

Abstract
L-type Ca channels (LTCC), which play a key role in cardiac excitation–contraction coupling, are located predominantly at the transverse (t-) tubules in ventricular myocytes. Caveolae and the protein caveolin-3 (Cav-3) are also present at the t-tubules and have been implicated in localizing a number of signaling molecules, including protein kinase A (PKA) and β2-adrenoceptors. The present study investigated whether disruption of Cav-3 binding to its endogenous binding partners influenced LTCC activity. Ventricular myocytes were isolated from male Wistar rats and LTCC current (ICa) recorded using the whole-cell patch-clamp technique. Incubation of myocytes with a membrane-permeable peptide representing the scaffolding domain of Cav-3 (C3SD) reduced basal ICa amplitude in intact, but not detubulated, myocytes, and attenuated the stimulatory effects of the β2-adrenergic agonist zinterol on ICa. The PKA inhibitor H-89 also reduced basal ICa; however, the inhibitory effects of C3SD and H-89 on basal ICa amplitude were not summative. Under control conditions, myocytes stained with antibody against phosphorylated LTCC (pLTCC) displayed a striated pattern, presumably reflecting localization at the t-tubules. Both C3SD and H-89 reduced pLTCC staining at the z-lines but did not affect staining of total LTCC or Cav-3. These data are consistent with the idea that the effects of C3SD and H-89 share a common pathway, which involves PKA and is maximally inhibited by H-89, and suggest that Cav-3 plays an important role in mediating stimulation of ICa at the t-tubules via PKA-induced phosphorylation under basal conditions, and in response to β2-adrenoceptor stimulation.
Keywords: t-tubules, Ca, Phosphorylation, β2-Adrenoceptors
Graphical abstract
Highlights
-
•
Basal L type calcium current was reduced by interfering with caveolin-3 binding.
-
•
L type calcium current is tonically regulated by PKA phosphorylation.
-
•
Interfering with caveolin-3 binding reduced beta2 adrenergic stimulation of ICa.
1. Introduction
The L-type Ca current (ICa) plays a key role in excitation–contraction coupling in cardiac myocytes: Ca influx via ICa triggers release of more Ca from adjacent sarcoplasmic reticulum (SR) to cause contraction, and loads the SR with Ca for subsequent release [1,2]. However ICa is not spatially uniform; it flows predominantly across the t-tubule membrane [3]. One reason for this localization appears to be that basal protein kinase A (PKA) activity stimulates ICa more at the t-tubules than at the surface membrane [4]: inhibition of PKA markedly inhibits basal ICa in intact ventricular myocytes, but has little effect on ICa after the t-tubules are uncoupled from the surface membrane (detubulation). This is consistent with previous work showing that many of the proteins constituting the β-adrenergic pathway are located predominantly at the t-tubules, and that β-adrenergic stimulation causes a greater increase of ICa in the t-tubules than at the surface membrane [5], suggesting better coupling of β-adrenergic signaling pathways to ICa at the t-tubules than at the cell surface.
Localization of β-adrenergic signaling is well-recognized: although the response to β1-adrenoceptor stimulation occurs throughout the cell, β2-stimulation causes local stimulation of ICa [6]. More recently it has been shown that β1-adrenoceptors are located across the entire cell surface, whereas β2-adrenoceptors are found predominantly at the t-tubules [7]. Thus, localization to the t-tubules of ICa stimulation by basal PKA activity may involve constitutive activity of the same pathways as β2-adrenergic stimulation.
However the mechanisms underlying this localization are unclear. Recent work suggests that caveolae play an important role in spatially restricting the response to β2-adrenergic stimulation [8,9]. Caveolae are invaginations of the cell membrane, whose internal surface is lined with the scaffolding protein caveolin, caveolin-3 (Cav-3) being the prevalent form in adult cardiac myocytes [10]. Caveolin binds a variety of signaling molecules, including PKA, via a scaffolding domain to form signaling complexes and regulate their activity. A sub-population of L-type Ca channels (LTCC) also co-localizes with caveolin in ventricular myocytes [11–13]. Thus it seems possible that co-localization of PKA and LTCC as a result of Cav-3 binding underlies localized activation of ICa by PKA activity. We have, therefore, investigated the role of Cav-3 on the phosphorylation and activation of ICa by PKA activity, by studying the effect of disrupting Cav-3 binding on ICa and Ca channel phosphorylation, and the response to altering PKA activity.
2. Materials and methods
2.1. Myocyte isolation and detubulation
Myocytes were isolated from the hearts of male Wistar rats (250–300 g). All procedures were performed in accordance with UK legislation and approved by the University of Bristol Ethics Committee. Animals were killed by cervical dislocation or under pentobarbitone anesthesia, the heart quickly excised, and Langendorff-perfused at 8 ml/min (37 °C), initially with Tyrode's solution (see below) plus 0.75 mM CaCl2 for 4 min, then with 0.9 mM EGTA for 4 min, and finally with 1 mg/ml collagenase (Worthington Corp) for 10 min. The left ventricle was then excised and shaken in collagenase-containing solution at 37 °C for 5–7 min, filtered, and centrifuged. The supernatant was discarded and the pellet re-suspended in Kraftbrühe solution, that contained (in mM): 90 Glutamic acid, 30 KCl, 10 HEPES, 1 EGTA, 5 Na pyruvate, 20 Taurine, 20 Glucose, 5 MgCl2, 5 Succinic acid, 5 Creatine, 2 Na2 ATP, 5 β-OH Butyric acid, pH 7.4 (KOH) and stored for 2–10 h before use on the day of isolation. Detubulation (DT) of myocytes (physical and functional uncoupling of the t-tubules from the surface membrane) was achieved using formamide-induced osmotic shock, as described previously [3].
