

Abstract
Phevor integrates phenotype, gene function, and disease information with personal genomic data for improved power to identify disease-causing alleles. Phevor works by combining knowledge resident in multiple biomedical ontologies with the outputs of variant-prioritization tools. It does so by using an algorithm that propagates information across and between ontologies. This process enables Phevor to accurately reprioritize potentially damaging alleles identified by variant-prioritization tools in light of gene function, disease, and phenotype knowledge. Phevor is especially useful for single-exome and family-trio-based diagnostic analyses, the most commonly occurring clinical scenarios and ones for which existing personal genome diagnostic tools are most inaccurate and underpowered. Here, we present a series of benchmark analyses illustrating Phevor’s performance characteristics. Also presented are three recent Utah Genome Project case studies in which Phevor was used to identify disease-causing alleles. Collectively, these results show that Phevor improves diagnostic accuracy not only for individuals presenting with established disease phenotypes but also for those with previously undescribed and atypical disease presentations. Importantly, Phevor is not limited to known diseases or known disease-causing alleles. As we demonstrate, Phevor can also use latent information in ontologies to discover genes and disease-causing alleles not previously associated with disease.
Introduction
Personal genome sequencing is dramatically changing the landscape of clinical genetics, but it also presents a host of challenges. Every sequenced exome presents the clinical geneticist with thousands of variants, any one of which might be responsible for the person’s illness. One approach to making sense of these data is to employ a whole-genome and whole-exome search tool such as ANNOVAR1 or the Variant Annotation, Analysis, Search Tool (VAAST)2,3 to identify disease-causing variants in an ab initio fashion. This is proving an effective approach for case-cohort analyses;4–8 likewise, sequencing additional family members can also improve diagnostic accuracy. Unfortunately, single affected individuals and small nuclear families are the most frequently encountered diagnostic scenarios in the clinic. Today’s whole-genome and whole-exome search and variant-prioritization tools are underpowered in these situations, limiting the number of successful diagnoses.2,9 In response, physicians and clinical genetics laboratories often attempt to narrow the list to a subset of candidate genes and alleles in light of an individual’s phenotype.10
Phenotype data are generally employed in an ad hoc fashion in which clinicians and geneticists choose genes and alleles as candidates on the basis of their expert knowledge. No general standards, procedures, or validated best practices yet exist. Moreover, genes not previously associated with the phenotype are not considered—often preventing the discovery of gene-disease associations. The potential impact of false positives and negatives on diagnostic accuracy is obviously considerable. Algorithmic means of prioritizing genes and variants in light of phenotype data are thus badly needed. In response, we have created Phevor, the Phenotype Driven Variant Ontological Re-ranking tool.
Phevor works by combining the outputs of widely used variant-prioritization tools with knowledge resident in diverse biomedical ontologies, such as the Human Phenotype Ontology (HPO),11 the Mammalian Phenotype Ontology (MPO),12 the Disease Ontology (DO),13 and the Gene Ontology (GO)14 (Figure S1, available online). Ontologies are graphical representations of the knowledge, such as gene functions or human phenotypes, in a given domain. Ontologies organize this knowledge by using directed acyclic graphs wherein concepts (terms) are nodes in the graph and the logical relationships obtained between them are modeled as edges, for example, “deaminase activity” (node) is_a (edge) “catalytic activity” (node).14 Ontology terms (nodes) are used to “annotate” biological data, rendering the data machine readable and traversable via the ontologies’ relationships (edges). For example, annotating a gene with the term “deaminase activity” makes it possible to deduce that the same gene encodes a protein with “catalytic activity.” In recent years, many biomedical ontologies have been created for the management of biological data.15–17
Phevor propagates an individual’s phenotype information across and between biomedical ontologies. This process enables Phevor to accurately reprioritize candidates identified by variant-prioritization tools in light of knowledge contained in the ontologies. As we show, Phevor can also discover emergent gene properties and latent phenotype information by combining ontologies, further improving its accuracy.
Phevor does not replace existing prioritization tools; rather, it provides a general means of improving every tool’s performance. As we demonstrate, Phevor substantially improves the accuracy of widely used variant-prioritization tools such as SIFT,18 conservation-based tools such as PhastCons,19 and genome-wide search tools such as VAAST2,3 and ANNOVAR.1 Phevor also outperforms tools such as PHIVE (Phenotypic Interpretation of Variants in Exomes),20 which combines a fixed-variant filtering approach with mouse phenotype data.
Phevor also differs from tools such as Phenomizer21 and sSAGA (Symptom- and Sign-Assisted Genome Analysis)10 in that it does not postulate a set of fixed associations between genes, phenotypes, and diseases. Rather, Phevor dynamically integrates knowledge resident in multiple biomedical ontologies into the variant-prioritization process. This enables Phevor to improve diagnostic accuracy not only for established disease phenotypes but also for previously undescribed and atypical disease presentations.
Importantly, Phevor also provides a means of integrating ontologies that contain knowledge not explicitly linked to phenotype (such as the GO) into the variant-prioritization process. As we demonstrate, Phevor can use information latent in such ontologies to discover disease-causing alleles in genes not previously associated with disease.
Phevor is especially useful for single-exome and family-trio-based diagnostic analyses, the most commonly occurring clinical scenarios and ones for which existing sequenced-based diagnostic tools are most inaccurate and underpowered. Here, we describe the algorithm underlying Phevor and present benchmark analyses illustrating Phevor’s performance characteristics. We also present three Utah Genome Project clinical applications in which Phevor was used to identify both known disease-causing alleles and ones not previously associated with disease.
Material and Methods
Phenotype and Candidate-Gene Information
Phevor can improve diagnostic accuracy by using phenotype and candidate-gene information derived from multiple sources. In the simplest scenario, users provide a tab-delimited list of terms describing the phenotype(s) drawn from the HPO.11 Alternatively, the list can consist of terms from the DO,13 MPO,12 GO,14 or Online Mendelian Inheritance in Man (OMIM). Lists containing terms from more than one ontology are also permitted. Users may also employ the online tool Phenomizer21 to describe an individual’s phenotype and to assemble a list of candidate genes. Phenomizer provides the physician with a means of producing a phenotype description for use with Phevor. The Phenomizer report can be downloaded to the user’s computer and passed directly to Phevor. See Figure S1 for more information.
Assembling a Gene List
Biomedical-ontology annotations are now readily available for many human and model-organism genes. Probably the best-known example is the GO. Currently, over 18,000 human genes have been annotated with GO terms.14 In addition, at last count, over 2,800 human genes have been annotated with HPO terms.11 Phevor employs these annotations to associate ontology concepts (nodes) to genes and vice versa. Consider the following example of a phenotype description consisting of two HPO terms: “hypothyroidism” (HP:0000812) and “abnormality of the intestine” (HP:0002242). If genes have previously been annotated to these two nodes in the ontology, Phevor saves those genes in an internal list. In cases where no genes are annotated to a user-provided ontology term, Phevor traverses that ontology by beginning at the provided term and proceeding toward the ontology’s root(s) until it encounters a node with annotated genes, and then it adds those genes to the list. At the end of this process, the resulting gene list is then used for seeding nodes in the other ontologies, e.g., the GO, MPO, and DO.
