelly DP. Fatty acid oxidation enzyme gene expression is downregulated in the failing heart. Circulation. 1996;94:2837–2842. doi: 10.1161/01.cir.94.11.2837. [DOI] [PubMed] [Google Scholar]
- 79.Osorio JC, Stanley WC, Linke A, Castellari M, Diep QN, Panchal AR, Hintze TH, Lopaschuk GD, Recchia FA. Impaired myocardial fatty acid oxidation and reduced protein expression of retinoid x receptor-alpha in pacing-induced heart failure. Circulation. 2002;106:606–612. doi: 10.1161/01.cir.0000023531.22727.c1. [DOI] [PubMed] [Google Scholar]
- 80.Narula J, Pandey P, Arbustini E, Haider N, Narula N, Kolodgie FD, Dal Bello B, Semigran MJ, Bielsa-Masdeu A, Dec GW, Israels S, Ballester M, Virmani R, Saxena S, Kharbanda S. Apoptosis in heart failure: Release of cytochrome c from mitochondria and activation of caspase-3 in human cardiomyopathy. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:8144–8149. doi: 10.1073/pnas.96.14.8144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Opie LH, Knuuti J. The adrenergic-fatty acid load in heart failure. Journal of the American College of Cardiology. 2009;54:1637–1646. doi: 10.1016/j.jacc.2009.07.024. [DOI] [PubMed] [Google Scholar]
- 82.Triposkiadis F, Karayannis G, Giamouzis G, Skoularigis J, Louridas G, Butler J. The sympathetic nervous system in heart failure physiology, pathophysiology, and clinical implications. Journal of the American College of Cardiology. 2009;54:1747–1762. doi: 10.1016/j.jacc.2009.05.015. [DOI] [PubMed] [Google Scholar]
- 83.Pilz S, Scharnagl H, Tiran B, Wellnitz B, Seelhorst U, Boehm BO, Marz W. Elevated plasma free fatty acids predict sudden cardiac death: A 6.85-year follow-up of 3315 patients after coronary angiography. European heart journal. 2007;28:2763–2769. doi: 10.1093/eurheartj/ehm343. [DOI] [PubMed] [Google Scholar]
- 84.Newman RJ. Comparison of the antilipolytic effect of metoprolol, acebutolol, and propranolol in man. British medical journal. 1977;2:601–603. doi: 10.1136/bmj.2.6087.601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wallhaus TR, Taylor M, DeGrado TR, Russell DC, Stanko P, Nickles RJ, Stone CK. Myocardial free fatty acid and glucose use after carvedilol treatment in patients with congestive heart failure. Circulation. 2001;103:2441–2446. doi: 10.1161/01.cir.103.20.2441. [DOI] [PubMed] [Google Scholar]
- 86.Arnlov J, Lind L, Zethelius B, Andren B, Hales CN, Vessby B, Lithell H. Several factors associated with the insulin resistance syndrome are predictors of left ventricular systolic dysfunction in a male population after 20 years of follow-up. American heart journal. 2001;142:720–724. doi: 10.1067/mhj.2001.116957. [DOI] [PubMed] [Google Scholar]
- 87.Battiprolu PK, Hojayev B, Jiang N, Wang ZV, Luo X, Iglewski M, Shelton JM, Gerard RD, Rothermel BA, Gillette TG, Lavandero S, Hill JA. Metabolic stress-induced activation of foxo1 triggers diabetic cardiomyopathy in mice. The Journal of clinical investigation. 2012;122:1109–1118. doi: 10.1172/JCI60329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Jaswal JS, Keung W, Wang W, Ussher JR, Lopaschuk GD. Targeting fatty acid and carbohydrate oxidation--a novel therapeutic intervention in the ischemic and failing heart. Biochimica et biophysica acta. 2011;1813:1333–1350. doi: 10.1016/j.bbamcr.2011.01.015. [DOI] [PubMed] [Google Scholar]