2.2. Solutions
The Tyrode's solution used for cell isolation contained (in mM): 130 NaCl, 5.4 KCl, 0.4 NaH2PO4, 4.2 HEPES, 10 Glucose, 1.4 MgCl2, 20 Taurine, 10 Creatinine, and pH 7.4 (NaOH). For patch-clamp experiments, cells were superfused with a solution that contained (in mM): 133 NaCl, 5 KCl, 1 MgSO4, 1 CaCl2, 1 Na2HPO4, 10 Glucose, 10 HEPES, and pH 7.4 (NaOH); 5 CsCl was either added or used to replace KCl, to inhibit K currents. The pipette solution contained (in mM): 110 CsCl, 20 TEACl, 0.5 MgCl2, 5 Mg-ATP, 5 BAPTA, 10 HEPES, 0.4 GTP-Tris, and pH 7.2 (CsOH); BAPTA buffers bulk cytoplasmic Ca and Ca within the dyad, and thus inhibits Ca-dependent inactivation of ICa [5,14]. For immunocytochemistry, the PBS solution contained (in mM): 3.8 NaH2PO4, 16.2 Na2HPO4, 150 NaCl, and pH 7.4 (NaOH).
PKA was inhibited using 20 μM H-89. Selective β2-adrenoceptor stimulation was achieved using the β2-agonist zinterol (1–10 μM) in combination with the specific β1-adrenoceptor blocker atenolol (10 μM); cells were superfused with atenolol for at least 4 min before application of zinterol. The specific β2-adrenoceptor blocker ICI 118,551 was used to verify the specific agonist effect of zinterol. A membrane-permeable TAT-tagged peptide (C3SD; [15]) was used to disrupt binding of Cav-3 to its partner proteins as described previously [16,17]; this peptide corresponds to the 20-residue scaffolding domain of Cav-3 linked by 4 glycine residues to the 11-residue trans-activating transcriptional activator sequence from HIV-1; a scrambled version of this peptide (Scram) with 100% amino acid identity but only 60% sequence identity that has been shown not to displace Cav-3 binding was used as control [17]. Ventricular myocytes were incubated in 1 μM peptide diluted in either isolation Tyrode's or Kraftbrühe solution for 30–60 mins at 37 °C. Cells were then washed in control buffer or used immediately for electrophysiological recordings. In some experiments, an antibody against Cav-3 (25 μg/ml, SC-16229, Santa Cruz Biotechnology, Inc.) was incorporated in the pipette solution to disrupt Cav-3 function. All experiments were performed at room temperature.
2.3. Recording ICa
Myocytes were placed in a chamber mounted on the stage of a Nikon Diaphot inverted microscope. Membrane currents and cell capacitance were recorded using the whole-cell patch-clamp technique, using an Axopatch 200B (Axon Instruments) and a Digidata 1322A A/D converter (Axon Instruments). pClamp 10 software (Axon Instruments) was used for data acquisition and analysis. Patch pipette resistance was typically 2–4 MΩ when filled with pipette solution. Pipette capacitance and series resistance were compensated by > 70%. The voltage protocol used to elicit ICa was: from a holding potential of − 80 mV, a 100 ms step depolarization to − 40 mV was applied to inactivate the sodium current, followed by a step depolarization to voltages ranging between − 50 and + 80 mV (10 mV steps) for 500 ms, before repolarization to − 80 mV, at a frequency of 0.2 Hz. To monitor the response to drug application a continuous train of step depolarizations to 0 mV (0.2 Hz) was used.
2.4. Analysis of ICa
The amplitude of ICa was measured as the difference between peak inward current and current at the end of the depolarizing pulse, normalized to cell capacitance and expressed as current density (pA/pF). Rundown of ICa was monitored for 12 min before application of H-89 (Fig. 3), 5 mins before application of ICI-118,551 (data presented in text) and 2 mins before application of zinterol (Fig. 6). An exponential function was fitted to the decay in the amplitude of ICa and extrapolated to the end of the drug application to estimate current rundown. Current–voltage relationships measured in the presence of H-89 were corrected for this rundown (control − 5.4 ± 3.2% and C3SD − 14 ± 2.4%). Inactivation of ICa was quantified by fitting the decay of the current from its peak to steady state (at the test voltage) to a double exponential function (pClamp 10) to give “fast” (τfast) and “slow” (τslow) time constants. ICa (measured at 0 mV) was converted to conductance (g) as follows:
| (1) |
where the reversal potential for ICa (Erev) was calculated for each cell from the current–voltage relationship. Activation curves were fitted with a modified Boltzmann equation:
| (2) |
where gmax represents the maximum L-type Ca2 + conductance, E0.5 is the membrane potential at which 50% of gmax is activated and k is a slope factor.
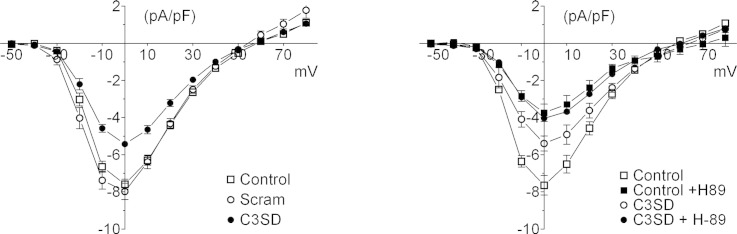
Fig. 3.

The effect of C3SD-peptide on the PKA–dependence of basal ICa. A: representative current records of ICa elicited by a step depolarization to 0 mV taken from an untreated (control, left) and a C3SD-treated (C3SD, right) myocyte in the presence and absence of H-89 (20 μM, 8 min, denoted by the arrow). Inset in A shows the voltage protocol. B: time course of the effect of H-89 (20 μM, denoted by the black bar) in reducing the peak amplitude of ICa elicited by a step depolarization to 0 mV in control (open circles, n = 5) and C3SD-treated cells (filled circles, n = 5). C: mean ICa–voltage relationship recorded in the absence (open symbols) and presence (closed symbols) of H-89 from control (circles, n = 5) and C3SD-treated (squares, n = 5) myocytes. D: mean data for the time course of ICa inactivation in the presence (+) and absence (−) of H-89 in control and C3SD-treated myocytes. E: mean data for the effect of H-89 on peak ICa amplitude elicited by a voltage clamp step to 0 mV with (n = 7) or without (n = 6) Cav3 antibody in the pipette. Statistical significance was assessed using Student's paired (D) or unpaired t-tests (E); * p < 0.05, **p < 0.01.
