

Abstract
The so called chromosome preparation is a procedure consisting of three strictly connected stages that enables to obtain chromosomes of quality suitable for cytogenetic analysis. Interestingly, experimental evidence strongly suggested that chromosome spreading and swelling (key processes that allow their counting and detailed structural analysis) are induced in the last fixative-evaporation stage by the interaction, mediated by acetic acid, between water from the environmental humidity, and the cytoplasmic matrix and the chromatin. However, since a considerable variation in the quality of chromosome preparations is observed, strongly depending on the environmental conditions in which the procedure takes place, a better comprehension of the mechanisms underlying chromosome preparation is required. To this aim, here we analysed intact lymphocytes before and at each stage of the chromosome preparation protocol by Fourier transform infrared (FTIR) spectroscopy, a technique widely used for the study not only of isolated biomolecules, but also of complex biological systems, such as whole cells. Interestingly, we found that the chromosome preparation protocol induces significant structural changes of cell proteins and DNA, in particular due to the interaction with acetic acid. Moreover, noteworthy, through the monitoring of changes in the water combination band between 2300 and 1800 cm–1, we provided evidence at molecular level of the crucial role of the bound water to the cytoplasmic matrix and to the chromatin in determining the chromosome spreading and swelling. Our FTIR results, therefore, underline the need to perform the last fixative-evaporation stage in standardized and optimized temperature and relative humidity conditions, thus providing chromosomes of high quality for the cytogenetic analysis that would lead in this way to more reliable results.
Key words: Chromosome preparation, chromosome spreading and swelling, DNA conformational transition, FTIR (micro)spectroscopy, protein, DNA hydration
Introduction
For over five decades the analysis of human chromosomes has been a well established procedure for obtaining prenatal and postnatal diagnosis. Nevertheless, the inconsistency in the quality of chromosome preparations still affects cytogenetic results, often rendering routine chromosome analysis difficult and time-consuming. The reason of it is probably due to the fact that the cell behaviour during the handling of chromosome preparation in cytogenetic laboratories is still poorly characterized and not yet completely understood. Interestingly, only in 2002 this problem became the subject of systematic studies by Claussen and colleagues.1
The so called chromosome preparation is a widely-used procedure that allows to obtain chromosomes suitable for cytogenetic analysis, starting from cells derived from any type of tissue that can be induced to replicate in culture (mitosis). Noteworthy, already in 1956 this procedure made it possible to establish the exact number of human chromosomes, that are 46 and not 48 as for long times was believed.2 The protocol (illustrated in detail in the following) consists essentially of three strictly connected stages: hypotonic treatment, prefixation, and methanol-acetic acid fixation followed by the evaporation of the fixative solution, by air drying. In particular, during the evaporation of the fixative solution mitotic cells dramatically swell and drag the chromosomes on the slide (chromosome spreading), making them well separated and enabling in this way their counting.2 Simultaneously, also the chromosomes swell, increasing their size up to five or more fold (chromosome swelling), and allowing thus the analysis of their structure by differential staining techniques.3 Since chromosome spreading and swelling play a key role to obtain samples of high quality for cytogenetic analysis, it is necessary to understand at molecular level the mechanisms that underlie these phenomena, in order to optimize the conditions that allow to standardize the chromosome preparation. The basic role played by the interaction, mediated by acetic acid, between on one hand the water deriving from the humidity of the environment in which the evaporation of the fixative solution occurs, and the cytoplasmic matrix and the chromatin on the other, prompted us to investigate through Fourier transform infrared (FTIR) spectroscopy each stage of the whole procedure, in order to obtain detailed information on the changes induced in particular by the final fixative-evaporation stage. Indeed, FTIR spectroscopy is a widely used technique for the study of the structural properties of isolated biomolecules, such as proteins, lipids, nucleic acids, and carbohydrates.4-12 Moreover, FTIR microspectroscopy, obtained coupling an infrared microscope to a FTIR spectrometer, is successfully applied to the study of complex biological systems, including intact cells, tissues, and whole model organisms.13-19 In particular, this non invasive and label free tool enables to obtain, within a single experiment, a unique molecular fingerprint of the sample under investigation, providing information on the composition and structure of the main cellular biomolecules.4-12 In addition, and of great relevance for the present study, also the infrared (IR) absorption of water can be monitored through specific water absorption bands, whose spectral features can vary when water interaction takes place with different bio-molecules.20-22 Here, we measured the IR absorption of intact lymphocytes before and after each stage of the chromosome preparation protocol. Firstly, significant changes in protein structures and DNA conformation were found to be induced by the treatment with acetic acid. Moreover, interestingly, this study made it possible to provide evidence of the crucial role of bound water to the cytoplasmic matrix and to chromatin in determining the chromosome spreading and swelling, through the identification of specific IR response due to the hydration of proteins and DNA.
