

Abstract
Tubulins, and microtubule polymers into which they incorporate, play critical mechanical roles in neuronal function during cell proliferation, neuronal migration, and postmigrational development: the three major overlapping events of mammalian cerebral cortex development. A number of neuronally expressed tubulin genes are associated with a spectrum of disorders affecting cerebral cortex formation. Such “tubulinopathies” include lissencephaly/pachygyria, polymicrogyria-like malformations, and simplified gyral patterns, in addition to characteristic extracortical features, such as corpus callosal, basal ganglia, and cerebellar abnormalities. Epilepsy is a common finding in these related disorders. Here we describe two unrelated individuals with infantile-onset epilepsy and abnormalities of brain morphology, harboring de novo variants that affect adjacent amino acids in a beta-tubulin gene TUBB2A. Located in a highly conserved loop, we demonstrate impaired tubulin and microtubule function resulting from each variant in vitro and by using in silico predictive modeling. We propose that the affected functional loop directly associates with the alpha-tubulin-bound guanosine triphosphate (GTP) molecule, impairing the intradimer interface and correct formation of the alpha/beta-tubulin heterodimer. This study associates mutations in TUBB2A with the spectrum of “tubulinopathy” phenotypes. As a consequence, genetic variations affecting all beta-tubulin genes expressed at high levels in the brain (TUBB2B, TUBB3, TUBB, TUBB4A, and TUBB2A) have been linked with malformations of cortical development.
Main Text
The correct formation of the cerebral cortex is critical to higher cognitive function in humans and requires an extremely coordinated sequence of events. Cerebral cortex development can be divided into three main stages: cell proliferation, neuronal migration, and postmigrational development.1 The importance of these precise and orchestrated events is reflected in the wide range of disease phenotypes that arise from their disruption, such as microcephaly (MIM 251200), lissencephaly (MIM 607432), and polymicrogyria (MIM 610031).1,2 Individuals with malformations of cortical development often display severe cognitive deficits and intractable epilepsy. Several genes are historically linked to migration disorders, however the recent emergence of tubulinopathies has provided new insights.1,3 Neuronally expressed tubulin genes associated with cortical malformations include TUBA1A (MIM 602529),4 TUBB2B (MIM 612850),5 TUBB3 (MIM 602661),6 TUBA8 (MIM 605742),7 TUBB (MIM 191130),8 TUBB4A (MIM 602662),9 and TUBG1 (MIM 191135).10
Alpha- and beta-tubulin proteins coassemble to form microtubule polymers, scaffold-like cytoskeletal structures implicated in a host of essential cellular functions.11–13 Microtubules are particularly important to neuronal function, especially during brain development when their dynamic properties are harnessed to perform specialized mechanical tasks such as mitotic division and proliferation of neuroblasts from their progenitors, migration of immature neurons from regions of proliferation in the ventricular and subventricular zones to the developing neocortex, and generation of axonal fibers that extend away from the cell body to mediate neuronal communication.13,14
We have previously described an overlapping spectrum of cortical phenotypes stemming from mutations in neuron-specific alpha- and beta-tubulin genes, ranging from complete agyria (smooth cortical surface) to a mildly simplified pattern of cortical gyration.3 In addition to cortical dysgeneses, extracortical abnormalities characteristic of tubulin gene involvement consist of agenesis/hypoplasia of the corpus callosum, dysmorphic basal ganglia and cerebellar hypoplasia.3,15,16 The breadth of cortical phenotypes that result from tubulin gene variation supports the notion that each tubulin isotype has unique, complex spatiotemporal-expression patterns and subtly dissimilar roles in microtubule polymer function within neuronal patterning and networking.17
In an ongoing genetic screening program of malformations of cerebral cortical development, we performed whole-exome sequencing (WES) in individuals in whom clinically available molecular genetic tests had failed to detect a mutation. The genetic analysis performed was subject to informed consent procedures and approved by the Institutional Review Boards at Seattle Children’s Hospital and University of Rochester Medical Center. We present two unrelated children harboring de novo missense variations in the beta-tubulin gene TUBB2A (MIM 615101) (Genomic RefSeq accession: NC_000006.11; mRNA RefSeq accession: NM_001069.2), located 70 kb telomeric to TUBB2B on chromosome 6p (with 98% mRNA sequence homology). Clinical records, electroencephalogram (EEG) tracings and reports, and magnetic resonance imaging (MRI) scans were reviewed retrospectively.