Fig. 6.

The effect of C3SD-peptide on the β2-adrenoceptor-mediated ICa stimulation. A, B: representative ICa records taken from an untreated (control, A) and a C3SD-treated myocyte (B) showing the effects of β2-adrenoceptor stimulation (zinterol 1, 3 & 10 μM). The inset in A shows the voltage protocol. C: mean peak ICa elicited by voltage-clamp steps to 0 mV (0.2 Hz) in control (n = 8, open symbols) and C3SD-treated (n = 7, closed symbols) cells. Atenolol (10 μM) was applied at least 4 min before the application of zinterol; at each concentration of zinterol used in control cells, the maximum effect on ICa was reached within 3 mins. D: mean data for the zinterol-mediated increase in ICa expressed as a percentage of control in untreated myocytes (open bars), in myocytes treated with the β2-adrenoceptor antagonist, ICI-118,551 (gray bars, 100 nM, n = 6) or treated with C3SD-peptide (filled bars). ***, p < 0.001 (Bonferroni post hoc analysis, 2-way ANOVA).
The zinterol-induced increase in ICa amplitude is expressed as a percentage of the amplitude of ICa in the presence of atenolol just before application of zinterol.
2.5. Immunocytochemistry
Phosphorylation of LTCC was investigated using a polyclonal antibody to the pore forming subunit (α1c) phosphorylated at Ser1928, generated as described previously ([18], anti-Cav1.2 phosphoserine-1928, A010-70, Badrilla Ltd.). This antibody provides a read-out of PKA-dependent phosphorylation of the LTCC α1c-subunit [19]. Myocytes were stained with this antibody using standard techniques. In brief, cells in 1 ml Kraftbrühe solution were placed on poly-l-lysine coated coverslips and left for 45 min. Cells were then fixed with 100 μl 4% paraformaldehyde for 10 min and permeabilized using 100 μl 0.1% triton X-100 in PBS for 10 min. Non-specific binding was blocked using 100 μl 10% normal goat serum in PBS for 20 min. Cells were incubated with anti-phospho-Ser1928 antibody (1:100 dilution in PBS with 10% normal goat serum) overnight at 4 °C, and with fluorescent secondary antibody (FITC conjugate goat anti-rabbit IgG 1:200 dilution in PBS) for a further 40 min. Coverslips were rinsed in PBS between each stage. The same protocol was used to label Cav-3 (anti-caveolin-3 antibody, 610420, BD Transduction Laboratories™) and total LTCC (phosphorylated + non-phosphorylated; L-type Ca2 + CP α1C (N-17) Antibody, rabbit, SC-16229, Santa Cruz Biotechnology, Inc.).
2.6. Confocal imaging
An x-y image was recorded from the center of immuno-labeled cells using a confocal laser scanning microscope (Leica SP5-AOBS, Leica Microsystems, Wetzlar, Germany) attached to an inverted epifluorescence microscope (Leica DM I6000) with excitation at 488 nm (using a 100 mW Ar laser) and fluorescence intensity collected between 500 and 572 nm. All settings were kept constant on a given day.
2.7. Image analysis
Staining intensity was analyzed using ImageJ software (v1.46, nih). Due to the variation in staining intensity, it is difficult to compare absolute staining intensities between images. However the distribution of staining was determined, by quantifying the bands of transverse staining using a fast Fourier transform (FFT) of the image. The power of the FFT was normalized and the amplitude of the first harmonic taken as a measure of the intensity of staining at the z-lines. The reciprocal of the spatial frequency provided a measure of the average inter-band distance.
2.8. Statistics
Data are expressed as mean ± SEM. Statistical analysis was performed using GraphPad Prism (GraphPad Software Inc.). Paired and unpaired t-tests and analysis of variance (1 or 2 way ANOVA) were used as appropriate with the Bonferroni post hoc test where appropriate. Statistical significance was taken as p < 0.05.
3. Results
3.1. Effect of disrupting Cav-3 binding on ICa and Ca channel phosphorylation
To test the hypothesis that protein binding to Cav-3 is necessary for basal stimulation of ICa by phosphorylation of LTCC, we investigated the effect of C3SD and its scrambled control (Scram) on ICa and LTCC phosphorylation: the C3SD peptide is designed to disrupt normal Cav-3-dependent protein localization and co-localization by competing with endogenous Cav-3 for binding partners.
Fig. 1A shows representative traces of ICa recorded from an untreated myocyte (left) and from Scram- (middle) and C3SD-treated (right) myocytes. Comparison of the middle and left panels shows that Scram-peptide had no apparent effect on either the magnitude or time-course of ICa. However, the traces in the right panel show that C3SD markedly reduced ICa peak amplitude. Fig. 1B shows mean ICa–voltage relationships in the three conditions. Although incubation of myocytes with either Scram or C3SD did not significantly alter cell capacitance (control, 179 ± 12 pF, n = 11; Scram, 170 ± 22 pF, n = 9; C3SD, 166 ± 11 pF, n = 15, ns, 1-way ANOVA), incubation with C3SD-peptide significantly reduced ICa density (p < 0.0001, 2 way ANOVA) across the voltage range − 20 to + 20 mV. Fig. 1C shows that the amplitude of ICa (recorded at 0 mV) was not altered by incubation with Scram peptide but was significantly reduced by incubation with C3SD (p < 0.001 vs Scram). Similarly, Fig. 1D shows that maximal conductance (gmax) was reduced in C3SD-incubated myocytes as compared to Scram-incubated cells (p < 0.05, 1-way ANOVA). There was no difference in the voltage-dependence of activation of g between the three conditions (E0.5: control, − 15.2 ± 0.7 mV, n = 11; Scram, − 17.0 ± 1.3 mV, n = 9; C3SD, − 14.9 ± 1.3 mV, n = 15, ns, 1-way ANOVA). The time course of inactivation of ICa was not significantly altered by incubation with either Scram or C3SD. Under the recording conditions used, Ca-dependent inactivation is inhibited [20]. Thus, voltage-dependent inactivation was not altered by these peptides.
Fig. 1.