Materials and Methods
Human lymphocyte culture and their isolation from red blood cells
Four mL of human peripheral blood were equally distributed in four flasks, each one containing 9 mL of a specific medium added with phytohaemagglutinin and heparin (Chromosome kit P, EuroClone®). After 72 h of incubation at 37°C, Colcemid at final concentration of 0.04 µg/mL was added in each culture 4 h before cell collection. Cell cultures from each flask were then centrifuged at 1000 × g for 10 min, the supernatant was discarded and the cell pellet was then resuspended vigorously in 2 mL of medium.
The samples were diluted with an equal volume of 0.9% NaCl aqueous solution and 4 mL of Lympholyte-H (Cedarlane) were carefully added. We should note that since Lympholyte-H has a higher density compared to the cell suspension, cells form a layer above the Lympholyte-H with a well distinct interface. Samples were, therefore, centrifuged for 20 min at 800 × g, and the resulting cultured lymphocyte layer was carefully transferred to a new centrifuge tube and diluted with an equal volume of 0.9% NaCl solution. After centrifugation at 1000 × g for 10 min, the supernatant was discarded and the cultured lymphocyte pellet was washed twice again with 0.9% NaCl solution. After centrifugation at 1000 × g for 10 min, the supernatant was discarded and the pellet of the first flask was used as a control for FTIR analysis.
Cultured lymphocyte hypotonic treatment
Each of the remaining cultured lymphocyte pellets was carefully resuspended in 5 mL of 75 mM KCl and maintained for 10 min at room temperature, as hypotonic treatment. After centrifugation at 1000 × g for 10 min, the supernatant was discarded and the pellet of the second flask was used for FTIR analysis.
Cultured lymphocyte prefixation stage
Each pellet of the remaining flasks was vigorously resuspended in 5 mL of Ibraimov solution (5% acetic acid in distilled water)23 and maintained at room temperature for 5 min, as prefixation stage. After centrifugation at 1000 × g for 10 min, the supernatant was discarded and the pellet of the third flask was submitted to FTIR analysis.
Cultured lymphocyte fixative-evaporation stage
Cell pellet of the last remaining flask was resuspended in 5 mL of fixative solution (methanol:glacial acetic acid 2:1) and maintained for 10 min at room temperature. After centrifugation at 1000 × g for 10 min, the supernatant was discarded and this stage was repeated twice. At the end, ~0.2 mL of freshly prepared fixative were added to cell pellet. The sample was then analysed by FTIR microspectroscopy.
To evaluate the mitotic index (number of cells in mitoses on number of cell nuclei) and to verify the chromosome spreading and swelling, one drop of this final preparation was then dripped onto a wet slide, let evaporate at 23°C and at 50% of relative humidity, then inspected by a phase contrast microscope at 20x magnification. In particular, the mitotic index, showing chromosome spreading and swelling of good quality for cytogenetic analyses, was 26%.
FTIR microspectroscopy analysis
FTIR microspectroscopy was performed on control lymphocytes, before any cell treatments, and after each stage of the chromosome preparation described above. For each sample, with the exception of that after the last fixative-evaporation stage, about 3-5 μL of the cell suspension were deposited onto a BaF2 window and dried at room temperature for ~1 h to eliminate the excess of water. The IR measurement of the sample after the last fixative took place, instead, at 23°C and at 50% of relative humidity accurately monitored (DATALOGGER ECOLOG TH1, Elpro-Buchs AG, Buchs, Switzerland). In particular, 50% of humidity was produced by paper stripes soaked in water, opportunely placed on the microscope stage, under the plastic cylinder that is normally used to protect the sample from external water vapour.
FTIR spectra were acquired in transmission mode, between 4000 and 800 cm–1, using a Varian 610-IR infrared microscope coupled to the Varian 670-IR FTIR spectrometer (both from Varian Australia Pty Ltd., Mulgrave, Australia), equipped with a mercury cadmium telluride (MCT) nitrogen-cooled detector. The side of the variable aperture of the microscope was adjusted from ~50 to 100 μm). Measurements were performed at 2 cm-1 spectral resolution; 25 KHz scan speed, triangular apodization, and by the accumulation of 512 scan coadditions. In this way, IR absorption spectra with an excellent signal-to-noise ratio (peak-to-peak noise of ~0.5 mA at 2000 cm–1) were obtained. Spectra were corrected, when necessary, for the residual water vapour absorption by subtraction of the vapour spectrum (Supplementary Figure 1). Moreover, the spectra affected by Mie scattering were corrected using the algorithm developed by Bassan and colleagues24 and the spectral range of the correction - originally from 4000 to 1000 cm–1 - was extended in our laboratory down to 800 cm–1. For comparison, we reported in Supplementary Figure 2 the spectra before the Mie scattering correction.