- 89.Park TS, Goldberg IJ. Sphingolipids, lipotoxic cardiomyopathy, and cardiac failure. Heart failure clinics. 2012;8:633–641. doi: 10.1016/j.hfc.2012.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sodi-Pallares D, Testelli MR, Fishleder BL, Bisteni A, Medrano GA, Friedland C, De Micheli A. Effects of an intravenous infusion of a potassium-glucose-insulin solution on the electrocardiographic signs of myocardial infarction. A preliminary clinical report. Am J Cardiol. 1962;9:166–181. doi: 10.1016/0002-9149(62)90035-8. [DOI] [PubMed] [Google Scholar]
- 91.Fath-Ordoubadi F, Beatt KJ. Glucose-insulin-potassium therapy for treatment of acute myocardial infarction: An overview of randomized placebo-controlled trials. Circulation. 1997;96:1152–1156. doi: 10.1161/01.cir.96.4.1152. [DOI] [PubMed] [Google Scholar]
- 92.Diaz R, Goyal A, Mehta SR, Afzal R, Xavier D, Pais P, Chrolavicius S, Zhu J, Kazmi K, Liu L, Budaj A, Zubaid M, Avezum A, Ruda M, Yusuf S. Glucose-insulin-potassium therapy in patients with st-segment elevation myocardial infarction. JAMA : the journal of the American Medical Association. 2007;298:2399–2405. doi: 10.1001/jama.298.20.2399. [DOI] [PubMed] [Google Scholar]
- 93.Mehta SR, Yusuf S, Diaz R, Zhu J, Pais P, Xavier D, Paolasso E, Ahmed R, Xie C, Kazmi K, Tai J, Orlandini A, Pogue J, Liu L. Effect of glucose-insulin-potassium infusion on mortality in patients with acute st-segment elevation myocardial infarction: The create-ecla randomized controlled trial. JAMA : the journal of the American Medical Association. 2005;293:437–446. doi: 10.1001/jama.293.4.437. [DOI] [PubMed] [Google Scholar]
- 94.Cottin Y, Lhuillier I, Gilson L, Zeller M, Bonnet C, Toulouse C, Louis P, Rochette L, Girard C, Wolf JE. Glucose insulin potassium infusion improves systolic function in patients with chronic ischemic cardiomyopathy. European journal of heart failure. 2002;4:181–184. doi: 10.1016/s1388-9842(01)00222-7. [DOI] [PubMed] [Google Scholar]
- 95.Kalay N, Ozdogru I, Gul A, Yucel Y, Cetinkaya Y, Inanc MT, Dogan A, Kaya MG, Eryol NK. Effects of intermittent and long-term glucose-insulin-potassium infusion in patients with systolic heart failure. Experimental and clinical cardiology. 2008;13:85–88. [PMC free article] [PubMed] [Google Scholar]
- 96.Bell DS. Heart failure: The frequent, forgotten, and often fatal complication of diabetes. Diabetes care. 2003;26:2433–2441. doi: 10.2337/diacare.26.8.2433. [DOI] [PubMed] [Google Scholar]
- 97.Suskin N, McKelvie RS, Burns RJ, Latini R, Pericak D, Probstfield J, Rouleau JL, Sigouin C, Solymoss CB, Tsuyuki R, White M, Yusuf S. Glucose and insulin abnormalities relate to functional capacity in patients with congestive heart failure. European heart journal. 2000;21:1368–1375. doi: 10.1053/euhj.1999.2043. [DOI] [PubMed] [Google Scholar]
- 98.Nikolaidis LA, Elahi D, Hentosz T, Doverspike A, Huerbin R, Zourelias L, Stolarski C, Shen YT, Shannon RP. Recombinant glucagon-like peptide-1 increases myocardial glucose uptake and improves left ventricular performance in conscious dogs with pacing-induced dilated cardiomyopathy. Circulation. 2004;110:955–961. doi: 10.1161/01.CIR.0000139339.85840.DD. [DOI] [PubMed] [Google Scholar]