Effect of C3SD-peptide on the basal amplitude of ICa. A: representative family of current records taken from an untreated (control) ventricular myocyte (left) and myocytes incubated in scrambled (Scram, middle) and C3SD-peptide (C3SD, right). The inset shows the voltage protocol. B: mean peak ICa–voltage relationship recorded from untreated (open square, n = 11), Scram-treated (open circle, n=9) and C3SD-treated (filled circle, n=15) myocytes. C: mean data for the peak ICa amplitude elicited by a voltage-clamp step to 0 mV for the three treatment groups. There is a significant difference in the ICa amplitude between the three groups (1-way ANOVA, p < 0.001), Bonferroni post hoc analysis shows no statistically significant difference between the control and Scram-treated myocytes and a significant difference between the C3SD-treated myocytes and control- (p < 0.001) and Scram-treated myocytes (p < 0.001). D: voltage-dependence of ICa activation. Maximal conductance was reduced by C3SD (see text), there was no difference in the voltage-dependence of activation of g, E0.5 (see text) or slope factor (k: control, − 4.8 ± 0.2 mV, n = 11; Scram, − 4.9 ± 0.1 mV, n = 9; C3SD, − 4.9 ± 0.2 mV, n = 15, ns, 1-way ANOVA) between the three conditions. Solid lines represent fits to Eq. (2).
To investigate whether the effect of C3SD on ICa could be due to direct block of LTCC, we studied its effect on the response of ICa to the LTCC agonist Bay K 8644. Bay K increased ICa in both control and C3SD-treated cells, so that in the presence of Bay K, ICa at 0 mV was not significantly different in the two groups of cells (control: − 8.3 ± 0.5 to − 9.6 ± 0.5, pA/pF, n = 6; C3SD: − 6.6 ± 0.4 to − 9.9 ± 1.1, n = 6, pA/pF), thereby demonstrating that C3SD is not acting as an LTCC blocker. Importantly, since basal ICa was smaller in C3SD-treated cells, the increase in current caused by Bay K was greater in these cells (control, 16 ± 5% vs C3SD, 47 ± 9%, p < 0.02). This is consistent with C3SD causing dephosphorylation of LTCC, thereby decreasing LTCC open probability (Po): since Bay K increases Po in a phosphorylation-independent manner, ICa would be expected to be the same in the presence of Bay K 8644 in control and in C3SD-treated cells. Thus, the reduction in the amplitude of ICa following treatment of cells with C3SD is consistent with the idea that Cav-3 plays a role in activation of ICa under basal conditions.
To investigate further whether activation of ICa by Cav-3 is due to channel phosphorylation, we used a phospho-specific antibody against the pore-forming LTCC α1c-subunit to determine whether C3SD also altered LTCC phosphorylation, and compared this with labeling of Cav-3 and total LTCC.
Fig. 2A shows confocal images of representative cardiac myocytes stained with antibodies against Cav-3 (left), total (i.e. phosphorylated and non-phosphorylated α1c-subunit) LTCC (middle), or phosphorylated LTCC (pLTCC, right) under control conditions (top row), and following incubation with either Scram (middle row) or C3SD (bottom row). Under normal conditions, the majority of staining for all three proteins occurred in clear transverse striations, presumably at the t-tubules. The images in the left and middle columns, and the corresponding mean data in Fig. 2C, show that neither Scram nor C3SD had any discernible effect on the distribution of staining for Cav-3 or total LTCC. Similarly, Scram-peptide had no significant effect on the distribution of pLTCC labeling (Fig. 2C); however, C3SD caused a significant decrease in the intensity of the transverse striations observed following staining for pLTCC (p < 0.001 vs Scram, Fig. 2C). Thus it appears unlikely that the effect of C3SD on ICa is due to changes in the distribution of Cav-3 or LTCC, but the data are consistent with the notion that the decrease in ICa caused by C3SD is due to reduced channel phosphorylation at the t-tubules.
Fig. 2.

The effect of C3SD on the expression and localization of caveolin, and non-phosphorylated and phosphorylated forms of LTCC. A: representative images of myocytes stained with anti-caveolin (Cav-3, left column), total (phosphorylated + non-phosphorylated) LTCC (tLTCC, middle column) and phosphorylated LTCC (pLTCC, right column) antibodies, taken from untreated (top row), scram- (middle row) or C3SD-treated (lower row) myocytes. Scale bar represents 50 μm. B: representative power spectrum of a longitudinal scan of an antibody-stained cell. Spectrum corresponds to the control tLTCC image (top, middle column). C: Mean z-line staining intensity in control and in Scram- and C3SD-treated cells for Cav-3 (n = control 4, Scram 7, C3SD 10), Total LTCC (tLTCC, n = control 6, Scram 7, C3SD 6) and phosphorylated LTCC (pLTCC, n = control 22, Scram 15, C3SD 20). **, p < 0.01; ***, p < 0.001; Bonferroni post hoc analysis, 1-way ANOVA.
3.2. Effect of PKA inhibition on C3SD-dependent changes of ICa and channel phosphorylation
To investigate the role of PKA in the changes in basal ICa and phosphorylation described in Section 3.1, we studied the effect of the PKA inhibitor H-89 on ICa and LTCC phosphorylation in the absence and presence of C3SD.
The recordings in Fig. 3A show the effect of H-89 (20 μM) on ICa (elicited by a voltage-clamp step to 0 mV) in an untreated (left) and C3SD-treated (right) myocyte. H-89 caused a significant decrease in the amplitude of ICa in both cell groups (Fig. 3B; after 8 min: control: from − 7.6 ± 0.5 pA/pF to − 3.6 ± 0.5 pA/pF, n = 5, p < 0.001; C3SD-treated: from − 5.4 ± 0.4 pA/pF to − 3.7 ± 0.3 pA/pF, n = 5, p < 0.001). However, the decrease was greater in control than in C3SD-treated cells (interaction, p < 0.0001, 2-way ANOVA); H-89 decreased ICa by 53 ± 3% in control cells, and by 22 ± 11% in C3SD-treated cells (p < 0.05). The effects of H-89 and C3SD on the amplitude of ICa were not however summative, so that the amplitude of ICa in the presence of H-89 was not significantly different in the absence or presence of C3SD. This is illustrated in Fig. 3C, which shows the effect of H-89 on the mean current–voltage relationship in each group of cells. These data show that C3SD decreased ICa amplitude (p < 0.001, 2-way ANOVA) but that in the presence of H-89 ICa was not significantly different between untreated and C3SD-treated cells (ns, 2-way ANOVA). The observation that ICa density was not reduced further by H-89 in the presence of C3SD than in control suggests that the actions of H-89 and C3SD share a common pathway, which is maximally inhibited by H-89 but not by C3SD.