Subject LR05-160 was a male infant; the product of a 36.5-week gestation pregnancy, who presented with infantile spasms at 5 months of age. EEG showed hypsarrhythmia. Treatment with adrenocorticotropin (ACTH) caused side effects, and his medication was changed to topiramate. Brain MRI revealed a diffuse simplified gyral pattern with reduced volume of white matter, globular basal ganglia, and thalami, moderately enlarged third and lateral ventricles, thin and dysmorphic corpus callosum, mild brainstem hypoplasia with a flat pons, mild cerebellar vermis hypoplasia, and mildly enlarged posterior fossa (Figures 1A–1D). His subsequent course was significant for continued seizures and global developmental delay. At 8 months, his occipitofrontal head circumference (OFC) was between the 10th and 25th centiles. Chromosome analysis, fluorescence in situ hybridization for 17p13.3 deletion, and sequencing of DCX (MIM 300121) and PAFAH1B1 (MIM 605006) (encoding for doublecortin and LIS1 microtubule-associated proteins, respectively) were normal.
Figure 1.
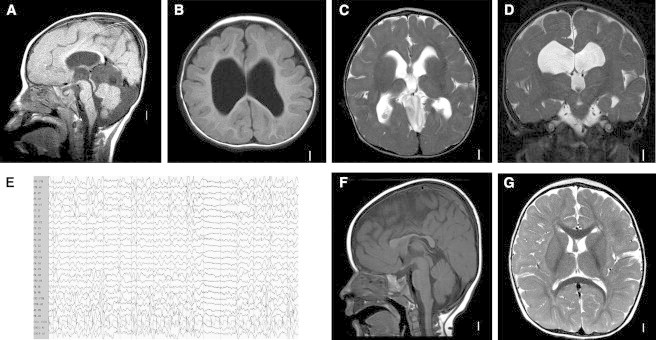
Magnetic Resonance Images and Electroencephalography Recordings of Affected Individuals
Representative MRI of subject LR05-160 showing rounded corpus callosum and enlarged third ventricle on sagittal views (A), simplified gyral pattern on T1 axial (B), globular basal ganglia and thalami with dilated lateral ventricles on axial (C) and coronal T2 (D) views. Subject DB12-007 had persistent multifocal epileptiform discharges at 11 months of age (E). The MRI findings of DB12-007 were less severe, with dysmorphic corpus callosum (F) but normal gyral pattern, basal ganglia, and thalami (G).
We performed WES of peripheral blood DNA from proband and both parents by using the Agilent SureSelect 50 Mb whole-exome capture kit, and sequence was generated on an Illumina HiSeq machine. Sequence was aligned to hg19 by using BWA v.0.6.1, and single-nucleotide variants (SNVs) and indels were called by using GATK v.1.3. Annotation of variants was performed with SeattleSeq Annotation 134. A de novo heterozygous c.741C>G [p.Asn247Lys] variation was identified in TUBB2A (Figure 2). Validation was achieved by standard bidirectional Sanger methods (primer sequences provided in Table S1 available online).
Figure 2.
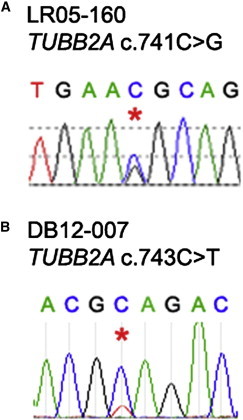
Heterozygous Mutations in TUBB2A Identified by Whole-Exome Sequencing
Asterisks highlight (A) a c.741C>G variation in LR05-160 resulting in a p.Asn247Lys substitution and (B) a c.743C>T change (p.Ala248Val) identified in DB12-007. Both were confirmed to be de novo by bidirectional Sanger sequencing of the parents.