Phevor relates different ontologies via their common gene annotations (Figure S2). Deleterious alleles in ABCB11, for example, are known to cause intrahepatic cholestasis, a fact captured by HPO’s annotation of ABCB11 to the node “intrahepatic cholestasis” (HP:0001406). In the GO, ABCB11 is annotated to “canalicular bile acid transport” (GO:0015722) and “bile acid biosynthetic process” (GO:0006699). Phevor uses the common gene (in this case, ABCB11) to relate the HPO node HP:0001406 to GO nodes GO:0015722 and GO:0006699. As we explain below, this process allows Phevor to extend its search to include additional genes with functions similar to those of ABCB11.
Ontology Propagation
Once Phevor identifies a set of starting nodes for each ontology, i.e., those provided by the user in their phenotype list (e.g., HP:0001406) or those derived from it by the cross-ontology linking procedure described in the preceding paragraph (e.g., GO:0015722 and GO:0006699), it next propagates this information across each ontology by means of a process we term ontological propagation. Consider the example shown in Figure S3. Here, two seed nodes in some ontology have been identified, and in both cases gene A has been previously annotated to both nodes. Each seed node is assigned a value of 1, and this information is then propagated across the ontology as follows. If we proceed from each seed node toward its children, each time an edge is crossed to a neighboring node, the current value of the previous node is divided by 2. For example, if the starting seed node has two children, its value is divided in half for each child, so in this case, both children receive a value of ½. This process is continued until a terminal leaf is encountered. The original seed scores are also propagated upward to the root node(s) of the ontology by means of the same procedure (Figure S3B). In practice, there can be many seed nodes. In such cases, intersecting threads of propagation are first added together, and the process of propagation then proceeds as previously described. One interesting consequence of this process is that nodes far from the original seeds can attain high values, greater even than those of any of the starting seed nodes. The phenomenon is illustrated by the darker red nodes in Figure S3C, in which propagation has identified two additional gene candidates, B and C, not associated with the original seed nodes.
From Node to Gene
Upon completion of propagation (Figure S3C), Phevor renormalizes each node’s value to between 0 and 1 by dividing it by the sum of all node scores in the ontology. Phevor next assigns each gene annotated to the ontology a score corresponding to the maximum score of any node in the ontology to which it is annotated. This process is repeated for each ontology; thus, genes annotated to more than one ontology will have a score from each. These scores are added to produce a final sum score for each gene and renormalized again to a value between 0 and 1. Consider a set of genes drawn from the HPO and assigned gene scores by the process described in the preceding paragraphs. Consider also a similar list of human genes derived from propagation across the GO. Simply summing each gene’s HPO and GO scores and renormalizing again by the total sum of sums will combine these lists.
Rational Expansion of Candidate-Gene Lists
The ontological propagation and combination procedures described above enable Phevor to extend the original HPO-derived gene list into an expanded candidate-gene list that can also include genes not annotated to the HPO. Recall that during propagation across an ontology, intersecting threads can cause nodes to have scores that equal or even exceed those of any original seed nodes. Thus, a gene not yet associated with a particular human disease can become an excellent candidate if it is annotated to an HPO node located at an intersection of phenotypes associated with other diseases or has GO functions, locations, and/or processes similar to those of known disease-genes annotated to HPO. Phevor also employs the MPO, allowing it to leverage model-organism phenotype information, and the DO, which provides it with additional information pertaining to human genetic disease. Thus, Phevor’s approach provides an automatic and rational means of expanding a candidate-gene list derived from a starting list of phenotype or gene-function terms to leverage knowledge contained in diverse biomedical ontologies. In the paragraphs below, we explain how gene sum scores are combined with the outputs of variant-prioritization tools for improving the accuracy of sequence-based diagnosis.
Combining Ontologies and Variant Data
Upon completion of all ontology propagation, combination, and gene-scoring steps described in the preceding paragraphs, genes are ranked by their gene sum scores; then, their percentile ranks are combined with variant and gene-prioritization scores as follows. Phevor first calculates a disease association score for each gene,
| (Equation 1) |
where is the percentile rank of the renormalized gene sum score as derived from the ontological combination and propagation procedures described in Figures S2 and S3 and is the gene’s percentile rank provided by the external variant-prioritization or search tool, e.g., ANNOVAR, SIFT, or PhastCons (except for VAAST, in which case its reported p values are used directly). Phevor then calculates , a second score summarizing the weight of evidence that the gene is not involved with the individual’s illness, i.e., neither the variants nor the gene is involved in the individual’s disease:
| (Equation 2) |
The Phevor score (Equation 3) is the log10 ratio of the disease association score and the healthy association score ,
| (Equation 3) |
These scores are distributed normally (data not shown). The performance benchmarks presented in the Results provide an objective basis for evaluating the utility of .
Sequencing Procedures
To sequence exome DNA, we used the Agilent SureSelect(XT) Human All Exon V5+UTRs targeted enrichment system. The whole genome of the STAT1 proband was sequenced (see Results for details). An Illumina HiSeq instrument programmed to perform 101-cycle paired-end sequencing was used for all cases.
Sanger Sequence Validation
Putative disease-causing mutations identified by exome sequencing were validated by Sanger sequencing in the DNA Sequencing Core Facility at the University of Utah. We also used DNA from probands and parents to validate inheritance patterns or confirm de novo mutations in all of the cases presented. PCR primers were designed and optimized and subsequently amplified. Sequencing was performed via capillary sequencing.
Variant-Calling Procedures
According to the best practices described by the Broad Institute,23 sequence reads were aligned with the Burrows-Wheeler Aligner, PCR duplicates were removed, and indel realignment was performed with the Genome Analysis Toolkit (GATK). Variants were joint called with the GATK UnifiedGenotyper in conjunction with 30 CEU (Utah residents with ancestry from northern and western Europe from the CEPH collection) Genome BAM files from the 1000 Genomes Project.24 For the benchmarking experiments, only single-nucleotide variants (SNVs) were used because not every variant-prioritization tool can score indels and splice-site variants. The case-study analyses searched SNVs, splice-site variants, and indels.
Benchmarking Procedures
We inserted known, disease-causing alleles into otherwise healthy (background) exomes. These exomes were sequenced to 50× coverage on an Illumina HiSeq (see sequencing procedures above) and jointly called with 30 CEU genomes drawn from the 1000 Genomes Project.24 Known disease-associated genes were randomly selected (without replacement) from the Human Gene Mutation Database (HGMD). For each gene in the HGMD, damaging SNV alleles were randomly selected (without replacement) from all recorded damaging alleles at that locus. The damaging allele was added to the target exome(s) VCF file(s), and the quality metrics of the closest mapped variant were attached to it. Damaging alleles were inserted into the appropriate number of healthy exomes depending on the inheritance model (e.g., two copies of the same allele for recessive and one for dominant). This process was repeated 100 times for 100 different, randomly selected genes with established disease associations; the entire process was then repeated 99 more times for determining margins of error. All prioritization tools (SIFT, PhastCons, ANNOVAR, and VAAST) were run with their default settings, except that dominant or recessive inheritance was specified for the VAAST and ANNOVAR runs because these two tools allow users to do so. For purposes of comparison, for the VAAST and ANNOVAR runs, the maximum minor allele frequency (MAF) cutoff was set to 1%, ANNOVAR’s default setting. We also explored running ANNOVAR with different MAF cutoffs but found that overall performance was best with this value. ANNOVAR was run with the “clinical variant flag” option enabled so as not to exclude known disease-causing variants present in dbSNP 135 from consideration. PHIVE20 was run from the Exomiser program. For these runs, the MAF cutoff was set to 1%, and the “remove dbSNP” and “pathogenic variant flag” options were set to “no.”