H-89 significantly reduced the slow, but not the fast, component of ICa inactivation in both control and C3SD-treated myocytes (p < 0.001, 2 way ANOVA) (Fig. 3D).
To investigate an alternative method of inhibiting Cav-3 function, we incorporated anti-Cav-3 antibody in the pipette solution and measured ICa amplitude in the presence and absence of H-89. Fig. 3E shows that Cav-3 antibody decreased the amplitude of ICa (p < 0.05, 2-way ANOVA) and decreased the response to H-89, and that in the presence of H-89 the amplitude of ICa was similar in the presence or absence of Cav-3 antibody (ns, 2-way ANOVA). These data suggest that, as for C3SD-peptide, the effects of antibody and H-89 are not summative, and are consistent with those obtained using C3SD.
Fig. 4A shows confocal images of myocytes labeled with pLTCC-specific antibody in untreated (top row), Scram-treated (middle row) and C3SD-treated (bottom row) cells in the absence (left column) or presence (right column) of H-89 (20 μM). As described in Section 3.1 (above), under control conditions, the majority of staining occurred in clear transverse striations, presumably at the t-tubules; this staining pattern was not significantly affected by Scram, but C3SD-peptide caused a significant decrease in the intensity of the transverse striations (Fig. 4B). Inhibition of PKA with H-89 caused a similar decrease in the intensity of the transverse striations, which was not significantly altered further by either Scram or C3SD (Fig. 4B). These data are consistent with the observed changes of ICa: C3SD decreased ICa and the t-tubule-associated staining, and both were decreased further by H-89 to a level that was not significantly different from H-89 alone. This is consistent with the idea that the observed changes of ICa are due to changes in LTCC phosphorylation.
Fig. 4.

The effect of C3SD and PKA inhibition on LTCC α1c-subunit phosphorylation. A: representative images of ventricular myocytes stained with anti-pLTCC antibodies taken from untreated (top row), scram- (middle row) or C3SD-treated (lower row) in the absence (left column) or presence (right column) of H-89 (20 μM). Scale bar represents 50 μm. B: mean z-line staining intensity in the absence (Control: n = untreated 22, Scram 15, C3SD 20) and presence of H-89 (H-89: n = untreated 26, Scram 6, C3SD 9). ***, p < 0.001 (Bonferroni post hoc analysis, 2-way ANOVA).
To investigate whether the effects of C3SD on ICa are functionally localized to the t-tubules, we studied the response of ICa to C3SD in detubulated myocytes, in which the t-tubules are physically and functionally uncoupled from the surface membrane, so that only ICa from the cell surface is recorded. Representative examples of ICa at 0 mV from a control cell (left), a detubulated cell (middle) and a C3SD-treated detubulated cell (right) are shown in Fig. 5A. Fig. 5B shows the mean current density–voltage relations for control, detubulated and C3SD-treated detubulated cells. As reported previously, detubulation causes a decrease of ICa density across the voltage range and reduced whole-cell capacitance (Fig. 5C). More interestingly, C3SD had no effect on ICa in detubulated cells, demonstrating that the decrease of ICa caused by C3SD in intact cells occurs in the t-tubules (ICa at 0 mV: control, − 6.8 ± 0.3 pA/pF, n = 11; DT, − 4.7 ± 0.2 pA/pF, n = 5, p < 0.01 vs control; DT + C3SD, − 4.2 ± 0.4 pA/pF, n = 6, p < 0.0001 vs control).
Fig. 5.

The effect of C3SD-peptide on basal ICa in detubulated myocytes. A: representative records of ICa elicited by a step depolarization to 0 mV in an untreated (control, left), a formamide-treated (DT, middle), and a formamide and C3SD-treated (DT + C3SD, right) myocyte. B: mean ICa–voltage relationships for untreated (circle, n = 11), formamide-treated (open square, n = 5) and formamide and C3SD-treated (closed square, n = 6) myocytes. C: Cell capacitance was reduced by ~ 30% from 283 ± 22 pF in control (n = 11) to 167 ± 11 pF in formamide-treated cells (n = 11, as C3SD had no effect on cell capacity, data from formamide-treated cells were pooled; p < 0.05, Student's unpaired t-test).
3.3. Effect of PKA stimulation on C3SD-dependent changes of ICa
The results described above suggest that basal phosphorylation of LTCC at the t-tubules by PKA, and thus basal ICa stimulation, depends on Cav-3 binding with its partner proteins via the scaffolding domain. We therefore investigated whether C3SD also alters the response of ICa to stimulation of β2-adrenoceptors, which are located predominantly at the t-tubules [7] and cause local stimulation of ICa [6], since they may involve the same pathways as those underlying basal phosphorylation (see Introduction).