Subject DB12-007 was a term infant girl who presented with hypotonia at 4 months of age. At 11 months an EEG showed frequent multifocal epileptiform discharges (Figure 1E), and electrographic seizures characterized by brief bursts of diffuse spike and polyspike activity followed by amplitude attenuation. Clinically, the seizures were subtle vertical eye movements. She was treated with valproic acid and topiramate. The seizures resolved and she was weaned off antiepileptics at 2 years of age. Brain MRI revealed a dysmorphic corpus callosum (Figures 1F and 1G) but normal basal ganglia and thalami. At last follow-up at 3 years of age, she was nonverbal and nonambulatory with generalized hypotonia. Her OFC was on the 25th centile.
Subject DB12-007 and both parents initially had WES performed from blood-derived DNA in a clinical laboratory. However, when no pathogenic variants in genes known to harbor mutations in similar phenotypes were found, the family approached ARP for data reanalysis. BAM files were obtained, and variants were recalled with GATK v.2.3-9. Annotation of variants was performed with SeattleSeq Annotation 137. A de novo heterozygous c.743C>T (p.Ala248Val) TUBB2A variant was identified (Figure 2), confirmed by Sanger methods.
De novo, autosomal-recessive, X-linked nonsynonymous SNVs and frameshift indels were identified with the publicly available SOLVE-Brain package, which was in turn used to annotate genes with candidate variants for brain expression. In both families, common variants were identified by filtering against the NHLBI Exome Variant Server, as well as dbSNP version 137. Exome read-depth metrics are summarized in Table S2. A 63-fold variation in TUBB2A mRNA content correlating with presence of two regulatory polymorphisms (c.–101C>T; c.–157A>G) has been previously demonstrated.18 According to our exome data, neither LR05-160 nor DB12-007 carry either of these polymorphisms. No potentially pathogenic variations were identified in any other genes in the affected individuals.
Both TUBB2A amino-acid substitutions affect highly conserved residues in a critical GTP-binding site (Figure 5). Neither variant was listed in online polymorphism databases including dbSNP, 1000 Genomes, and the NHLBI Exome Variant Server. Both p.Asn247Lys and p.Ala248Val are predicted to be “not tolerated” by SIFT (Scores 0 & 0.01, respectively) and highly likely to interfere with protein function by Align GVGD (GD = 93.88 [Class C65] & GD = 65.28 [Class C65]). PolyPhen-2 predicts p.Asn247Lys to be “probably damaging” (score 0.999) but p.Ala248Val as benign (0.004).
Figure 5.
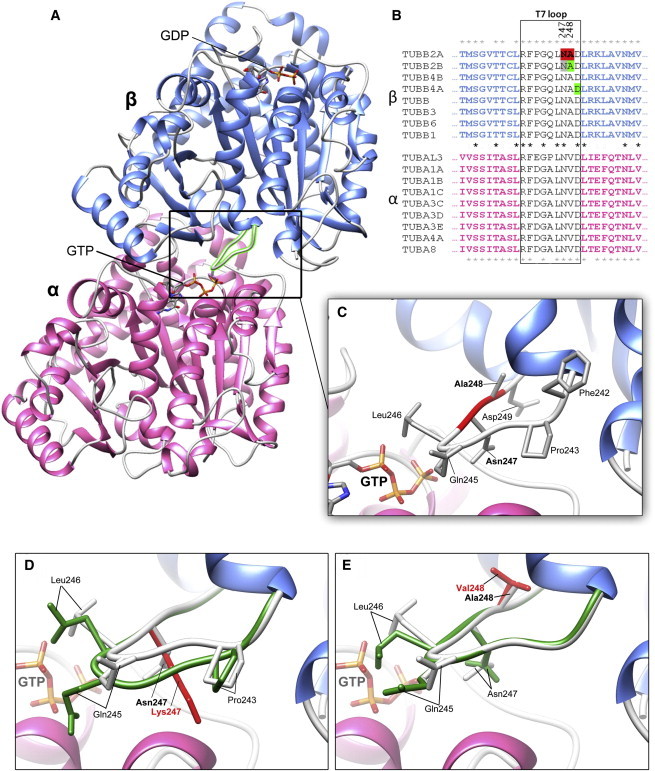
The Highly Conserved TUBB2A “T7” Loop Is Positioned at the Intradimer Interface
Structural predictions of WT and variant TUBB2A were generated via a previously described homology modeling pipeline.28 The best homology attained for this model was based on 41% identity of a beta-tubulin template (PDB: 4I4T).29
(A) Asn247 and Ala248 are located in the “T7” loop (box) between 7th and 8th alpha-helix of the β-subunit. This loop is located at the intradimer interface (between subunits of the heterodimer) in close proximity to GTP bound to the α-subunit.