Second derivative analysis25 of the corrected spectra was performed (after a 13-point smoothing of the measured spectra) by the Savitzky-Golay method (3rd polynomial, 9 smoothing points), using the GRAMS/32 software (Galactic Ind. Corp., Salem, NH, USA). For each cell pellet deposited onto a BaF2 window, several measurements were repeated by selecting different areas on the same sample through the variable diaphragm aperture of the infrared microscope. We should add that three independent experiments were performed to evaluate the reproducibility of the results.
Results and Discussion
To try to comprehend, at molecular level, the effects of the interaction (mediated by acetic acid) between on one hand water (deriving from the humidity of the environment in which the evaporation of the fixative solution occurs) and the cytoplasmic matrix and the chromatin on the other, that leads to chromosome spreading and swelling,1,26,27 we monitored the IR response of intact cultured lymphocytes before treatment and after each stage of the protocol, from the hypotonic to the final fixative evaporation one.
As shown in Figure 1, the FTIR spectrum of intact cultured lymphocytes is due to the overlapping absorption of the different cell biomolecules. In particular, as illustrated in the figure, the spectral range between 3050-2800 cm–1 is dominated by the absorption of the lipid acyl chains, that also occurs between 1500-1350 cm–1, while the ester carbonyl absorbs around 1740 cm–1.9,10,12 Furthermore, the spectral range between 1700-1500 cm–1 is dominated by the amide I and amide II bands, mainly due to the C=O stretching and the NH bending of the peptide bond, respectively. In particular, the amide I band gives information on the protein secondary structures.5,8,10 Moreover, in the complex spectral range between 1250-1000 cm–1 the sugar-phosphate vibrations sensitive to nucleic acid backbone conformation and the absorption of the phospholipid phosphate group overlap with the IR response of the C-O group of glycogen and other carbohydrates.6,11,12,19 Finally, the IR absorption between 1000-800 cm–1 is mostly due to vibrational modes of nucleic acid sugar vibrations, sensitive to their conformation.6,7
Figure 1.
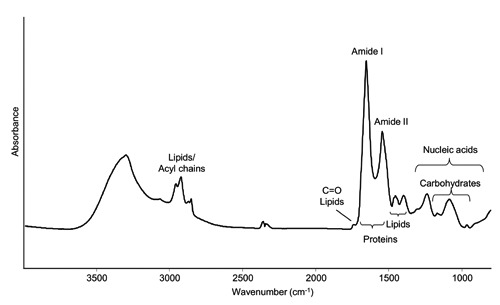
FTIR absorption spectrum of intact cultured lymphocytes, after correction of the Mie scattering. The absorption of the most important biomolecules is indicated.
Effects of acetic acid on cell proteins
To evaluate the effects of the treatment for chromosome preparation on the cell protein content, we firstly focused our attention on the spectral range between 1700-1600 cm–1, mainly due to the amide I band, particularly sensitive to changes in the protein secondary structures.4,5,8,10
We should note that in this spectral range also the absorption of other molecules occurs, including nucleic acids. However, as also indicated by the appreciable intensity of the amide II band around 1545 cm–1 (Figure 1 and Figure 2 A,B,C), the absorption of proteins dominates the IR response at each stage of the chromosome preparation protocol.
Figure 2.
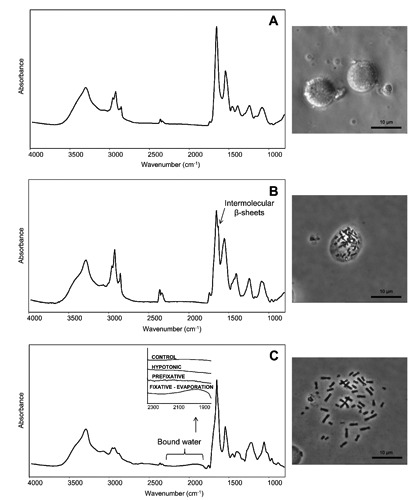
FTIR absorption spectra of cultured lymphocytes at each stage of chromosome preparation: A) after the hypotonic treatment; B) after prefixation stage with Ibraimov solution; C) after the fixative treatment with a solution of methanol:glacial acetic acid 2:1 followed by the final evaporation. The spectra are shown after correction of the Mie scattering and smoothing. The phase contrast microscopy images acquired upon the different procedure stages are also reported. In C, a magnification of the spectral range between 2300 and 1800 cm–1 due to bound water is also reported.