Ethical Statement
Procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national), and proper informed consent was obtained.
Results
Benchmark Analyses
Figure 1A summarizes the ability of four different variant tools—SIFT, ANNOVAR, PhastCons, and VAAST— to use a single affected individual’s exome to identify recessive disease-causing alleles within a known disease-associated gene. These four tools were selected so as to include two prominent conservation-based variant-prioritization tools (SIFT and PhastCons) and two prominent genome-wide search tools (ANNOVAR and VAAST). SIFT18 is a tool for amino acid conservation and functional prediction, PhastCons19 is a tool for sequence-conservation identification, ANNOVAR1 filters on variant frequencies to search genomes for disease-casing alleles, and VAAST2,3 is a probabilistic disease-associated-gene finder that uses information on variant frequency and amino acid conservation. To assemble these data, we inserted two copies of a known disease-causing allele randomly selected from HGMD25 (see Material and Methods for details) into a single target exome and repeated the process 100 times for 100 different genes with known disease associations in order to determine margins of error. For these analyses, we used only SNVs, excluding indels and other types of variants because not every variant-prioritization tool can score them.
Figure 1.
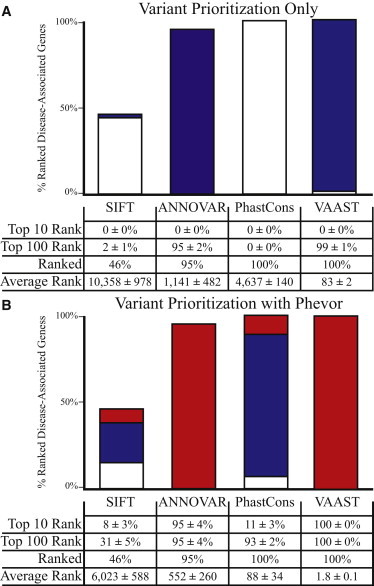
Variant Prioritization for Known Disease-Causing Alleles
Performance comparisons of four different variant-prioritization tools before (A) and after (B) postprocessing them with Phevor. Two copies of a known disease-causing allele were randomly selected from HGMD and spiked into a single target exome at the reported genomic location; hence, these results model simple, recessive diseases. This process was repeated 100 times for 100 different, randomly selected already disease-associated genes for determining margins of error. Bar charts show the percentage of time for which the disease-associated gene was ranked among the top ten candidates genome-wide (red) or among the top 100 candidates (blue); white denotes a rank greater than 100 in the candidate list. For the Phevor analyses in (B), each tool’s output files were fed to Phevor along with phenotype report containing the HPO terms annotated to each disease-associated gene. The table below the bar charts summarizes this information in more detail. Bars do not reach 100% because of false negatives, i.e., not every tool is able to prioritize every disease-causing allele. When the target gene’s disease-causing alleles were unscored or predicted to be benign by a tool, the gene was placed at the midpoint of the list of the 22,107 annotated human genes.
The heights of the bars in Figure 1A summarize the percentage of the 100 trials in which the prioritization tool scored the known disease-causing allele. Importantly, the percentages in Figure 1A include all scored alleles whether or not they were scored as deleterious. For example, SIFT scored 46% of the known disease-causing variants as either deleterious or tolerated. It was unable to score the remaining 54% of the alleles. ANNOVAR scored 95% of the alleles, and VAAST and PhastCons scored every allele. These percentages vary because not every tool is capable of scoring every potential disease-causing variant. The reasons vary from tool to tool and case to case. SIFT, for example, cannot score alleles located in poorly conserved coding regions of genes.26
The colors of the bars in Figure 1A summarize the percentage of time the gene with the inserted disease-causing alleles was ranked among the top ten candidates genome-wide (red) or among the top 100 candidates (blue); white denotes a rank greater than 100 in the candidate list. The table in Figure 1A summarizes this information in more detail. ANNOVAR, for example, ranked 95% of the genes spiked with known disease-causing alleles as potentially damaged and judged the remainder of these genes as containing only nondeleterious alleles. Of the 95% of damaged genes it detected, on average it ranked all of them within the top 100 candidates genome-wide. For the 5% of genes that ANNOVAR failed to rank, we assigned a rank of 11,141—the midpoint of the annotated 22,107 human genes. Hence, the average rank was much lower: 3,653. VAAST, by comparison, ranked every gene and identified the disease-causing allele among the top 100 candidates 99% of the time and gave an average rank of 83 genome-wide. Note that in 100 runs of 100 different test cases, no tool ever placed the gene containing the disease-causing alleles among the top ten candidates. Figure 1A thus illustrates a basic fact of personal genome analysis: using only a single affected exome, today’s tools are underpowered to identify the damaged gene and its disease-causing variants.
Figure 1B summarizes the results of using Phevor to reanalyze the same SIFT, ANNOVAR, PhastCons, and VAAST output files in Figure 1A. For these analyses, each tool’s output files were provided to Phevor along with a phenotype report containing the HPO terms annotated to every gene with inserted disease-causing alleles. These phenotype descriptions are provided in Table S1. As can be seen, Phevor dramatically improved the performance of every tool benchmarked in Figure 1A. For the 95% of genes ranked by ANNOVAR, all were among the top ten candidates, and Phevor improved the average rank for ANNOVAR from 3,653 to 552. Similar trends were seen for SIFT. Phevor showed even better improvements for PhastCons and VAAST outputs. The average rank for VAAST, for example, improved from 83 to 1.8, and the gene containing the disease-causing alleles was ranked among the top ten genes100% of the time. Phevor performed best on VAAST outputs because VAAST has a lower false-negative rate than SIFT and ANNOVAR (Figure 1A). This is because Phevor only improves the ranks of prioritized genes; it doesn’t rerank genes previously determined by a tool to harbor no deleterious alleles.
Results for dominant disease are provided in Figure S4. As would be expected, benchmarks for dominant diseases showed the same trends in that every tool exhibited lower power than for recessive cases. However, Phevor still markedly improved power. Using VAAST outputs, Phevor ranked the gene containing the disease-causing variant in the top ten candidates 93% of the time.
Collectively, these results demonstrate that Phevor can improve the power of widely used variant-prioritization tools. Recall, however, that the HPO provides a list of ∼2,800 known human genes, each annotated to one or more HPO nodes, and that Phevor uses this information during the ontology combination and propagation steps described in Figures S2 and S3 (see Material and Methods). In light of this fact, the question naturally arises as to how much Phevor depends on the fact that the gene with the disease-causing allele(s) has been previously annotated to an ontology. Figure 2 addresses this issue.
Figure 2.