The representative recordings of ICa in Fig. 6 show that the concentration-dependent increase of ICa caused by the β2-adrenergic agonist zinterol in control myocytes (Fig. 6A) was inhibited in C3SD-treated myocytes (Fig. 6B). The mean data in Fig. 6C show that the response to 1 μM and to 3 μM zinterol was abolished in C3SD-incubated cells. The concentration-dependent response to zinterol was also inhibited by the β2-adrenoceptor antagonist, ICI-118,551 (Fig. 6D). The reason for the small, statistically non-significant, increase of ICa produced by the highest concentration of zinterol in the presence of C3SD is unknown, but since this increase in ICa was not significantly different from that observed in the presence of the β2-antagonist, ICI-118,551 (Fig. 6D), the data are consistent with a role for Cav-3 in the localization of signaling via the β2-adrenoceptor. Thus, these data are similar to those reported for mouse ventricular myocytes, which showed inhibition of β2-adrenergic stimulation of ICa following disruption of caveolae using Cav-3 siRNA [11]. The data therefore suggest that the Cav-3-localized β2-adrenoceptor pathway increased ICa by approximately 40%, consistent with previous reports from rat ventricular myocytes (e.g. [21]). Applied alone, ICI-118,551 (5 min, 100 nM) did not significantly reduce basal ICa amplitude (from − 9.6 ± 1.5 pA/pF, to − 10.1 ± 1.6 pA/pF, n = 6, p = ns). These data suggest that Cav-3 binding plays a role in coupling β2-adrenoceptors to ICa and are thus consistent with the idea that basal stimulation of ICa, which is also disrupted by C3SD (above), may be due to tonic activity of PKA associated with the β2-adrenoceptor pathway at the t-tubules.
4. Discussion
The present data show that interventions designed to disrupt protein binding to Cav-3 decrease ICa in basal conditions and in response to β2-adrenergic stimulation, and that these decreases are associated with decreased LTCC phosphorylation at the t-tubules. This is consistent with the hypothesis that Cav-3 plays an important role in the stimulation of ICa by PKA-induced phosphorylation at the t-tubules under basal conditions, and in response to β2-adrenoceptor stimulation.
4.1. Localization of the response to C3SD
Previous work has shown that stimulation of ICa by PKA occurs predominantly at the t-tubules in basal conditions and in response to β-adrenergic stimulation. Similarly, β2-adrenoceptors are localized at the t-tubules, and stimulation of such receptors is known to cause a localized increase of ICa. However the mechanisms underlying this localization of activity are less clear, although Cav-3 has been implicated.
The present work shows that a peptide that competes with endogenous Cav-3 for proteins that normally bind to its scaffolding domain [15–17] causes a marked decrease in basal ICa. A scrambled peptide, with 100% amino acid identity but only 60% sequence identity was without effect on basal ICa, demonstrating the specificity of action of the C3SD peptide. Moreover, a similar decrease was observed when anti-Cav-3 antibody was included in the pipette solution. These data are compatible with the ICa decrease being due to an effect on Cav-3. The peptide had no effect on the distribution of staining for Cav-3 or tLTCC. Thus, the observed effects are unlikely to be due to changes in the distribution of these proteins, nor is the distribution of LTCC apparently determined by its binding to the Cav-3 scaffolding domain (or vice versa), at least on the time-scale of the current study, in which myocytes were exposed to C3SD for a relatively short period in order to affect dynamic events such as phosphorylation, rather than longer term changes. However, the decrease of ICa was associated with decreased t-tubular staining for pLTCC, and C3SD did not decrease ICa in detubulated cells. Interestingly, the ICa rundown observed in voltage-clamp experiments, which has been suggested to be due to reduced LTCC phosphorylation, was also increased by C3SD. The simplest explanation of these data is that C3SD alters Cav-3 binding, thereby disrupting PKA activity and/or localization, by either direct or indirect interactions, thus disrupting localized LTCC phosphorylation by basal PKA activity at the t-tubules, decreasing t-tubular ICa. Interestingly, inhibition of Cav-3 binding or PKA activity (Figs. 2 & 4) also appeared to increase pLTCC signal intensity at the surface membrane. This may reflect more uniform staining, since the surface membrane will be present throughout the depth of the confocal slice, whereas the t-tubules are smaller, and unlikely to be present throughout the slice, resulting in a smaller signal from the t-tubules than from the surface membrane for the same density of pLTCC staining. More uniform pLTCC density in the presence of C3SD and H-89 would be consistent with disruption of local phosphorylation at the t-tubules by these agents, as suggested above. This also emphasizes the localization of staining to the t-tubules in the absence of these agents, when little surface staining was observed.
In some experiments we investigated the effect of C3SD on the distribution of PKA and adenylyl cyclase 5/6; although staining was observed at the t-tubules and surface membrane, C3SD had little apparent effect on the localization of PKA or adenylyl cyclase 5/6 to the t-tubules (not shown). However, the scale of disruption required to alter the functional interaction between PKA and LTCC may be smaller than can be resolved using immunohistochemistry and confocal microscopy.
Although images of Cav-3 protein staining indicate that Cav-3 may not be restricted to the z-lines, the functional data from detubulated cells suggest that both PKA-dependent [4] and Cav-3-dependent (Fig. 5) modulation of ICa are confined to the t-tubules, and that the Cav-3 observed at the surface membrane is not involved in such modulation of ICa. This is of interest, given previous work showing localization of β2-receptors at the t-tubules [7] and the observation that treatment with C3SD increases β2-adrenoceptor-dependent phosphorylation of phospholamban [15], consistent with our hypothesis that Cav-3 underlies localization of β2-adrenoceptor activity, and indicates that Cav-3 plays a role in controlling PKA-dependent phosphorylation of other proteins.
In ventricular myocytes, the majority (~ 90%) of LTCC are located at the dyad, a sub-population of LTCC co-localizes with caveolin [11–13], and a caveolin-linked inhibitor can inhibit ICa [22]. However it is not clear whether C3SD is acting on dyadic and/or extra-dyadic ICa. In support of the former, it has been suggested that Cav-3 forms a scaffold in the dyad that complexes with Cav1.2 and is essential for the inotropic effects of PKA [23], compatible with the current observations. In contrast, Cav-3 has been reported to be absent from the dyad, and caveolin-targeted Cav1.2 inhibition causes only a small (~ 15%) decrease of ICa and no change in contractility, which is unexpected if dyadic ICa is being affected [24]. However, the decrease of ICa observed in the present study when Cav-3 (or PKA) function is disrupted is greater than would be expected from the non-junctional fraction of LTCC (~ 10%); this may be due to up-regulation of extra-dyadic LTCC or to coupling to dyadic LTCC: although direct coupling of Cav-3 to PKA and its target proteins is one model, there are other possibilities; for example, Cav-3 could tether PKA, or part of the PKA-activating pathway, outside the dyad, but sufficiently close to regulate dyadic LTCC phosphorylation. It is notable that the ICa decrease due to PKA inhibition is mirrored by a decrease in the Ca-transient amplitude in intact myocytes, but that neither ICa nor the Ca-transient is affected by PKA inhibition in detubulated cells [4]. Thus the change in the Ca-transient appears to be due to a change in the function of t-tubular proteins, the most likely candidate being ICa, which is unexpected if extra-dyadic LTCC were the only target for localized PKA activity.