As reported in Figure 3, we analysed the second derivative spectra which make it possible to resolve the components that overlap under the amide I broad band.25 In particular, control cells were characterized mainly by a spectral component at 1655 cm–1, due to the alpha-helix and unordered structures of the whole cell protein content. Moreover, low intensity absorptions respectively at 1691 cm–1 and 1641 cm–1 were observed, both assigned to intramolecular beta sheet structures.
Figure 3.
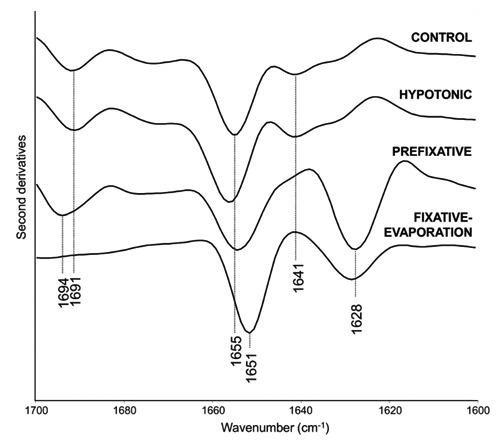
FTIR second derivative spectra of intact lymphocytes, and at each stage of the procedure for chromosome preparation, in the amide I band. Spectra have been normalized at the tyrosine peak around 1515 cm–1.
Hypotonic treatment
The spectral features described for the control cells were found to be very similar for cells after the hypotonic treatment with KCl (Figure 3), that, as reported by Claussen and co-workers,1 causes a movement of the chromosomes from the centre to the periphery into the mitotic sphere, with a minor increase of the cell volume.
Prefixation stage
Interestingly, the amide I band profile significantly changed following the first acetic acid treatment with Ibraimov23 (Figure 3 and Supplementary Figure 3), that destroys the cell membranes, enabling the visualisation of chromosomes, which start to appear like little black sticks, under a phase contrast microscope (Figure 2B).1 In particular, at this stage we observed a decrease of the whole cell native proteins, as indicated by the marked decrease in intensity of the band around 1655 cm–1. This response was also accompanied by the appearance of a new component at 1628 cm–1, that can be assigned to intermolecular beta sheets, typical of protein aggregates, and by the upshift of the 1691 cm–1 band to 1694 cm–1, again due to protein aggregates.4,8 Interestingly, the decrease in intensity of the native secondary structures indicates that most of the cell proteins unfold and aggregate following the first acetic acid treatments, as expected.28 These unfolding and aggregation processes are likely induced by modifications of the protein environment, such as the variation of pH toward acidic values.
Fixative-evaporation stage
Noteworthy, after the final treatment with the fixative solution (2:1 of methanol:glacial acetic acid), the evaporation of the solution occurs; in particular, methanol evaporates firstly,1 leaving acetic acid to react with water from the environmental humidity and cell proteins. This process induces a dramatic cell volume increase, followed by the spreading and the swelling of the chromosomes, which also increase their dimensions up to five or more fold.1,3 In particular, at this stage the cytoplasmic matrix disruption occurs, leading to the spreading of well-defined chromosomes (Figure 2C). We should note that in this final stage we monitored mainly the IR absorption of the cytoplasmic proteins and of the chromosomes, made up essentially of histones and DNA.
Interestingly, this treatment led to important differences in the spectral features of the amide I band (Figure 3), that was characterized by a component around 1651 cm–1 which can be assigned both to unordered structures and/or alpha-helices typical of histones in complex with DNA.29,30 Moreover, a band around 1628 cm–1 was also found, of lower intensity and broader compared to the prefixation stage, that can be mainly due to protein aggregates.4,8
We should also note that the component at ~1694 cm–1 observed in the prefixation stage disappeared, suggesting that in this last stage the formation of aggregates likely with parallel intermolecular beta sheet structures is induced. 31-33
Effects of acetic acid on DNA
Acetic acid treatment affected not only protein structures, as discussed above, but also DNA conformations. In Figure 4 we reported a comparison among the IR spectrum of control cells, that of cells after the prefixation stage, and the IR response of the sample after the last fixative-evaporation stage, between 1300-800 cm–1, where the response of nucleic acids dominates the spectra. In particular, the IR response of DNA in the control cells is characterized by the components at ~1236 cm–1 (PO2-antisymmetric stretching), 1170 cm–1 (sugarphosphate backbone vibration), 1083 cm-1 (PO2- symmetric stretching), and 966 cm–1 with a shoulder around 955 cm–1 (C-C stretching of DNA backbone).6
Figure 4.