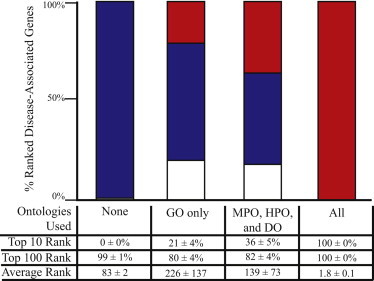
Variant Prioritization for Genes Previously Unassociated with Disease
The procedure used in Figure 1B was repeated, but instead the disease-associated gene’s ontological annotations were removed from all but the specified ontologies prior to running Phevor. For economic reasons, only VAAST results are shown. Removing all the disease-associated gene’s annotations from all ontologies mimics the case of a previously unreported allele in a gene with unknown GO function, process, and cellular location and no previous association with a known disease or phenotype. This is equivalent to running VAAST alone (“none”), and the leftmost bar chart and table column summarize these results. The right-hand bar and table column (“All”) summarize the results of running VAAST and Phevor with the current ontological annotations of the disease-associated gene. The “GO only” column reports the results of removing the disease-associated gene’s phenotype annotations, depicting discovery success with only GO ontological annotations. This column models the ability of Phevor to identify a disease association when that gene is annotated to GO but has no disease, human, or model-organism phenotype annotations. In contrast The “MPO, HPO, and DO” column assays the impact of removing a gene’s GO annotations but leaving its disease, human, and model-organism phenotype annotations intact.
Figure 2 employs the same procedure as in Figure 1, but instead the disease-associated gene was removed from one or more of the ontologies prior to running Phevor. This made it possible to evaluate Phevor’s ability to improve the ranks of a gene containing disease-causing alleles in the absence of any ontological assignments (i.e., as if the gene had never before been associated with a disease, function, or phenotype). For these benchmarks, we investigated not only the impact of simultaneously masking the gene’s HPO, MPO, and DO phenotype annotations but also its GO annotations. Because of space limitations, the results of these experiments are only shown for VAAST outputs (Figure 2).
As can be seen, removing the gene from one or more ontologies did decrease Phevor’s power to identify the gene but did not eliminate it, demonstrating that Phevor gained power by combining multiple ontologies. Removing the target gene from the GO and using only the three phenotype ontologies (HPO, MPO, and DO), Phevor still ranked the target gene in the top ten candidates 36% of the time and in the top 100 candidates 82% of the time. By comparison, using VAAST alone, Phevor ranked the target gene among the top 10 and 100 candidates 0% and 99% of the time, respectively. The 18% false-negative rate was an artifact of the benchmark procedure and resulted from removing the gene from the GO. In brief, because the majority of human genes (18,824) are already annotated to the GO, the prior expectation is that any new gene playing a role in the disease is more likely to be annotated to the GO than not, causing Phevor to prefer candidates already annotated to the GO in this benchmarking scenario.
Similar trends were seen with using the GO14 alone. This time, removing the gene from the MPO, HPO, and DO, Phevor placed the gene containing the disease-causing alleles among the top ten candidates 21% of the time and among the top 100 candidates 80% of the time—still much better than when it used VAAST alone. Recall that for this analysis, Phevor was only provided with a phenotype description—not GO terms—and that the gene containing the inserted disease-causing alleles was removed from every ontology containing any phenotype data, e.g., the, HPO, DO, and MPO. Thus, this rank increase (e.g., from 0% to 21% in the top ten) was solely the result of Phevor’s ability to integrate the GO into a phenotype-driven prioritization process, demonstrating that Phevor can use the GO to aid in the discovery of disease-causing alleles in genes not previously associated with a given disease phenotype. Collectively, these results demonstrate that a significant portion of Phevor’s power is derived from its ability to relate phenotype concepts in the HPO to gene function, process, and location concepts modeled by the GO.
Figure 2 demonstrates that Phevor improved the performance of the variant-prioritization tool even when the disease-causing alleles were located within genes with no prior disease association. This is possible because even when the gene containing the (novel) disease allele(s) is absent from the HPO, Phevor can nonetheless assign it a high score for disease association after information associated with its paralogs is propagated by Phevor from the HPO to GO. This is a complex point, and an illustration is helpful. Consider the case for two potassium-transporter-encoding genes, A and B. Deleterious alleles in one (A) are known to cause cardiomyopathy, whereas gene B has no disease associations as of yet. If genes A and B are both annotated in the GO as “potassium transporters,” when Phevor propagates the HPO associations of gene A to the GO, the GO node “potassium transporter” will receive some score, which in turn will be propagated to gene B. Thus, even though gene B is absent from the HPO, its Phevor disease association score will increase because of its GO annotation. This illustrates the simplest of cases. Many more-complex scenarios are possible. For example, genes A and B might be annotated to different nodes in the GO, in which case gene B’s disease association score would increase proportionally after propagation across the GO.
Figure 3 compares the relative performance of Phevor to PHIVE,20 an online tool that uses ANNOVAR in conjunction with mouse phenotype data to improve ANNOVAR’s prioritization accuracy. PHIVE is accessible through the Exomiser.20 For this benchmark, repeating the process in Figure 1, we once again inserted two copies of a known disease-causing allele randomly selected from HGMD25 (see Material and Methods for details) into a target exome and repeated the process for 100 different genes. The left column in Figure 3 provides a breakdown of the results when ANNOVAR was used alone, the middle column reports the results of uploading these same 100 exomes with their unprioritized variants to the Exomiser, and the right column shows the results for the same 100 exomes with the use of ANNOVAR and Phevor. Although the Exomiser did increase the percentage of cases for which the target gene was located in the top 10 and top 100 candidates in comparison to ANNOVAR alone, it did so at the expense of additional false negatives. In contrast, Phevor obtained much better power on the same data set (right column in Figure 3) without incurring any additional false negatives. Phevor was, however, ultimately limited by ANNOVAR’s false-negative rate. This limitation can be overcome simply by means of using VAAST reports instead of ANNOVAR reports, in which case Phevor places 100% of the target genes among the top ten candidates (c.f. Figure 1B).
Figure 3.
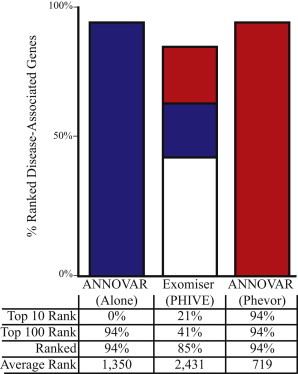
Comparison of Phevor to the Exomiser’s PHIVE
Comparison of disease-allele-identification success rates for Phevor and the PHIVE methodology, which is available through the Exomiser. The Exomiser is based upon ANNOVAR’s filtering logic; thus, the Phevor comparison uses ANNOVAR as the variant-prioritization tool. Shown are the results of 100 searches of known recessive disease-associated genes. Identical variant files and phenotype descriptions were given to Exomiser + PHIVE and ANNOVAR + Phevor. Bar charts show the percentage of time for which the target, i.e., disease-associated, gene was ranked among the top ten candidates genome-wide (red) or among the top 100 candidates (blue); white denotes a rank greater than 100 in the candidate list. The table below the bar charts summarizes this information in more detail. Bars do not reach 100% because of false negatives, i.e., the tool reported the disease-causing allele to be nondeleterious; these cases were placed at the midpoint of the list of 22,107 annotated human genes.
Next, we sought to determine the impact of atypical disease presentation upon Phevor’s accuracy. The term atypical presentation refers to cases in which an individual has a known genetic disease but does not present with the typical disease phenotype. Reasons include previously unreported alleles in known genes, previously undescribed combinations of alleles, ethnicity (genetic-background effects), environmental influences, and in some cases, multiple genetic diseases presenting in the same individual to produce a compound phenotype.27 Atypical presentation resulting from previously unreported disease-associated alleles in known genes and compound phenotypes due to multiple disease-causing alleles are emerging as a common occurrence in personal genome-driven diagnosis;9,27,28 thus, Phevor’s performance in such situations is of interest.