(B) Asn247 and Ala248 are highlighted (red) in the TUBB2A amino-acid sequence. The T7 loop (box) is highly conserved, not only between β-tubulins isotypes, but also throughout α- and β-tubulin families (conserved residues indicated by asterisks). Asn247 is preserved throughout tubulin evolution, suggesting that it is critical to nucleotide binding at both the α- and β-subunits. Ala248 is conserved throughout β-tubulin, but not α-tubulin isotypes, suggesting that the alanine plays a critical role in GTP binding at the intradimer, but not interdimer, interfaces (between adjacent heterodimers). Support for pathogenicity of variants at the T7 loop is provided by the recently described TUBB2B p.Ala248Val and TUBB4A p.Asp249Asn variants (positions highlighted in green),9,16 as well as a TUBB2B p.Asn247Ser substitution identified in mice (shown in gray).19
(C) The Asn247 side chain extends into the intradimer interface. Substitution of a polar asparagine residue with a basic lysine residue is likely to directly affect electrostatic interactions at this interface. The methyl side chain of Ala248 does not extend toward the α-tubulin subunit and substitution of two nonpolar residues at this position would suggest a more modest influence on intradimer interactions.
(D and E) Closer examination of the T7 loop highlights the conformational changes predicted for the (D) p.Asn247Lys and (E) p.Ala248Val substitutions (variant T7 loops shown in dark green; WT loops shown in white). p.Asn247Lys is predicted to have a notable effect on T7 loop conformation, likely to result in spacial conflicts along the interface. The consequences of p.Ala248Val appear to be more subtle, because no marked changes to loop conformation are predicted. Rearrangements are not exclusive to residues 247 and 248, however; Gln245 and Leu246 side chains appear to assume orientations in both variant models when compared to WT, as well as Pro243 in p.Asn247Lys.
To investigate the functional consequences of p.Asn247Lys and p.Ala248Val in vitro, wild-type (WT) and variant TUBB2A expression constructs were transfected into human embryonic kidney 293 (HEK293) cells. Both variant proteins demonstrate impaired ability to coassemble with endogenous alpha-tubulin subunits and integrate into microtubule polymers (Figure 3). TUBB2A p.Asn247Lys does not incorporate into the microtubule polymer network but remains unpolymerized throughout the cytoplasm in vitro. This suggests that the substitution prevents correct protein processing through several possible mechanisms such as folding, heterodimer formation, or integration into growing microtubule polymers. A p.Asn247Ser substitution in TUBB2B has recently been identified in the brdp mouse model with cortical development abnormalities.19 This highlights Asn247 as a critical residue to tubulin function, especially during the development of the cerebral cortex. Interestingly, the authors demonstrated the successful incorporation of the variant protein into microtubule polymers in vitro. This suggests that the introduction of a large basic lysine side chain at this position in TUBB2A p.Asn247Lys might significantly affect intradimer and/or GTP interactions, subsequently influencing alpha/beta-tubulin heterodimer formation and coassembly into microtubules.
Figure 3.