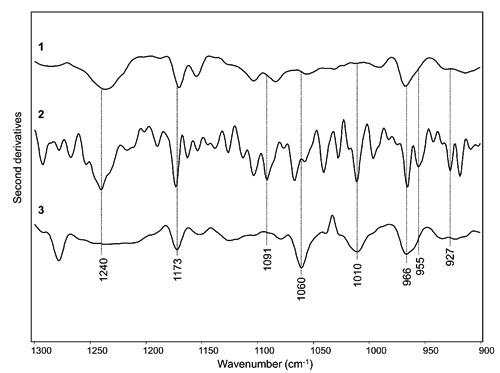
FTIR second derivative spectra of (1) intact lymphocytes (control); (2) sample after prefixative stage; (3) sample after the fixative-evaporation stage between 1300-900 cm–1. Spectra are the same of Figure 3, and they have been displayed after normalization at the tyrosine peak around 1515 cm–1.
Prefixation stage
Noteworthy, dramatic changes were observed in the spectral features of the sample measured after the first acetic acid treatment (i.e., after the prefixation stage). In particular, an intensity increase occurred for the ~1173 cm–1 (sugar-phosphate backbone vibration), and 966 cm–1 (C-C stretching of DNA backbone) bands. In addition, a well resolved component at 955 cm–1, due to the C-C stretching of DNA backbone, appeared together with a new band at 927 cm–1. Interestingly, the coexistence of the three bands at ~966 cm–1, 955 cm–1, and 927 cm–1 suggests the presence of DNA in Z conformation6. Moreover, a new intense spectral component appeared at 1010 cm–1 due to furanose, interestingly found strongly enhanced in Z-form DNA.6 These results, overall, indicate that the first acetic acid treatment induced a rearrangement of DNA conformation, that indeed appears to be mainly in Z form. Furthermore, the remarkable intensity increase of several IR bands could be due to an at least partial denaturation of DNA, that underlies the transition to the Z conformation.
Fixative-evaporation stage
Interestingly, the DNA response after the last fixative-evaporation stage was found to further change compared to that observed in the previous stage. In particular, the 1240 cm–1 component observed in control and prefixative stages almost disappeared, while the 1173 cm–1 band decreased in intensity. On the contrary, a new intense component at 1060 cm–1 appeared, that together with the well resolved band at 1010 cm–1 can be assigned to furanose, indicating again the presence of DNA mainly in Z-form.6
As discussed above, the presence of the left-handed Z conformation is likely due to the treatment with acetic acid. Indeed, it has been reported that the acid fixation induces the polynucleotide protonation, that in turn drives the transition toward Z conformation.34,35 Noteworthy, through immunofluorescence analysis it has been also reported that acetic acid fixation leads to the transition to Z-conformation in chromosomes, as a result of torsional stress in DNA.36
Water absorption following the final fixative-evaporation stage
In order to obtain information on possible interactions (mediated by acetic acid) among chromatin, cytoplasmic matrix and the water of the surrounding environment in which the last fixative stage took place (at 23°C and 50% of relative humidity), we analyzed the IR absorption spectra measured before and after each stage of the treatment (Figure 1 and Figure 2 A,B,C). In particular, we focused our attention on the spectral range between ~2300 and 1800 cm–1, where the broad combination absorption band of water occurs (Figure 2C, inset).21 Indeed, we focused mainly on this band since this spectral range is free from other absorptions. Interestingly, while in the spectra of the samples measured before (Figure 1), and after the hypotonic (Figure 2A) and prefixation (Figure 2B) treatments the IR response of water is negligible, after the last acetic acid treatment the spectral profile between ~2300 and 1800 cm–1 changed (Figure 2C). In particular, at this stage an IR absorption due to bound water appears, as better shown in the spectra magnification of Figure 2C. Indeed, the peak position of the combination band, lower compared to that of bulk water,21 strongly suggests that water was interacting with other molecules. Concerning this point, we should underline that, as mentioned above, this last stage took place in an environment with high humidity (about 50%), a prerequisite to ensure a good chromosome quality.1
Moreover, we should note the appearance of a shoulder around 1710 cm–1, due to the COOH absorption, that indicates the presence of acetic acid traces.37 Interestingly, the observed water absorption appears to be due to the hydration of the cytoplasmic proteins and of chromatin proteins and/or DNA, a process that is favoured by the treatment with acetic acid. In particular, acetic acid could interact with proteins and DNA making them more accessible to the water of the surrounding environment.28 In this way, water could easily bind not only to the proteins of the cytoplasm, but also to the proteins and/or DNA of the chromatin. These processes, altogether, are likely responsible for the chromosome spreading, favoured in particular by the hydration of the cytoplasmic matrix that, by its swelling, causes in turn the chromosome spreading on the slide, enabling in this way their counting.2 Moreover, the absorption of water