Although a truly thorough investigation of atypical presentation lies outside the scope of the current study, Figure 4 addresses its impact on Phevor for case cohorts of one, three, and five unrelated individuals by showing the same benchmarking methodology as in Figure 1. For this experiment, however, we randomly replaced each target gene’s HPO-based phenotype description with another’s, thereby mimicking an extreme scenario of atypical presentation and/or misdiagnosis whereby each individual presents with not only an atypical phenotype but also one normally associated with some other known genetic disease. Unsurprisingly, this significantly affected Phevor’s diagnostic accuracy. Using VAAST outputs for a single affected individual, Phevor declined in accuracy from ranking the damaged gene in the top ten candidates genome-wide in 100% of the cases to ranking it in only 26% of the cases. More surprising is that Phevor was still able to improve on VAAST’s performance alone, a phenomenon resulting again from Phevor’s use of the GO (as in Figure 3) and a point that we address in more detail in our Discussion.
Figure 4.
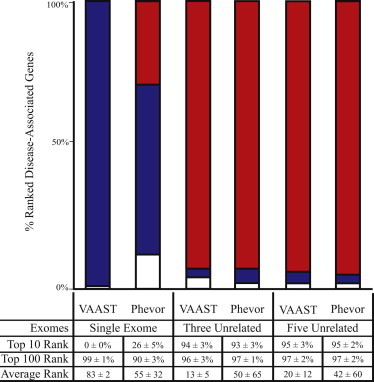
Phevor Accuracy and Atypical Disease Presentation
In order to evaluate the impact of incorrect diagnosis or atypical phenotypic presentation on Phevor’s accuracy, we repeated the analysis shown in Figure 1; this time, we randomly shuffled the phenotype descriptions for each gene at runtime and used the same phenotype descriptions for every member of a case cohort. For economic reasons, only VAAST results are shown. The results of running VAAST with and without Phevor for case cohorts of one, three, and five unrelated individuals are shown. As would be expected, providing Phevor with incorrect phenotype data significantly affected its diagnostic accuracy. For a single affected individual, Phevor declined in accuracy from ranking the damaged gene in the top ten candidates genome-wide in 100% of the cases to ranking it in 26% of cases. Nevertheless, Phevor was still able to improve upon VAAST’s performance alone. Phevor placed 95% of the damaged genes in the top ten candidates with cohorts of three and five unrelated affected individuals, despite the misleading phenotype data, given that the additional statistical power provided by VAAST increasingly outweighed the incorrect prior probabilities provided by Phevor.
The remaining columns in Figure 4 measure the impact of increasing the size of the case cohort. As can be seen, with three or more unrelated individuals all with the same (shuffled) atypical phenotypic presentation, Phevor performed very well, even when the phenotype information was misleading. Thus, these results demonstrate how Phevor’s ontology-derived scores, e.g., in Equations 1 and 2, are gradually overridden in the face of increasing sequence-based experimental evidence to the contrary—a clearly desirable behavior.
Application of Phevor in the Clinic
We present three recent Utah Genome Project cases in which we employed Phevor in tandem with ANNOVAR and VAAST to identify disease-causing alleles in individuals with an undiagnosed disease of a most likely genetic cause. All three applications of Phevor involved either small families or single affected individuals—scenarios for which, as we have shown, existing prioritization tools are underpowered. These analyses thus demonstrate Phevor’s utility by using real clinical examples.
A Gene-Disease Association for NFKB2
We identified a family affected by autosomal-dominant, early-onset hypogammaglobulinemia with variable autoimmune features and adrenal insufficiency. Blood samples were obtained from the affected mother, the unaffected father, and their two affected children (family A). Blood was also obtained from a fourth, unrelated affected individual with the same phenotype (family B). Sequencing was performed as described in Chen et al.,4 and variant annotation was performed with the VAAST Annotation Tool (VAT).3
Exome data from the four individuals in family A and the affected individual in family B were then analyzed with VAAST.2,3 In family A, this analysis identified a deletion (c.2564delA [RefSeq accession number NM_002502.4], resulting in p.Lys855Serfs∗7) in NFKB2 (MIM 164012). VAAST identified a second NFKB2 allele (c.2557C>T [p.Arg853∗]) in family B. Subsequent immunoblot analysis and immunofluorescence microscopy of transformed B cells from affected individuals showed that the NFKB2 mutations affect phosphorylation and proteasomal processing of the p100 NFKB2 to its p52 derivative and, ultimately, p52 nuclear translocation.4
Figure 5A shows the results of running ANNOVAR (top left panel) and VAAST (top right panel) on the union of all variants identified in the affected children and mother from family A and the affected individual from family B. The x axes of the Manhattan plots in Figure 5A are the genomic coordinates of the candidate genes. The y axes show the log10 value of the ANNOVAR score, VAAST p value, or Phevor score depending upon the method. For purposes of comparison to VAAST, we transformed the ANNOVAR scores to frequencies by dividing the number of candidates by the total number of annotated human genes—hence the “shelf” of candidates in the ANNOVAR plot at y = 1.14 (about 13.8% of human genes). Both ANNOVAR and VAAST identified a number of equally likely candidate genes. NFKB2 (shown in red) was among them in both analyses.
Figure 5.
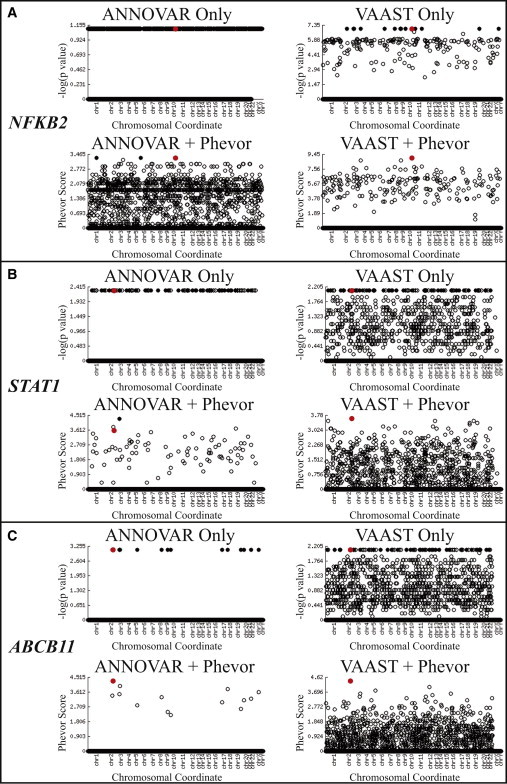
Phevor Analyses of Three Clinical Cases
Plotted on the x axes of each Manhattan plot are the genomic coordinates of the candidate genes. The y axes show the log10 value of the ANNOVAR score, VAAST p value, or Phevor score depending upon the panel. Black, filled circles denote top ranked gene(s), all of which had either the same ANNOVAR score or the same VAAST p value. Red circles denote the gene containing disease-causing allele(s). For purposes of comparison to VAAST, we transformed the ANNOVAR scores to frequencies by dividing the number of gene candidates identified by ANNOVAR by the total number of annotated human genes.
(A) Phevor identified NFKB2 as a disease-associated gene. (Top) Results of running ANNOVAR (left) and VAAST (right) on the union of variants identified in affected members of family A and those in the affected individual from family B. Both ANNOVAR and VAAST identified a large number of equally likely candidate genes. NFKB2 (shown in red) was among them in both cases. (Bottom) Phevor identified a single best candidate, NFKB2, by using the VAAST output, and NFKB2 was ranked second with the ANNOVAR output (two other genes were tied for first place).