- 99.Poornima I, Brown SB, Bhashyam S, Parikh P, Bolukoglu H, Shannon RP. Chronic glucagon-like peptide-1 infusion sustains left ventricular systolic function and prolongs survival in the spontaneously hypertensive, heart failure-prone rat. Circulation. Heart failure. 2008;1:153–160. doi: 10.1161/CIRCHEARTFAILURE.108.766402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Zhao T, Parikh P, Bhashyam S, Bolukoglu H, Poornima I, Shen YT, Shannon RP. Direct effects of glucagon-like peptide-1 on myocardial contractility and glucose uptake in normal and postischemic isolated rat hearts. The Journal of pharmacology and experimental therapeutics. 2006;317:1106–1113. doi: 10.1124/jpet.106.100982. [DOI] [PubMed] [Google Scholar]
- 101.Munaf M, Pellicori P, Allgar V, Wong K. A meta-analysis of the therapeutic effects of glucagon-like peptide-1 agonist in heart failure. International journal of peptides. 2012;2012:249827. doi: 10.1155/2012/249827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Thrainsdottir I, Malmberg K, Olsson A, Gutniak M, Ryden L. Initial experience with glp-1 treatment on metabolic control and myocardial function in patients with type 2 diabetes mellitus and heart failure. Diabetes & vascular disease research : official journal of the International Society of Diabetes and Vascular Disease. 2004;1:40–43. doi: 10.3132/dvdr.2004.005. [DOI] [PubMed] [Google Scholar]
- 103.Nikolaidis LA, Mankad S, Sokos GG, Miske G, Shah A, Elahi D, Shannon RP. Effects of glucagon-like peptide-1 in patients with acute myocardial infarction and left ventricular dysfunction after successful reperfusion. Circulation. 2004;109:962–965. doi: 10.1161/01.CIR.0000120505.91348.58. [DOI] [PubMed] [Google Scholar]
- 104.Sokos GG, Nikolaidis LA, Mankad S, Elahi D, Shannon RP. Glucagon-like peptide-1 infusion improves left ventricular ejection fraction and functional status in patients with chronic heart failure. Journal of cardiac failure. 2006;12:694–699. doi: 10.1016/j.cardfail.2006.08.211. [DOI] [PubMed] [Google Scholar]
- 105.Halbirk M, Norrelund H, Moller N, Holst JJ, Schmitz O, Nielsen R, Nielsen-Kudsk JE, Nielsen SS, Nielsen TT, Eiskjaer H, Botker HE, Wiggers H. Cardiovascular and metabolic effects of 48-h glucagon-like peptide-1 infusion in compensated chronic patients with heart failure. Am J Physiol Heart Circ Physiol. 2010;298:H1096–1102. doi: 10.1152/ajpheart.00930.2009. [DOI] [PubMed] [Google Scholar]
- 106.Scirica BM, Bhatt DL, Braunwald E, Steg PG, Davidson J, Hirshberg B, Ohman P, Frederich R, Wiviott SD, Hoffman EB, Cavender MA, Udell JA, Desai NR, Mosenzon O, McGuire DK, Ray KK, Leiter LA, Raz I. Saxagliptin and cardiovascular outcomes in patients with type 2 diabetes mellitus. N Engl J Med. 2013;369:1317–1326. doi: 10.1056/NEJMoa1307684. [DOI] [PubMed] [Google Scholar]
- 107.White WB, Cannon CP, Heller SR, Nissen SE, Bergenstal RM, Bakris GL, Perez AT, Fleck PR, Mehta CR, Kupfer S, Wilson C, Cushman WC, Zannad F. Alogliptin after acute coronary syndrome in patients with type 2 diabetes. N Engl J Med. 2013;369:1327–1335. doi: 10.1056/NEJMoa1305889. [DOI] [PubMed] [Google Scholar]
- 108.Richter G, Feddersen O, Wagner U, Barth P, Goke R, Goke B. Glp-1 stimulates secretion of macromolecules from airways and relaxes pulmonary artery. The American journal of physiology. 1993;265:L374–381. doi: 10.1152/ajplung.1993.265.4.L374. [DOI] [PubMed] [Google Scholar]
- 109.Nystrom T, Gutniak MK, Zhang Q, Zhang F, Holst JJ, Ahren B, Sjoholm A. Effects of glucagon-like peptide-1 on endothelial function in type 2 diabetes patients with stable coronary artery disease. American journal of physiology. Endocrinology and metabolism. 2004;287:E1209–1215. doi: 10.1152/ajpendo.00237.2004. [DOI] [PubMed] [Google Scholar]