also by chromatin is responsible for the simultaneous dramatic chromosome swelling, that makes it possible to evaluate in detail theirstructure.3 Indeed, following the last fixative-evaporation stage the IR signal of the sample is dominated by the protein absorption, as indicated by the high amide I and amide II bands, but displays also a significant contribution of DNA between 1300-900 cm–1 (Figures 4 and 5). In Figure 5, we reported, for comparison, also the spectra of bovine albumin and of DNA from calf thymus, taken as model samples, after the fixative-evaporation treatment. As shown, the hydration, indicated by the presence in particular of the combination band of water, occurs in both samples. These results indicate, therefore, that in the case of chromosomes both DNA and chromatin proteins could bind water molecules, leading to the chromosome swelling. We should add that we performed also a control experiment in which the sample after the last stage was measured with the same experimental setup used for the previous stages, namely in absence of any hydration produced by the paper stripes soaked in water. Unexpectedly, the combination water band was also detectable, even if at low intensity due to the reduction of humidity (data not shown). To confirm the results obtained by the inspection of the water combination band, we analysed also the IR response between 3700-3100 cm–1, due to the OH stretching of water21 (Supplementary Figure 4). We should note that, contrarily to the spectral range between 2300-1800 cm–1 where the water combination band falls, in the 3700-3100 cm–1 spectral range the overlapping absorption of different biomolecules, including proteins and DNA, occurs (Figure 5). Interestingly, the spectral profile of this spectral component was found to change after the last fixative-evaporation stage. In particular, a more pronounced shoulder around 3455 cm–1 was observed (Supplementary Figure 4A), more evident after the control spectrum subtraction (Supplementary Figure 4B).
Figure 5.
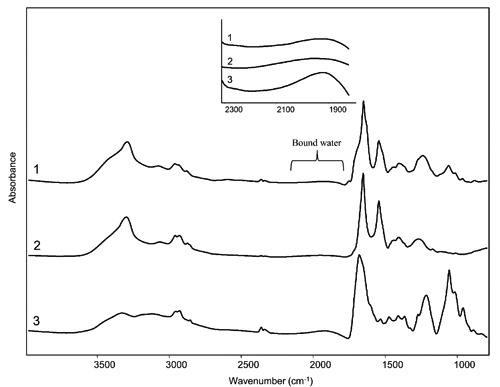
FTIR absorption spectra of cultured lymphocytes (1), albumin (2) and calf thymus DNA (3) after the fixative treatment with a solution of methanol:glacial acetic acid 2:1 followed by evaporation. The spectra are shown after correction of the Mie scattering and smoothing. A magnification of the spectral range between 2300 and 1800 cm–1 due to bound water is also reported.
Moreover, to clarify the crucial role of acetic acid in mediating the binding of water molecules by the cytoplasmic matrix and chromatin, we should add that we observed similar results following a more aggressive protocol, whose last step consisted of the sample treatment with an aqueous solution of acetic acid up to 60%.
Conclusions and Perspectives
Our FTIR study firstly highlighted that the chromosome preparation protocol leads to cell protein and DNA structural changes. In particular, the acetic acid treatments in the prefixative and in the last fixative-evaporation stages induced protein aggregation, with likely an intermolecular parallel beta sheet orientation after the last addition of acetic acid. Moreover, acetic acid was also found to promote DNA conformational transition to Z-form starting from the prefixative stage, a result in agreement with what already reported in literature.34-36 In addition, the FTIR results unequivocally provided evidence that the key factor leading to the spreading and swelling of chromosomes is the hydration of cytoplasmic proteins and of the chromosome components mediated by the acetic acid treatment, that makes proteins and DNA more accessible to water deriving from the humidity of the environment where the fixative solution evaporation occurs. In particular, the IR spectral features of the sample after the last fixative-evaporation stage were characterized by the water combination band between 2300-1800 cm–1, indicating that water was interacting with other molecules, namely DNA and proteins of the cytoplasmic matrix and of chromatin, as discussed above.
Our FTIR results overall underline that the last chromosome preparation stage has to take place in an environment at controlled temperature and relative humidity that, in our experiments, were of 23°C and of 50%, respectively. For this reason, this study highlights the need to control the inner temperature and relative humidity to perform the fixative-evaporation stage in standardized environmental conditions. This issue can be achieved using in cytogenetic laboratories a properly designed drying chamber, since experimental evidence showed that the use of this facility in the routine of chromosome preparations has dramatically reduced the variability of the results, constantly providing chromosomes of high quality that makes the cytogenetic analysis easier and more reliable.