(B) Phevor identified a de novo variant in STAT1 as responsible for a previously undescribed phenotype in an already disease-associated gene. (Top) Results of running ANNOVAR (left) and VAAST (right) on the single affected individual’s exome. Both ANNOVAR and VAAST identified multiple candidate genes. STAT1 (shown in red) was among them in both cases. (Bottom) Phevor identified a single best candidate, STAT1, by using the VAAST output. STAT1 was the third best candidate with the ANNOVAR output.
(C) Phevor identified a mutation in ABCB11, a known disease-associated gene. (Top) Results of running ANNOVAR (left) and VAAST (right) on the single affected child’s exome. Both ANNOVAR and VAAST identified a number of equally likely candidate genes. ABCB11 (shown in red) was among them. (Bottom) Phevor identified a single best candidate, ABCB11, by using the ANNOVAR and VAAST outputs.
The lower panel of Figure 5A presents the results of postprocessing these same ANNOVAR and VAAST output files with the use of Phevor, as well as a Phenomizer-derived, HPO-based phenotype description consisting of terms “recurrent infections” (HPO:0002719) and “abnormality of humoral immunity” (HPO:0005368). Phevor identified a single best candidate, NFKB2, by using the VAAST output, and the same gene ranked second with the ANNOVAR output. Functional follow-up studies established NFKB2—hence the noncanonical NF-κB signaling pathway—as a genetic etiology for this primary immunodeficiency syndrome.4 Thus, these analyses demonstrate Phevor’s ability to identify a human gene not currently associated with a disease or phenotype in the HPO, DO, or MPO.
An Atypical Phenotype Caused by a Dominant Allele of STAT1
The proband is a 12-year-old male with severe diarrhea in the context of intestinal inflammation, total villous atrophy, and hypothyroidism. He required total parenteral nutrition to support growth, resulting in multiple hospitalizations for central-line-associated bloodstream infections. During multidisciplinary comprehensive clinical evaluation, we considered a diagnosis of X-linked immunodysregulation, polyendocrinopathy, and enteropathy (IPEX syndrome [MIM 304790]), but clinical sequencing of FOXP3 (MIM 300292) and IL2RA (MIM 147730), genes associated with IPEX,29,30 revealed no pathologic variants. His clinical picture was life threatening, warranting hematopoietic stem cell transplantation despite diagnostic uncertainty. Prior to pretransplant myeloablation, DNA was obtained from the proband and both parents. Figure 5B shows the results of ANNOVAR and VAAST analysis using the proband’s exome. As was the case for NFKB2, both ANNOVAR and VAAST were underpowered to distinguish the disease-associated gene and causative alleles from a background of other likely candidates. Phevor analyses of these same data, together with a phenotype description consisting of the HPO terms “hyopthryoidism” (HP:0000812), “paronychia” (HP:0001818), “autoimmunity” (HP:0002960), and “abnormality of the intestine” (HP:0002242), identified a single gene, STAT1 (MIM 600555), as the third-ranked candidate in the ANNOVAR outputs and the best candidate in the VAAST analyses (lower panels of Figure 5B). Subsequent analyses of the proband’s parents determined that the top-scoring variant in the VAAST-Phevor run was a single STAT1 de novo mutation (c.1154C>T [RefSeq NM_139266.2]) affecting the DNA-binding region of STAT1 (p.Thr385Met [RefSeq NP_009330.1]). We confirmed the variant by Sanger sequencing.
Multiple-protein-sequence alignment shows conservation across phyla at this amino acid position (data not shown). Moreover, gain-of-function mutations in STAT1 cause immune-mediated human disease,31 and STAT1 encodes a transcription factor that regulates FOXP3.32 Functional studies have indicated that this mutation leads to an overexpression of STAT1,32–34 suggesting gain-of-function mutation as a mechanism. Supporting this conclusion are recent studies reporting that this same allele causes chronic mucocutaneous candidiasis35 and an IPEX-like syndrome.32 These results highlight Phevor’s ability to use only a single affected exome to identify a mutation located in a known disease-associated gene and producing an atypical phenotype.
A Mutation in a Known Disease-Associated Gene, ABCB11
The proband is a 6-month-old infant with an undiagnosed liver disease phenotypically similar to progressive familial intrahepatic cholestasis.36 To identify mutations in the proband, we performed exome sequencing on the affected individual and both parents. Sequencing and bioinformatics processing were performed as described in the Material and Methods.
For these Phevor analyses, a single HPO phenotype term was used: “intrahepatic cholestasis” (HP:0001406). As shown in Figure 5C, Phevor analysis identified a single candidate gene (ABCB11) in the proband’s exome sequence.
Mutations in ABCB11 (MIM 603201) are known to cause progressive familial intrahepatic cholestasis type 2. The variants identified by VAAST, and supported as causative by Phevor, form a compound heterozygote in the proband. We confirmed these variants by Sanger sequencing (see Material and Method). The paternal variant (c.3332T>C [RefSeq NM_003742.2], leading to p.Phe1111Ser) and the maternal variant (c.890A>G [p.Glu297Gly]) are both considered highly damaging by SIFT. The maternal variant is known to cause intrahepatic cholestasis,37 whereas the paternal mutation is not currently associated with disease. These results demonstrate Phevor’s ability to use only a single affected exome to identify a previously unreported mutation located in a known disease-associated gene and present in trans to a known disease-causing allele.
Discussion
We have presented a series of benchmark and clinical applications demonstrating that Phevor provides an effective means of improving the diagnostic power of widely used variant-prioritization tools. These results demonstrate that Phevor is especially useful for single-exome and small, family-based analyses, the most commonly occurring clinical scenarios and ones for which existing variant-prioritization tools are most inaccurate and underpowered.
As we have shown, Phevor’s ability to improve the accuracy of variant-prioritization tools is the result of its ability to relate phenotype and disease concepts in ontologies such as the HPO and DO to gene function, process, and location concepts modeled by the GO. This allows Phevor to model key genetic-disease features that are not taken into account by existing methods that employ phenotype information for variant prioritization.10,20 For example, paralogous genes often produce similar diseases38 because they have similar functions, operate in similar biological processes, and are located in the same cellular compartments.
Phevor scores take into account not only the evidence that a gene is associated with an individual’s illness but also the evidence that it is not. In typical whole-exome searches, every variant-prioritization tool identifies many genes harboring what it considers to be deleterious mutations. Often the most damaging of them are found in genes without any known phenotype associating them with the disease of interest; moreover, in practice, highly deleterious alleles are often false-positive variant calls. Phevor successfully downweights these genes and alleles, causing the target gene to climb in rank as an indirect result. This phenomenon is well illustrated by the fact that Phevor improved the accuracy of variant-prioritization and genome-wide search tools even when provided with an incorrect phenotype description, e.g., Figure 4. This result underscores the consistency of Phevor’s approach; it also has some important implications, namely that the lack of previous disease association, weak phylogenetic conservation, and the lack of GO annotations for a gene are (weak) prima facie evidence against disease association.