Conversely, if C3SD predominantly affects extra-dyadic LTCC, this suggests that these channels are responsible for the changes of ICa induced by inhibiting basal PKA activity and in response to β2-adrenergic stimulation (see below). This could explain why β2-stimulation has little effect on the systolic Ca-transient, but raises the question what purpose is served by stimulating extra-dyadic LTCC.
4.2. The role of PKA in the response to C3SD
The role of PKA in the changes of phosphorylation caused by C3SD was investigated using the PKA inhibitor H-89 and β2-adrenoceptor stimulation. H-89 had a qualitatively similar, but quantitatively larger, inhibitory effect on ICa amplitude as C3SD. Importantly, the inhibitory effects of the two interventions were not summative, so that ICa in the presence of C3SD and H-89 was the same as in the presence of H-89 alone. This suggests that their inhibitory effects on ICa may share a common pathway.
The effect of H-89 on the distribution of LTCC phosphorylation at Ser1928, a PKA-specific site [19], was also the same as for C3SD, causing a marked decrease in transverse staining. Taken together, these data suggest that H-89 and C3SD decrease LTCC phosphorylation via PKA, and hence reduce ICa, although this is likely to be by different mechanisms; H-89 has a direct inhibitory effect on PKA, whereas C3SD is more likely to uncouple PKA or component(s) of the PKA pathway from LTCC. Nevertheless the lack of summation is consistent with both working via PKA. The smaller effect of C3SD may be because PKA remains active and at least some remains in the vicinity of the LTCC, although an effect of H-89 on other protein kinases cannot be entirely ruled out [25].
The observation that both agents decrease phosphorylation at the t-tubules is interesting because β2-adrenoceptors have recently been reported to be localized predominantly at the t-tubules. This localization could underlie localization of ICa stimulation by basal and stimulated PKA activity at the t-tubules: both basal and β2-adrenoceptor-stimulated ICa were inhibited by C3SD, consistent with the idea that the two responses share a common pathway.
In support of this idea, t-tubules are extensively labeled by antibody to Cav-3 (this study and [26]), consistent with electron microscopy showing caveolae in t-tubules, and recent work suggests that caveolae play an important role in spatially restricting the response to β2-adrenergic stimulation: disruption of caveolae converts the normal localized response to β2-adrenoceptor stimulation to one that occurs throughout the cell [8]. The association of cAMP-related signaling proteins with LTCC and Cav-3, via the A-kinase anchoring protein AKAP79/150, and the presence of phosphodiesterases, have been suggested to underlie localization of cAMP-signaling, and it has recently been suggested that caveolae not only limit cAMP production but also maintain SR phosphatase activity, so that their disruption increases the cAMP signal and allows SR protein phosphorylation [15]. These data are compatible with present work showing that Cav-3 appears to modulate PKA-induced LTCC phosphorylation, and hence ICa, at the t-tubules. Since the initial submission of this paper, similar conclusions have been reached by Timofeyev et al. [27].
Disclosure statement
None declared.
Acknowledgments
We are grateful to Professor John Colyer for providing the LTCC antibodies and to Dr Sarah Calaghan for information and advice about the C3SD peptide. This work was supported by the British Heart Foundation.
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial-No Derivative Works License, which permits non-commercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
Contributor Information
Andrew F. James, Email: a.james@bristol.ac.uk.
Clive H. Orchard, Email: clive.orchard@bristol.ac.uk.
References
- 1.Beuckelmann D.J., Wier W.G. Mechanism of release of calcium from sarcoplasmic reticulum of guinea-pig cardiac cells. J Physiol Lond. 1988;405:233–255. doi: 10.1113/jphysiol.1988.sp017331. [233–55] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fabiato A. Simulated calcium current can both cause calcium loading and trigger calcium release from the sarcoplasmic reticulum of a skinned canine cardiac Purkinje cell. J Gen Physiol. 1985;85:291–320. doi: 10.1085/jgp.85.2.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kawai M., Hussain M., Orchard C.H. Excitation–contraction coupling in rat ventricular myocytes after formamide-induced detubulation. Am J Physiol. 1999;277:H603–H609. doi: 10.1152/ajpheart.1999.277.2.H603. [DOI] [PubMed] [Google Scholar]
- 4.Chase A., Colyer J., Orchard C.H. Localised Ca channel phosphorylation modulates the distribution of L-type Ca current in cardiac myocytes. J Mol Cell Cardiol. 2010;49:121–131. doi: 10.1016/j.yjmcc.2010.02.017. [DOI] [PubMed] [Google Scholar]
- 5.Shan J., Xie W., Betzenhauser M., Reiken S., Chen B.-X., Wronska A. Calcium leak through ryanodine receptors leads to atrial fibrillation in 3 mouse models of catecholaminergic polymorphic ventricular tachycardia. Circ Res. 2012;111:708–717. doi: 10.1161/CIRCRESAHA.112.273342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guzadhur L., Jiang W., Pearcey S.M., Jeevaratnam K., Duehmke R.M., Grace A.A. The age-dependence of atrial arrhythmogenicity in Scn5a +/− murine hearts reflects alterations in action potential propagation and recovery. Clin Exp Pharmacol Physiol. 2012;39:518–527. doi: 10.1111/j.1440-1681.2012.05706.x. [DOI] [PubMed] [Google Scholar]