Acknowledgments
D.A. acknowledges a postdoctoral fellowship of the University of MilanoBicocca. The authors wish to thank Paolo Tortora and Antonino Natalello of the University of Milano Bicocca, and EuroClone® for support and discussions.
References
- 1.Claussen U, Michel S, Muhlig P, Westermann M, Grummt UW, Kromeyer-Hauschild K, Liehr T. Demystifying chromosome preparation and the implications for the concept of chromosome condensation during mitosis. Cytogenet Genome Res 2002;98:136-46 [DOI] [PubMed] [Google Scholar]
- 2.Tjio JH, Levan A. The chromosome number of man. Hereditas 1956;42:1-6 [Google Scholar]
- 3.Hliscs R, Muhlig P, Claussen U. The spreading of metaphases is a slow process which leads to a stretching of chromosomes. Cytogenet Cell Genet 1997;76:167-71 [DOI] [PubMed] [Google Scholar]
- 4.Ami D, Ricagno S, Bolognesi M, Bellotti V, Doglia SM, Natalello A. Structure, stability, and aggregation of β-2 microglobulin mutants: insights from a Fourier transform infrared study in solution and in the crystalline state. Biophys J 2012;102:1676-84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barth A. Infrared spectroscopy of proteins. Biochim Biophys Acta 2007;1767:1073-101 [DOI] [PubMed] [Google Scholar]
- 6.Banyay M, Sarkar M, Gräslund A. A library of IR bands of nucleic acids in solution. Biophys Chem 2003;104:477-88 [DOI] [PubMed] [Google Scholar]
- 7.Banyay M, Gräslund A. Structural effects of cytosine methylation on DNA sugar pucker studied by FTIR. J Mol Biol 2002;324:667-76 [DOI] [PubMed] [Google Scholar]
- 8.Seshadri S, Khurana R, Fink AL. Fourier transform infrared spectroscopy in analysis of protein deposits. Methods Enzymol 1999;309:559-76 [DOI] [PubMed] [Google Scholar]
- 9.Arrondo JLR, Goni FM. Infrared studies of protein-induced perturbation of lipids in lipoproteins and membranes. Chem Phys Lipids 1998;96:53-68 [DOI] [PubMed] [Google Scholar]
- 10.Tamm LK, Tatulian SA. Infrared spectroscopy of proteins and peptides in lipid bilayers. Q Rev Biophys 1997;30:365-429 [DOI] [PubMed] [Google Scholar]
- 11.Kacuráková M, Mathlouthi M. FTIR and laser-Raman spectra of oligosaccharides in water: characterization of the glycosidic bond. Carbohydr Res 1996;284:145-57 [DOI] [PubMed] [Google Scholar]
- 12.Casal HL, Mantsch HH. Polymorphic phase behaviour of phospholipid membranes studied by infrared spectroscopy. Biochim Biophys Acta 1984;779:381-401 [DOI] [PubMed] [Google Scholar]
- 13.Caine S, Heraud P, Tobin MJ, McNaughton D, Bernard CC. The application of Fourier transform infrared microspectroscopy for the study of diseased central nervous system tissue. Neuroimage 2012;59:3624-40 [DOI] [PubMed] [Google Scholar]
- 14.Sandt C, Féraud O, Oudrhiri N, Bonnet ML, Meunier MC, Valogne Y, et al. Identification of spectral modifications occurring during reprogramming of somatic cells. PLoS One 2012;7:e30743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ami D, Mereghetti P, Natalello A, Doglia SM, Zanoni M, Redi CA, et al. FTIR spectral signatures of mouse antral oocytes: molecular markers of oocyte maturation and developmental competence. Biochim Biophys Acta 2011;1813:1220-9 [DOI] [PubMed] [Google Scholar]
- 16.Heraud P, Nga ES, Caine S, Yu QC, Hirst C, Mayberry R, et al. Fourier transform infrared microspectroscopy identifies early lineage commitment in differentiating human embryonic stem cells. Stem Cell Res 2010;4:140-7 [DOI] [PubMed] [Google Scholar]
- 17.Ami D, Natalello A, Zullini A, Doglia SM. Fourier transform infrared microspectroscopy as a new tool for nematode studies. FEBS Lett 2004;576:297-300 [DOI] [PubMed] [Google Scholar]