The interplay of all of the above factors is well illustrated by the clinical applications we present from the Utah Genome Project. For these analyses, we employed Phevor in tandem with ANNOVAR and VAAST to identify disease-causing alleles. All three cases involved small case cohorts containing either related individuals or single affected exomes. For all of these cases, variant prioritization alone was insufficient to identify the causative alleles, whereas when combined with Phevor, these same data revealed a single candidate. These analyses demonstrate Phevor’s ability to use real clinical examples to identify a previously unreported mutation present in trans to a known disease-causing allele (ABCB11), dominant mutations in a gene not previously associated with the disease phenotype (NFKB2), and a de novo dominant allele located in an known disease-associated gene (STAT1) and producing an atypical phenotype. Collectively, these cases illustrate that Phevor can improve diagnostic accuracy for individuals with typical or atypical disease phenotypes and that it can also use information latent in ontologies to discover disease-causing alleles in genes not previously associated with human disease.
The incorporation of new ontologies and gene-pathway information into Phevor is an active area of development. Phevor can employ any variant-prioritization tool and any ontology, so long as the ontology has gene annotations and is available in OBO format.39 Over 50 biomedical ontologies, many satisfying both criteria, are available at the Open Biological and Biomedical Ontologies (OBO) Foundry. Thus, Phevor’s approach should also prove useful for nonmodel-organism and agricultural studies. Such applications raise interesting points. For the analyses presented here, we have used the MPO to leverage model-organism phenotype data to improve diagnostic power for humans. For application to model organisms, novel organisms, and agriculture, the HPO could be used in a manner analogous to that of the MPO in the analyses presented here, i.e., Phevor could provide a systematic means to bring human disease knowledge and human gene annotations to bear for nonmodel-organism and agricultural studies. A publicly available Phevor web server and test data sets are available online.
Acknowledgments
Phevor development was support by National Institute of General Medical Sciences (NIGMS) grant R01GM104390 to C.D.H., L.B.J., and M.Y. M.V.S. was supported by NIGMS grant R01GM104390 and by National Human Genome Research Institute (NHGRI) grant R44HG006579 to M.Y. and M.G.R. K.E. was supported by NHGRI grant R01HG004341. S.L.G. and C.D.H. were supported by NIH National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) grant R01DK084198. Additional support for S.L.G. was provided by NIDDK grant DK091374. Clinical sequencing and analyses were supported in part by the Utah Genome Project, the University of Utah’s Program in Personalized Health Care, and the Utah Science, Technology, and Research Center for Genetic Discovery. Research reported in this publication was also supported by grant 1ULTR001067 from the NIH National Center for Advancing Translational Sciences. We thank Linda Book, John Bohnsack, W. Daniel Jackson, and Michael Pulsipher for their assistance in enrolling subjects.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes, http://www.1000genomes.org/
Burrows-Wheeler Aligner (BWA), http://bio-bwa.sourceforge.net/
The Exomiser, https://www.sanger.ac.uk/resources/databases/exomiser/query/
Genome Analysis Toolkit (GATK), http://www.broadinstitute.org/gatk/
Human Gene Mutation Database (HGMD), http://www.hgmd.org
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
Open Biological and Biomedical Ontologies (OBO) Foundry, http://www.obofoundry.org
PhastCons, http://compgen.bscb.cornell.edu/phast/phastCons-HOWTO.html
Phevor Web, http://weatherby.genetics.utah.edu/cgi-bin/Phevor/PhevorWeb.html
SIFT, http://sift.jcvi.org/
Utah Genome Project, http://healthsciences.utah.edu/utah-genome-project/
The Variant Annotation, Analysis, and Search Tool (VAAST), http://www.yandell-lab.org/software/vaast.html
References
- 1.Wang K., Li M., Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hu H., Huff C.D., Moore B., Flygare S., Reese M.G., Yandell M. VAAST 2.0: improved variant classification and disease-gene identification using a conservation-controlled amino acid substitution matrix. Genet. Epidemiol. 2013;37:622–634. doi: 10.1002/gepi.21743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yandell M., Huff C., Hu H., Singleton M., Moore B., Xing J., Jorde L.B., Reese M.G. A probabilistic disease-gene finder for personal genomes. Genome Res. 2011;21:1529–1542. doi: 10.1101/gr.123158.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen K., Coonrod E.M., Kumánovics A., Franks Z.F., Durtschi J.D., Margraf R.L., Wu W., Heikal N.M., Augustine N.H., Ridge P.G. Germline mutations in NFKB2 implicate the noncanonical NF-κB pathway in the pathogenesis of common variable immunodeficiency. Am. J. Hum. Genet. 2013;93:812–824. doi: 10.1016/j.ajhg.2013.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ng S.B., Buckingham K.J., Lee C., Bigham A.W., Tabor H.K., Dent K.M., Huff C.D., Shannon P.T., Jabs E.W., Nickerson D.A. Exome sequencing identifies the cause of a mendelian disorder. Nat. Genet. 2010;42:30–35. doi: 10.1038/ng.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rope A.F., Wang K., Evjenth R., Xing J., Johnston J.J., Swensen J.J., Johnson W.E., Moore B., Huff C.D., Bird L.M. Using VAAST to identify an X-linked disorder resulting in lethality in male infants due to N-terminal acetyltransferase deficiency. Am. J. Hum. Genet. 2011;89:28–43. doi: 10.1016/j.ajhg.2011.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shirley M.D., Tang H., Gallione C.J., Baugher J.D., Frelin L.P., Cohen B., North P.E., Marchuk D.A., Comi A.M., Pevsner J. Sturge-Weber syndrome and port-wine stains caused by somatic mutation in GNAQ. N. Engl. J. Med. 2013;368:1971–1979. doi: 10.1056/NEJMoa1213507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McElroy J.J., Gutman C.E., Shaffer C.M., Busch T.D., Puttonen H., Teramo K., Murray J.C., Hallman M., Muglia L.J. Maternal coding variants in complement receptor 1 and spontaneous idiopathic preterm birth. Hum. Genet. 2013;132:935–942. doi: 10.1007/s00439-013-1304-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang Y., Muzny D.M., Reid J.G., Bainbridge M.N., Willis A., Ward P.A., Braxton A., Beuten J., Xia F., Niu Z. Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N. Engl. J. Med. 2013;369:1502–1511. doi: 10.1056/NEJMoa1306555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Saunders C.J., Miller N.A., Soden S.E., Dinwiddie D.L., Noll A., Alnadi N.A., Andraws N., Patterson M.L., Krivohlavek L.A., Fellis J. Rapid whole-genome sequencing for genetic disease diagnosis in neonatal intensive care units. Sci. Transl. Med. 2012;4:ra135. doi: 10.1126/scitranslmed.3004041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Robinson P.N., Köhler S., Bauer S., Seelow D., Horn D., Mundlos S. The Human Phenotype Ontology: a tool for annotating and analyzing human hereditary disease. Am. J. Hum. Genet. 2008;83:610–615. doi: 10.1016/j.ajhg.2008.