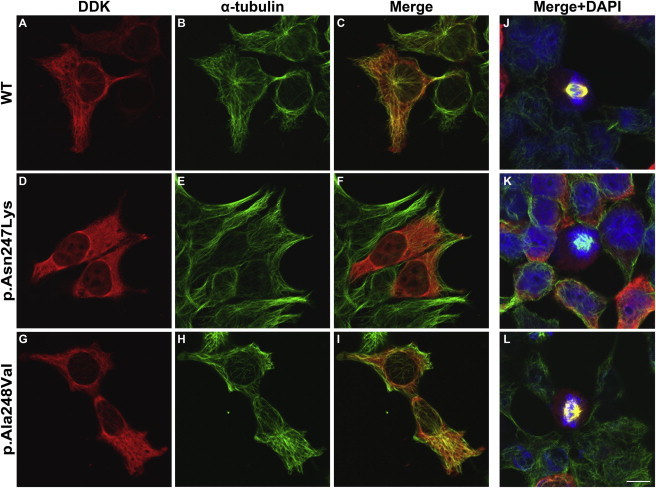
TUBB2A Variants Have Mixed Success Incorporating into Microtubule Polymers In Vitro
WT and variant TUBB2A expression constructs were transfected into HEK293 cells to observe incorporation into in vitro microtubule polymers during interphase (A–I) and mitosis (J–L). A WT C-terminally DDK-tagged human TUBB2A expression construct (OriGene Technologies; pCMV6-entry) was modified to reflect p.Asn247Lys and p.Ala248Val substitutions using QuikChange site-directed mutagenesis (Stratagene, UK) and entire transgene-coding regions were sequence validated following maxiprep yields (QIAgen). HEK293 cells, cultured in Dulbecco’s modified Eagle’s medium (GIBCO) supplemented with 10% fetal calf serum (GIBCO) and 1% penicillin/streptomycin (Sigma), were seeded (4 × 104 cells per well) on 16 mm poly-L-lysine (Sigma) coated glass coverslips. After 24 hr, TUBB2A expression constructs were transfected with TurboFectin 8.0 transfection reagent (OriGene). Twenty-four hours after transfection, HEK293 cells were methanol fixed and permeabilized with Phosphate-Buffered Saline (PBS; Sigma) plus 0.5% Triton X-100 (Sigma) to enable immunostaining of intracellular epitopes. Cells were sequentially stained with antibodies specific to transgenic and endogenous tubulin protein. First, transgenic TUBB2A was immunostained (A, D, G) with Anti-DDK (IgG2a) mouse monoclonal antibody (OriGene; 1:1,500 dilution), biotin-conjugated goat polyclonal secondary antibody to mouse immunoglobulin G2a (IgG2a) (Abcam) and a streptavidin-Alexa Fluor® 594 conjugate (Life Technologies). Following sufficient washes and a 1 hr blocking step, alpha-tubulin protein was stained (to detect endogenous microtubule network; B, E, H) with monoclonal anti-α-tubulin (IgG1) primary antibody (Sigma; 1:500 dilution) and a Cy2-conjugated goat anti-mouse IgG1 secondary antibody (Jackson ImmunoResearch Laboratories Inc.). Nuclear material was visualized with 4’,6-diamidino-2-phenylindole (DAPI; J–L). Immunostained cells were mounted onto glass slides using ProLong Gold Antifade Reagent (Life Technologies) and stored light-protected at 4°C overnight before confocal microscope image acquisition (Zeiss LSM 710 & Zen Software). All antibody dilutions, washes, and blocking steps were performed in PBS plus 0.1% Triton and 2% w/v Bovine Serum Albumin (Sigma) and negative controls were used to ensure no cross-reactivity of antibodies was observed. While TUBB2A p.Asn247Lys does not demonstrate microtubule integration (D, E, F, K), TUBB2A p.Ala248Val does incorporate into cytoskeletal network (G, H, I, L) but with less well defined polymer fibers in comparison with WT (A, B, C, J). Images were acquired blind to construct type and are representative of >95% of cells observed. Scale bar represents 10 μm.
TUBB2A p.Ala248Val does incorporate into the in vitro cytoskeleton network but with less well-defined microtubule polymers and an increased proportion of the visible variant beta-tubulin remaining unpolymerized in the cell cytoplasm in comparison with WT-expressing cells. This implies that p.Ala248Val has partial and subtle changes with at least some successfully folded and dimerized alpha/beta tubulin subunits. Consequently, the phenotype in this individual is most likely explained by a reduced-rate of heterodimer incorporation.