- 110.Yamamoto H, Lee CE, Marcus JN, Williams TD, Overton JM, Lopez ME, Hollenberg AN, Baggio L, Saper CB, Drucker DJ, Elmquist JK. Glucagon-like peptide-1 receptor stimulation increases blood pressure and heart rate and activates autonomic regulatory neurons. The Journal of clinical investigation. 2002;110:43–52. doi: 10.1172/JCI15595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Gros R, You X, Baggio LL, Kabir MG, Sadi AM, Mungrue IN, Parker TG, Huang Q, Drucker DJ, Husain M. Cardiac function in mice lacking the glucagon-like peptide-1 receptor. Endocrinology. 2003;144:2242–2252. doi: 10.1210/en.2003-0007. [DOI] [PubMed] [Google Scholar]
- 112.Fillmore N, Lopaschuk GD. Targeting mitochondrial oxidative metabolism as an approach to treat heart failure. Biochimica et biophysica acta. 2013;1833:857–865. doi: 10.1016/j.bbamcr.2012.08.014. [DOI] [PubMed] [Google Scholar]
- 113.Kantor PF, Lucien A, Kozak R, Lopaschuk GD. The antianginal drug trimetazidine shifts cardiac energy metabolism from fatty acid oxidation to glucose oxidation by inhibiting mitochondrial long-chain 3-ketoacyl coenzyme a thiolase. Circ Res. 2000;86:580–588. doi: 10.1161/01.res.86.5.580. [DOI] [PubMed] [Google Scholar]
- 114.Lopaschuk GD, Barr R, Thomas PD, Dyck JR. Beneficial effects of trimetazidine in ex vivo working ischemic hearts are due to a stimulation of glucose oxidation secondary to inhibition of long-chain 3-ketoacyl coenzyme a thiolase. Circ Res. 2003;93:e33–37. doi: 10.1161/01.RES.0000086964.07404.A5. [DOI] [PubMed] [Google Scholar]
- 115.Fragasso G, Perseghin G, De Cobelli F, Esposito A, Palloshi A, Lattuada G, Scifo P, Calori G, Del Maschio A, Margonato A. Effects of metabolic modulation by trimetazidine on left ventricular function and phosphocreatine/adenosine triphosphate ratio in patients with heart failure. European heart journal. 2006;27:942–948. doi: 10.1093/eurheartj/ehi816. [DOI] [PubMed] [Google Scholar]
- 116.Liu X, Gai Y, Liu F, Gao W, Zhang Y, Xu M, Li Z. Trimetazidine inhibits pressure overload-induced cardiac fibrosis through nadph oxidase-ros-ctgf pathway. Cardiovascular research. 2010;88:150–158. doi: 10.1093/cvr/cvq181. [DOI] [PubMed] [Google Scholar]
- 117.Belardinelli R, Solenghi M, Volpe L, Purcaro A. Trimetazidine improves endothelial dysfunction in chronic heart failure: An antioxidant effect. European heart journal. 2007;28:1102–1108. doi: 10.1093/eurheartj/ehm071. [DOI] [PubMed] [Google Scholar]
- 118.Fragasso G, Rosano G, Baek SH, Sisakian H, Di Napoli P, Alberti L, Calori G, Kang SM, Sahakyan L, Sanosyan A, Vitale C, Marazzi G, Margonato A, Belardinelli R. Effect of partial fatty acid oxidation inhibition with trimetazidine on mortality and morbidity in heart failure: Results from an international multicentre retrospective cohort study. International journal of cardiology. 2013;163:320–325. doi: 10.1016/j.ijcard.2012.09.123. [DOI] [PubMed] [Google Scholar]
- 119.Gao D, Ning N, Niu X, Hao G, Meng Z. Trimetazidine: A meta-analysis of randomised controlled trials in heart failure. Heart (British Cardiac Society) 2011;97:278–286. doi: 10.1136/hrt.2010.208751. [DOI] [PubMed] [Google Scholar]