- 7.Nikolaev V.O., Moshkov A., Lyon A.R., Miragoli M., Novak P., Paur H. β2-Adrenergic receptor redistribution in heart failure changes cAMP compartmentation. Science. 2010;327:1653–1657. doi: 10.1126/science.1185988. [DOI] [PubMed] [Google Scholar]
- 8.Calaghan S., Kozera L., White E. Compartmentalisation of cAMP-dependent signalling by caveolae in the adult cardiac myocyte. J Mol Cell Cardiol. 2008;45:88–92. doi: 10.1016/j.yjmcc.2008.04.004. [DOI] [PubMed] [Google Scholar]
- 9.Calaghan S., White E. Caveolae modulate excitation–contraction coupling and β2-adrenergic signalling in adult rat ventricular myocytes. Cardiovasc Res. 2006;69:816–824. doi: 10.1016/j.cardiores.2005.10.006. [DOI] [PubMed] [Google Scholar]
- 10.Balijepalli R.C., Kamp T.J. Caveolae, ion channels and cardiac arrhythmias. Prog Biophys Mol Biol. 2008;98:149–160. doi: 10.1016/j.pbiomolbio.2009.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Balijepalli R.C., Foell J.D., Hall D.D., Hell J.W., Kamp T.J. Localization of cardiac L-type Ca2 + channels to a caveolar macromolecular signaling complex is required for β2-adrenergic regulation. Proc Natl Acad Sci U S A. 2006;103:7500–7505. doi: 10.1073/pnas.0503465103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Scriven D.R.L., Dan P., Moore E.D.W. Distribution of proteins implicated in excitation–contraction coupling in rat ventricular myocytes. Biophys J. 2000;79:2682–2691. doi: 10.1016/S0006-3495(00)76506-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guzadhur L., Pearcey S., Duehmke R., Jeevaratnam K., Hohmann A., Zhang Y. Atrial arrhythmogenicity in aged Scn5a +/ΔKPQ mice modeling long QT type 3 syndrome and its relationship to Na+ channel expression and cardiac conduction. Pflugers Arch — Eur J Physiol. 2010;460:593–601. doi: 10.1007/s00424-010-0851-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.You Y., Pelzer D.J., Pelzer S. Modulation of L-type Ca2 + current by fast and slow Ca2 + buffering in guinea pig ventricular cardiomyocytes. Biophys J. 1997;72:175–187. doi: 10.1016/S0006-3495(97)78656-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.MacDougall D.A., Agarwal S.R., Stopford E.A., Chu H., Collins J.A., Longster A.L. Caveolae compartmentalise β2-adrenoceptor signals by curtailing cAMP production and activating phosphatase in the sarcoplasmic reticulum of the adult ventricular myocyte. J Mol Cell Cardiol. 2012;52:388–400. doi: 10.1016/j.yjmcc.2011.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Couet J., Li S., Okamoto T., Ikezu T., Lisanti M.P. Identification of peptide and protein ligands for the caveolin-scaffolding domain. J Biol Chem. 1997;272:6525–6533. doi: 10.1074/jbc.272.10.6525. [DOI] [PubMed] [Google Scholar]
- 17.Feron O., Dessy C., Opel D.J., Arstall M.A., Kelly R.A., Michel T. Modulation of the endothelial nitric-oxide synthase-caveolin interaction in cardiac myocytes. Implications for the autonomic regulation of heart rate. J Biol Chem. 1998;273:30249–30254. doi: 10.1074/jbc.273.46.30249. [DOI] [PubMed] [Google Scholar]
- 18.George C.H., Parthimos D., Silvester N.C. A network-oriented perspective on cardiac calcium signaling. Am J Physiol. 2012;303:C897–C910. doi: 10.1152/ajpcell.00388.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hulme J.T., Westenbroek R.E., Scheuer T., Catterall W.A. Phosphorylation of serine 1928 in the distal C-terminal domain of cardiac CaV1.2 channels during β1-adrenergic regulation. Proc Natl Acad Sci. 2006;103:16574–16579. doi: 10.1073/pnas.0607294103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sham J.S.K. Ca2 + release-induced inactivation of Ca2 + current in rat ventricular myocytes: evidence for local Ca2 + signalling. J Physiol Lond. 1997;500:285–295. doi: 10.1113/jphysiol.1997.sp022020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xiao R.P., Lakatta E.G. Beta 1-adrenoceptor stimulation and beta 2-adrenoceptor stimulation differ in their effects on contraction, cytosolic Ca2 +, and Ca2 + current in single rat ventricular cells. Circ Res. 1993;73:286–300. doi: 10.1161/01.res.73.2.286. [DOI] [PubMed] [Google Scholar]
- 22.Chen X., Bing Z., He J., Jiang L., Luo X., Su Y. Downregulation of peroxisome proliferator-activated receptor-γ expression in hypertensive atrial fibrillation. Clin Cardiol. 2009;32:337–345. doi: 10.1002/clc.20566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nichols C.B., Rossow C.F., Navedo M.F., Westenbroek R.E., Catterall W.A., Santana L.F. Sympathetic stimulation of adult cardiomyocytes requires association of AKAP5 with a subpopulation of L-type calcium channels. Circ Res. 2010;107:747–756. doi: 10.1161/CIRCRESAHA.109.216127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jayasinghe I.D., Cannell M.B., Soeller C. Organization of ryanodine receptors, transverse tubules, and sodium–calcium exchanger in rat myocytes. Biophys J. 2009;97:2664–2673. doi: 10.1016/j.bpj.2009.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Murray A.J. Pharmacological PKA, inhibition: all may not be what it seems. Sci Signal. 2008;1:re4. doi: 10.1126/scisignal.122re4. [DOI] [PubMed] [Google Scholar]
- 26.Scriven D.R.L., Klimek A., Asghari P., Bellve K., Moore E.D.W. Caveolin-3 is adjacent to a group of extradyadic ryanodine receptors. Biophys J. 2005;89:1893–1901. doi: 10.1529/biophysj.105.064212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Timofeyev V., Myers R.E., Kim H.J., Woltz R.L., Sirish P., Heiserman J.P. Adenylyl cyclase subtype-specific compartmentalization: differential regulation of L-type Ca2 + current in ventricular myocytes. Circ Res. 2013;112:1567–1576. doi: 10.1161/CIRCRESAHA.112.300370. [DOI] [PMC free article] [PubMed] [Google Scholar]