- 18.Choo L P, Wetzel DL, Halliday WC, Jackson M, LeVine SM, Mantsch HH. In situ characterization of betaamyloid in Alzheimer’s diseased tissue by synchrotron Fourier transform infrared microspectroscopy. Biophys J 1996;71:1672-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wong PT, Wong RK, Caputo TA, Godwin TA, Rigas B. Infrared spectroscopy of exfoliated human cervical cells: evidence of extensive structural changes during carcinogenesis. Proc Natl Acad Sci USA 1991;88:10988-92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Giuffrida S, Cottone G, Bellavia G, Cordone L. Proteins in amorphous saccharide matrices: structural and dynamical insights on bioprotection. Eur Phys J E Soft Matter 2013;36:79. [DOI] [PubMed] [Google Scholar]
- 21.Venyaminov SY, Prendergast FG. Water (H2O and D2O) molar absorptivity in the 1000-4000 cm-1 range and quantitative infrared spectroscopy of aqueous solutions. Anal Biochem 1997;248:234-45 [DOI] [PubMed] [Google Scholar]
- 22.Abbott T P, Nabetani H, Sessa DJ, Wolf WJ, Liebman MN, Dukor RK. Effects of bound water on FTIR spectra of glycinin. J Agric Food Chem 1996;44:2220-4 [Google Scholar]
- 23.Ibraimov AI. Chromosome preparations of human whole blood lymphocytes: an improved technique. Clin Genet 1983;24:240-2 [DOI] [PubMed] [Google Scholar]
- 24.Bassan P, Kohler A, Martens H, Lee J, Byrne H.J., Dumas P, et al. Resonant Mie scattering (RMieS) correction of infrared spectra from highly scattering biological samples. Analyst 2010;135:268-77 [DOI] [PubMed] [Google Scholar]
- 25.Susi H, Byler DM. Resolution-enhanced Fourier transform infrared spectroscopy of enzymes. Methods Enzymol 1986;130:290-311 [DOI] [PubMed] [Google Scholar]
- 26.Spurbeck JL, Zinsmeister AR, Meyer KG, Jalal SM. Dynamics of chromosome spreading. Am Journal of Med Genet 1996; 61:387-93 [DOI] [PubMed] [Google Scholar]
- 27.Lundsteen C, Lind AM. A test of a climate room for preparation of chromosome spreading. Clinical Genet 1985;28:260-2 [DOI] [PubMed] [Google Scholar]
- 28.Dzwolak W, Ravindrab R, Winter R. Hydration and structure - the two sides of the insulin aggregation process. Phys Chem Chem Phys 2004;6:1938-43 [Google Scholar]
- 29.Munishkina LA, Fink AL, Uversky VN. Conformational prerequisites for formation of amyloid fibrils from histones. J Mol Biol 2004;342:1305-24 [DOI] [PubMed] [Google Scholar]
- 30.Vila R, Ponte I, Collado M, Arrondo JL, Jiménez MA, Rico M, et al. DNA-induced alpha-helical structure in the NH2-terminal domain of histone H1. J Biol Chem. 2001;276:46429-35 [DOI] [PubMed] [Google Scholar]
- 31.Sarroukh R, Goormaghtigh E, Ruysschaert JM, Raussens V. ATR-FTIR: a “rejuvenated” tool to investigate amyloid proteins. Biochim Biophys Acta 2013;1828:2328-38 [DOI] [PubMed] [Google Scholar]
- 32.Natalello A, Prokorov VV, Tagliavini F, Morbin M, Forloni G, Beeg M, et al. Conformational plasticity of the GerstmannSträussler-Scheinker disease peptide as indicated by its multiple aggregation pathways. J Mol Biol 2008;381:1349-61 [DOI] [PubMed] [Google Scholar]
- 33.Barth A, Zscherp C. What vibrations tell us about proteins. Q Rev Biophys 2002;35:369-430 [DOI] [PubMed] [Google Scholar]
- 34.Tajmir-Riahi HA, Neault J F, Naoui M. Does DNA acid fixation produce left-handed Z structure? FEBS Lett 1995;370:105-8 [DOI] [PubMed] [Google Scholar]
- 35.Puppels GJ, Otto C, Greve J, Robert-Nicoud M, Arndt-Jovin DJ, Jovin TM. Raman microspectroscopic study of low-pH-induced changes in DNA structure of polytene chromosomes. Biochemistry 1994;33:3386-95 [DOI] [PubMed] [Google Scholar]
- 36.Hill RJ. Native salivary chromosomes of Drosophila melanogaster: retrospect and prospect. Aust J Biol Sci 1985;38:1-11 [PubMed] [Google Scholar]
- 37.Natalello A, Sasso F, Secundo F. Enzymatic transesterification monitored by an easy-to-use Fourier transform infrared spec-troscopy method. Biotechnol J 2013;8:133-8 [DOI] [PubMed] [Google Scholar]