- 120.Zhang L, Lu Y, Jiang H, Zhang L, Sun A, Zou Y, Ge J. Additional use of trimetazidine in patients with chronic heart failure: A meta-analysis. Journal of the American College of Cardiology. 2012;59:913–922. doi: 10.1016/j.jacc.2011.11.027. [DOI] [PubMed] [Google Scholar]
- 121.Masmoudi K, Masson H, Gras V, Andrejak M. Extrapyramidal adverse drug reactions associated with trimetazidine: A series of 21 cases. Fundamental & clinical pharmacology. 2012;26:198–203. doi: 10.1111/j.1472-8206.2011.01008.x. [DOI] [PubMed] [Google Scholar]
- 122.Ashrafian H, Horowitz JD, Frenneaux MP. Perhexiline. Cardiovascular drug reviews. 2007;25:76–97. doi: 10.1111/j.1527-3466.2007.00006.x. [DOI] [PubMed] [Google Scholar]
- 123.Lee L, Campbell R, Scheuermann-Freestone M, Taylor R, Gunaruwan P, Williams L, Ashrafian H, Horowitz J, Fraser AG, Clarke K, Frenneaux M. Metabolic modulation with perhexiline in chronic heart failure: A randomized, controlled trial of short-term use of a novel treatment. Circulation. 2005;112:3280–3288. doi: 10.1161/CIRCULATIONAHA.105.551457. [DOI] [PubMed] [Google Scholar]
- 124.Turcani M, Rupp H. Etomoxir improves left ventricular performance of pressure-overloaded rat heart. Circulation. 1997;96:3681–3686. doi: 10.1161/01.cir.96.10.3681. [DOI] [PubMed] [Google Scholar]
- 125.Lopaschuk GD, Wall SR, Olley PM, Davies NJ. Etomoxir, a carnitine palmitoyltransferase i inhibitor, protects hearts from fatty acid-induced ischemic injury independent of changes in long chain acylcarnitine. Circ Res. 1988;63:1036–1043. doi: 10.1161/01.res.63.6.1036. [DOI] [PubMed] [Google Scholar]
- 126.Schmidt-Schweda S, Holubarsch C. First clinical trial with etomoxir in patients with chronic congestive heart failure. Clinical science (London, England : 1979) 2000;99:27–35. [PubMed] [Google Scholar]
- 127.Holubarsch CJ, Rohrbach M, Karrasch M, Boehm E, Polonski L, Ponikowski P, Rhein S. A double-blind randomized multicentre clinical trial to evaluate the efficacy and safety of two doses of etomoxir in comparison with placebo in patients with moderate congestive heart failure: The ergo (etomoxir for the recovery of glucose oxidation) study. Clinical science (London, England : 1979) 2007;113:205–212. doi: 10.1042/CS20060307. [DOI] [PubMed] [Google Scholar]
- 128.Lionetti V, Linke A, Chandler MP, Young ME, Penn MS, Gupte S, d’Agostino C, Hintze TH, Stanley WC, Recchia FA. Carnitine palmitoyl transferase-i inhibition prevents ventricular remodeling and delays decompensation in pacing-induced heart failure. Cardiovascular research. 2005;66:454–461. doi: 10.1016/j.cardiores.2005.02.004. [DOI] [PubMed] [Google Scholar]
- 129.Bachmann E, Weber E. Biochemical mechanisms of oxfenicine cardiotoxicity. Pharmacology. 1988;36:238–248. doi: 10.1159/000138390. [DOI] [PubMed] [Google Scholar]
- 130.Young ME, Laws FA, Goodwin GW, Taegtmeyer H. Reactivation of peroxisome proliferator-activated receptor alpha is associated with contractile dysfunction in hypertrophied rat heart. The Journal of biological chemistry. 2001;276:44390–44395. doi: 10.1074/jbc.M103826200. [DOI] [PubMed] [Google Scholar]
- 131.Cook WS, Yeldandi AV, Rao MS, Hashimoto T, Reddy JK. Less extrahepatic induction of fatty acid beta-oxidation enzymes by ppar alpha. Biochemical and biophysical research communications. 2000;278:250–257. doi: 10.1006/bbrc.2000.3739. [DOI] [PubMed] [Google Scholar]