Following cold-induced depolymerization, TUBB2A p.Ala248Val demonstrates a diminished rate of reintegrating into growing microtubules in comparison to WT protein (Figure 4), supporting the notion that this substitution subtly perturbs polymer incorporation. Microtubule polymerization was not affected by the presence of the p.Asn247Lys amino-acid substitution (data not shown), offering confirmation that this variant does not integrate with the cytoskeletal network whatsoever. We additionally examined the rate of depolymerization during the incubation period at 4°C; however, neither p.Asn247Lys nor p.Ala248Val caused any notable changes in microtubule collapse (data not shown). This would suggest that, in the case of the p.Ala248Val substitution, incorporation of the variant subunit would not subsequently alter polymer stability sufficiently to induce microtubule depolymerization at an accelerated rate.
Figure 4.
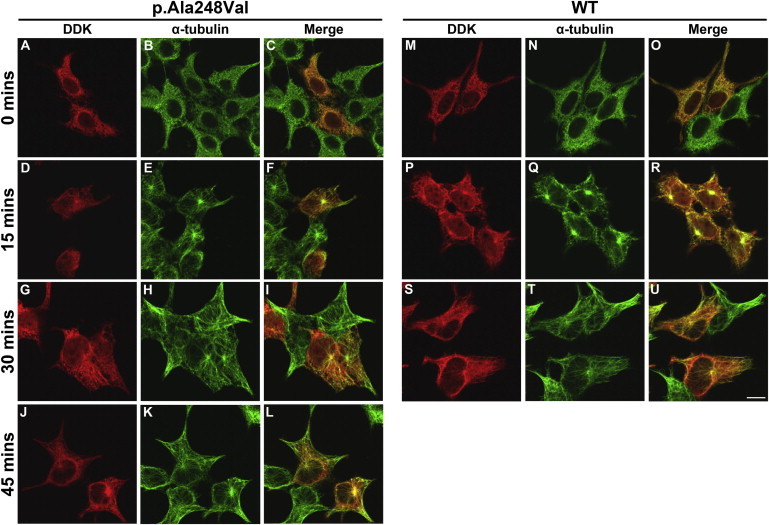
Effect of TUBB2A p.Ala248Val on Microtubule Reassembly following Cold-Induced Depolymerization
To examine the effect of either substitution on microtubule dynamics in vitro, HEK293 cells expressing WT and variant TUBB2A were transferred to 4°C to induce depolymerization of the microtubule polymer network. After 30 min, coverslips were returned to 37°C to promote cytoskeleton polymer rescue. At 0 (A–C, M–O), 15 (D–F, P–R), 30 (G–I, S–U), and 45 min (J–L), cells were removed and methanol fixed prior to immunocytochemical examination, to visualize the rate of microtubule growth in the presence of each TUBB2A construct. WT tubulin (M–U) demonstrates microtubule repolymerization at a roughly stoichiometric rate to endogenous tubulin. After 15 and 30 min (D–I), microtubule rescue in TUBB2A p.Ala248Val-expressing cells is visible in the endogenous tubulin, but with no fibers detectable in the DDK channel. TUBB2A p.Ala248Val incorporation is only evident after 45 min at 37°C (J–L). Images were acquired blind to construct type and are representative of >95% of cells observed. Scale bar represents 10 μm.
The two variants affect adjacent amino-acid residues on a loop located at the intradimer interface, i.e., between alpha- and beta-tubulin subunits within the same heterodimer (Figure 5A). This loop is highly conserved throughout both beta- and alpha-tubulin families, and extends to the prokaryotic ortholog FtsZ, highlighting a functionally critical motif for correct tubulin protein function.20 Upon dimerization, this loop interacts with the GTP nucleotide bound to the alpha-tubulin subunit.20,21 This interaction with GTP is critical in bringing together the alpha- and beta-tubulin subunits within a heterodimer and, because newly formed heterodimers cannot be released from tubulin-folding chaperone complexes in the absence of GTP at this position, it is therefore essential for subsequent incorporation into microtubule polymers.22
On closer inspection of this region (Figures 5D and 5E), the polar side chain of Asn247 is predicted to extend from this loop toward the GTP. The introduction of a relatively large, basic lysine group into the alpha/beta-tubulin interface is likely to affect both intradimer and GTP interactions. The smaller, nonpolar side chain of Ala248 points away from the GTP nucleotide and back toward the beta-subunit core, and substitution of this alanine with another nonpolar valine residue would produce a substantially milder impact on structure and function than p.Asn247Lys.