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Smith C.L., Eppig J.T. The Mammalian Phenotype Ontology as a unifying standard for experimental and high-throughput phenotyping data. Mamm. Genome. 2012;23:653–668. doi: 10.1007/s00335-012-9421-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schriml L.M., Arze C., Nadendla S., Chang Y.W., Mazaitis M., Felix V., Feng G., Kibbe W.A. Disease Ontology: a backbone for disease semantic integration. Nucleic Acids Res. 2012;40(Database issue):D940–D946. doi: 10.1093/nar/gkr972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ashburner M., Ball C.A., Blake J.A., Botstein D., Butler H., Cherry J.M., Davis A.P., Dolinski K., Dwight S.S., Eppig J.T., The Gene Ontology Consortium Gene ontology: tool for the unification of biology. Nat. Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Whetzel P.L., Noy N.F., Shah N.H., Alexander P.R., Nyulas C., Tudorache T., Musen M.A. BioPortal: enhanced functionality via new Web services from the National Center for Biomedical Ontology to access and use ontologies in software applications. Nucleic Acids Res. 2011;39(Web Server issue):W541–W545. doi: 10.1093/nar/gkr469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Smith B., Ashburner M., Rosse C., Bard J., Bug W., Ceusters W., Goldberg L.J., Eilbeck K., Ireland A., Mungall C.J., OBI Consortium The OBO Foundry: coordinated evolution of ontologies to support biomedical data integration. Nat. Biotechnol. 2007;25:1251–1255. doi: 10.1038/nbt1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Robinson P.N., Bauer S. Taylor & Francis; Boca Raton: 2011. Introduction to bio-ontologies. [Google Scholar]
- 18.Ng P.C., Henikoff S. Predicting the effects of amino acid substitutions on protein function. Annu. Rev. Genomics Hum. Genet. 2006;7:61–80. doi: 10.1146/annurev.genom.7.080505.115630. [DOI] [PubMed] [Google Scholar]
- 19.Siepel A., Bejerano G., Pedersen J.S., Hinrichs A.S., Hou M., Rosenbloom K., Clawson H., Spieth J., Hillier L.W., Richards S. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005;15:1034–1050. doi: 10.1101/gr.3715005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Robinson P.N., Köhler S., Oellrich A., Wang K., Mungall C.J., Lewis S.E., Washington N., Bauer S., Seelow D.S., Krawitz P., Sanger Mouse Genetics Project Improved exome prioritization of disease genes through cross-species phenotype comparison. Genome Res. 2014;24:340–348. doi: 10.1101/gr.160325.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Köhler S., Bauer S., Mungall C.J., Carletti G., Smith C.L., Schofield P., Gkoutos G.V., Robinson P.N. Improving ontologies by automatic reasoning and evaluation of logical definitions. BMC Bioinformatics. 2011;12:418. doi: 10.1186/1471-2105-12-418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., DePristo M.A. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Abecasis G.R., Altshuler D., Auton A., Brooks L.D., Durbin R.M., Gibbs R.A., Hurles M.E., McVean G.A., 1000 Genomes Project Consortium A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cooper D.N., Ball E.V., Krawczak M. The human gene mutation database. Nucleic Acids Res. 1998;26:285–287. doi: 10.1093/nar/26.1.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kumar P., Henikoff S., Ng P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009;4:1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 27.Roach J.C., Glusman G., Smit A.F., Huff C.D., Hubley R., Shannon P.T., Rowen L., Pant K.P., Goodman N., Bamshad M. Analysis of genetic inheritance in a family quartet by whole-genome sequencing. Science. 2010;328:636–639. doi: 10.1126/science.1186802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Boycott K.M., Vanstone M.R., Bulman D.E., MacKenzie A.E. Rare-disease genetics in the era of next-generation sequencing: discovery to translation. Nat. Rev. Genet. 2013;14:681–691. doi: 10.1038/nrg3555. [DOI] [PubMed] [Google Scholar]
- 29.Bennett C.L., Christie J., Ramsdell F., Brunkow M.E., Ferguson P.J., Whitesell L., Kelly T.E., Saulsbury F.T., Chance P.F., Ochs H.D. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat. Genet. 2001;27:20–21. doi: 10.1038/83713. [DOI] [PubMed] [Google Scholar]
- 30.Caudy A.A., Reddy S.T., Chatila T., Atkinson J.P., Verbsky J.W. CD25 deficiency causes an immune dysregulation, polyendocrinopathy, enteropathy, X-linked-like syndrome, and defective IL-10 expression from CD4 lymphocytes. J. Allergy Clin. Immunol. 2007;119:482–487. doi: 10.1016/j.jaci.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 31.Boisson-Dupuis S., Kong X.F., Okada S., Cypowyj S., Puel A., Abel L., Casanova J.L. Inborn errors of human STAT1: allelic heterogeneity governs the diversity of immunological and infectious phenotypes. Curr. Opin. Immunol. 2012;24:364–378. doi: 10.1016/j.coi.2012.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Uzel G., Sampaio E.P., Lawrence M.G., Hsu A.P., Hackett M., Dorsey M.J., Noel R.J., Verbsky J.W., Freeman A.F., Janssen E. Dominant gain-of-function STAT1 mutations in FOXP3 wild-type immune dysregulation-polyendocrinopathy-enteropathy-X-linked-like syndrome. J. Allergy Clin. Immunol. 2013;131:1611–1623. doi: 10.1016/j.jaci.2012.11.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sampaio E.P., Hsu A.P., Pechacek J., Bax H.I., Dias D.L., Paulson M.L., Chandrasekaran P., Rosen L.B., Carvalho D.S., Ding L. Signal transducer and activator of transcription 1 (STAT1) gain-of-function mutations and disseminated coccidioidomycosis and histoplasmosis. J. Allergy Clin. Immunol. 2013;131:1624–1634. doi: 10.1016/j.jaci.2013.01.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Takezaki S., Yamada M., Kato M., Park M.J., Maruyama K., Yamazaki Y., Chida N., Ohara O., Kobayashi I., Ariga T. Chronic mucocutaneous candidiasis caused by a gain-of-function mutation in the STAT1 DNA-binding domain. J. Immunol. 2012;189:1521–1526. doi: 10.4049/jimmunol.1200926. [DOI] [PubMed] [Google Scholar]
- 35.van de Veerdonk F.L., Plantinga T.S., Hoischen A., Smeekens S.P., Joosten L.A., Gilissen C., Arts P., Rosentul D.C., Carmichael A.J., Smits-van der Graaf C.A. STAT1 mutations in autosomal dominant chronic mucocutaneous candidiasis. N. Engl. J. Med. 2011;365:54–61. doi: 10.1056/NEJMoa1100102. [DOI] [PubMed] [Google Scholar]
- 36.Baghdasaryan A., Chiba P., Trauner M. Clinical application of transcriptional activators of bile salt transporters. Mol. Aspects Med. 2013 doi: 10.1016/j.mam.2013.12.001. Published online December 12, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Strautnieks S.S., Bull L.N., Knisely A.S., Kocoshis S.A., Dahl N., Arnell H., Sokal E., Dahan K., Childs S., Ling V. A gene encoding a liver-specific ABC transporter is mutated in progressive familial intrahepatic cholestasis. Nat. Genet. 1998;20:233–238. doi: 10.1038/3034. [DOI] [PubMed] [Google Scholar]
- 38.Yandell M., Moore B., Salas F., Mungall C., MacBride A., White C., Reese M.G. Genome-wide analysis of human disease alleles reveals that their locations are correlated in paralogous proteins. PLoS Comput. Biol. 2008;4:e1000218. doi: 10.1371/journal.pcbi.1000218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fauci A.S. Alterations in Gastrointestinal Function. In: Fauci A.S., Braunwald E., Kasper D.L., Hauser S.L., Longo D.L., Jameson J.L., Loscalzo J., editors. Harrison’s Principles of Internal Medicine. McGraw-Hill Medical; New York: 2008. p. 237. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.