- 132.Yue TL, Bao W, Jucker BM, Gu JL, Romanic AM, Brown PJ, Cui J, Thudium DT, Boyce R, Burns-Kurtis CL, Mirabile RC, Aravindhan K, Ohlstein EH. Activation of peroxisome proliferator-activated receptor-alpha protects the heart from ischemia/reperfusion injury. Circulation. 2003;108:2393–2399. doi: 10.1161/01.CIR.0000093187.42015.6C. [DOI] [PubMed] [Google Scholar]
- 133.Ogata T, Miyauchi T, Sakai S, Takanashi M, Irukayama-Tomobe Y, Yamaguchi I. Myocardial fibrosis and diastolic dysfunction in deoxycorticosterone acetate-salt hypertensive rats is ameliorated by the peroxisome proliferator-activated receptor-alpha activator fenofibrate, partly by suppressing inflammatory responses associated with the nuclear factor-kappa-b pathway. Journal of the American College of Cardiology. 2004;43:1481–1488. doi: 10.1016/j.jacc.2003.11.043. [DOI] [PubMed] [Google Scholar]
- 134.Labinskyy V, Bellomo M, Chandler MP, Young ME, Lionetti V, Qanud K, Bigazzi F, Sampietro T, Stanley WC, Recchia FA. Chronic activation of peroxisome proliferator-activated receptor-alpha with fenofibrate prevents alterations in cardiac metabolic phenotype without changing the onset of decompensation in pacing-induced heart failure. The Journal of pharmacology and experimental therapeutics. 2007;321:165–171. doi: 10.1124/jpet.106.116871. [DOI] [PubMed] [Google Scholar]
- 135.Sidell RJ, Cole MA, Draper NJ, Desrois M, Buckingham RE, Clarke K. Thiazolidinedione treatment normalizes insulin resistance and ischemic injury in the zucker fatty rat heart. Diabetes. 2002;51:1110–1117. doi: 10.2337/diabetes.51.4.1110. [DOI] [PubMed] [Google Scholar]
- 136.Yue TL, Bao W, Gu JL, Cui J, Tao L, Ma XL, Ohlstein EH, Jucker BM. Rosiglitazone treatment in zucker diabetic fatty rats is associated with ameliorated cardiac insulin resistance and protection from ischemia/reperfusion-induced myocardial injury. Diabetes. 2005;54:554–562. doi: 10.2337/diabetes.54.2.554. [DOI] [PubMed] [Google Scholar]
- 137.Shiomi T, Tsutsui H, Hayashidani S, Suematsu N, Ikeuchi M, Wen J, Ishibashi M, Kubota T, Egashira K, Takeshita A. Pioglitazone, a peroxisome proliferator-activated receptor-gamma agonist, attenuates left ventricular remodeling and failure after experimental myocardial infarction. Circulation. 2002;106:3126–3132. doi: 10.1161/01.cir.0000039346.31538.2c. [DOI] [PubMed] [Google Scholar]
- 138.Liu B, Liang G, Xu G, Liu D, Cai Q, Gao Z. Intervention of rosiglitazone on myocardium glut-4 mrna expression during ischemia-reperfusion injury in cardio-pulmonary bypass in dogs. Molecular and cellular biochemistry. 2013;373:279–284. doi: 10.1007/s11010-012-1501-x. [DOI] [PubMed] [Google Scholar]
- 139.Cariou B, Charbonnel B, Staels B. Thiazolidinediones and ppargamma agonists: Time for a reassessment. Trends in endocrinology and metabolism: TEM. 2012;23:205–215. doi: 10.1016/j.tem.2012.03.001. [DOI] [PubMed] [Google Scholar]
- 140.Panchal AR, Stanley WC, Kerner J, Sabbah HN. Beta-receptor blockade decreases carnitine palmitoyl transferase i activity in dogs with heart failure. Journal of cardiac failure. 1998;4:121–126. doi: 10.1016/s1071-9164(98)90252-4. [DOI] [PubMed] [Google Scholar]
- 141.Podbregar M, Voga G. Effect of selective and nonselective beta-blockers on resting energy production rate and total body substrate utilization in chronic heart failure. Journal of cardiac failure. 2002;8:369–378. doi: 10.1054/jcaf.2002.130238. [DOI] [PubMed] [Google Scholar]