In addition to the specific amino-acid residue changes, the conformational alterations resulting from the p.Asn247Lys substitution were more profound than those of p.Ala248Val (Figures 5D and 5E). The loop structure does not differ much as a result of the alanine to valine substitution, whereas there is a more marked change in configuration with the lysine substitution at position 247.
The TUBB2A p.Ala248Val substitution is expected to have much less profound structural consequences, supported by successful incorporation into microtubule polymers in vitro. It should be noted that alpha-tubulin proteins harbor a valine at the equivalent position within the T7 loop (Figure 5), which might explain the milder in vitro and in vivo consequences observed. The identification of an identical p.Ala248Val substitution in TUBB2B (99% amino-acid sequence homology) in individuals with polymicrogyric cortical malformations further supports pathogenicity of this amino-acid change in TUBB2A.16
The variants identified in TUBB2A contribute to the growing list of tubulin gene mutations associated with impaired brain development in humans. Mutations in all beta-tubulin genes expressed highly in the brain, i.e., TUBB2B, TUBB3, TUBB, TUBB4A, and TUBB2A, are currently linked to a recognizable spectrum of brain malformations involving the cortex, basal ganglia, corpus callosum, brainstem, and cerebellum.3,5,6,8,9 On the basis of the in vitro functional analysis and in silico predictive modeling, we propose a mechanism of disease involving impaired GTP interaction at the intradimer interface between alpha- and beta-tubulin subunits. Extensive investigation into the GTP-interacting properties of both variants would be further required to elucidate the exact pathogenic mechanism in affected individuals.
The phenotypes observed in these two children with TUBB2A mutations are less severe than reported in most other children with tubulinopathies,3 suggesting that this beta-tubulin isotype might assume a background role in neuronal function during brain development. Although malformations of the corpus callosum were seen in both individuals, only the more severely affected boy with the p.Asn247Lys substitution had malformation of the cerebral cortex (the simplified gyral pattern), basal ganglia, thalami, brainstem, and cerebellum. Similarly, two of the three individuals reported with TUBB mutations had a normal cortex (one had focal polymicrogyria), whereas TUBB2B and a subset of TUBB3 mutations are usually associated with a polymicrogyria-like cortical dysplasia (MIM 610031 and 614039).3,5,6,8 TUBA1A and TUBG1 variants are most commonly associated with more severe lissencephaly (also known as pachygyria) (MIM 611603 and 615412).3,10,15,23 Extensive polymicrogyria and optic atrophy (MIM 613180) was reported in four children from two consanguineous families with mutations in TUBA8.7 Abnormal basal ganglia morphology, callosal abnormalities, and cerebellar hypoplasia or dysgenesis are most commonly observed in all tubulinopathies.3,5,6,8,10,15,23 Mutations in TUBB3, which result in congenital fibrosis of the extraocular muscles (MIM 600638), have not been associated with a cortical dysplasia, but corpus callosum abnormalities and dysmorphic basal ganglia are frequently observed.24 Collectively, these associations demonstrate that tubulinopathies are associated with a wide and overlapping range of brain malformations.
The roles of individual tubulin isotypes within the intricate mechanisms of cerebrocortical development remain unclear, owing to extremely high levels of sequence and structural homologies, in addition to overlapping and often indistinguishable expression patterns. TUBB2A is thought to comprise around 30% of all beta-tubulin within the brain,25 however Breuss and colleagues8 have recently demonstrated that relative TUBB2A expression at both 13 and 22 weeks gestation in humans is notably less than TUBB2B, TUBB, and TUBB3. Similarly in mice, Tubb2a is expressed at a lower level than that of the orthologs of these genes throughout development, despite rising sharply around the time of birth. This again suggests that although being present throughout, this beta-tubulin isotype carries out relatively minor functions specific to brain development, concurrent with the mild brain phenotypes observed in affected individuals.
A specific role for TUBB2A in neuronal function might be its interaction with the E3 ubiquitin-protein ligase ZNRF1, which is thought to mediate regulation of neuritogenesis via interaction with the TUBB2A ortholog in mice.26 This ligase has also been linked to Wallerian degeneration, via a signaling pathway inducing microtubule reorganization.27 A link between cortical development and axonal degradation is unclear from the available data but could implicate a mechanism of microtubule-dependent axon guidance during latter stages of cortical organization and synaptogenesis.
Acknowledgments
The authors would like to thank the affected individuals and their families for participation in this study. We would also like to thank Owain Howell for providing immunofluorescent antibodies to perform the in vitro functional analysis. Research reported in this publication was supported by the (UK) National Institute of Social Care and Health Research (M.I.R., D.T.P.), the Wales Epilepsy Research Network (WERN – M.I.R., S.-K.C., D.T.P.) and Waterloo Foundation (M.I.R.), and by the (USA) National Institute for Neurological Disorders and Stroke (NINDS) of the National Institutes of Health under award numbers K08NS078054 (to A.R.P.), K12NS066098 (to L.E.S.), and R01NS050375 and R01NS058721 (to W.B.D.).
Contributor Information
Mark I. Rees, Email: m.i.rees@swansea.ac.uk.
William B. Dobyns, Email: wbd@u.washington.edu.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes, http://browser.1000genomes.org
World Health Organization Align GVGD, http://agvgd.iarc.fr/agvgd_input.php/
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
PolyPhen-2, http://www.genetics.bwh.harvard.edu/pph2/
RCSB Protein Data Bank, http://www.rcsb.org/pdb/
SeattleSeq Annotation 134, http://snp.gs.washington.edu/SeattleSeqAnnotation134/
SeattleSeq Annotation 137, http://snp.gs.washington.edu/SeattleSeqAnnotation137/
SOLVE-Brain, https://paciorkowski-lab.urmc.rochester.edu/solve_brain
References
- 1.Barkovich A.J., Guerrini R., Kuzniecky R.I., Jackson G.D., Dobyns W.B. A developmental and genetic classification for malformations of cortical development: update 2012. Brain. 2012;135:1348–1369. doi: 10.1093/brain/aws019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barkovich A.J., Kuzniecky R.I., Jackson G.D., Guerrini R., Dobyns W.B. A developmental and genetic classification for malformations of cortical development. Neurology. 2005;65:1873–1887. doi: 10.1212/01.wnl.0000183747.05269.2d. [DOI] [PubMed] [Google Scholar]
- 3.Cushion T.D., Dobyns W.B., Mullins J.G., Stoodley N., Chung S.K., Fry A.E., Hehr U., Gunny R., Aylsworth A.S., Prabhakar P. Overlapping cortical malformations and mutations in TUBB2B and TUBA1A. Brain. 2013;136:536–548. doi: 10.1093/brain/aws338. [DOI] [PubMed] [Google Scholar]
- 4.Keays D.A., Tian G., Poirier K., Huang G.J., Siebold C., Cleak J., Oliver P.L., Fray M., Harvey R.J., Molnár Z. Mutations in alpha-tubulin cause abnormal neuronal migration in mice and lissencephaly in humans. Cell. 2007;128:45–57. doi: 10.1016/j.cell.2006.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jaglin X.H., Poirier K., Saillour Y., Buhler E., Tian G., Bahi-Buisson N., Fallet-Bianco C., Phan-Dinh-Tuy F., Kong X.P., Bomont P. Mutations in the beta-tubulin gene TUBB2B result in asymmetrical polymicrogyria. Nat. Genet. 2009;41:746–752. doi: 10.1038/ng.380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Poirier K., Saillour Y., Bahi-Buisson N., Jaglin X.H., Fallet-Bianco C., Nabbout R., Castelnau-Ptakhine L., Roubertie A., Attie-Bitach T., Desguerre I. Mutations in the neuronal ß-tubulin subunit TUBB3 result in malformation of cortical development and neuronal migration defects. Hum. Mol. Genet. 2010;19:4462–4473. doi: 10.1093/hmg/ddq377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Abdollahi M.R., Morrison E., Sirey T., Molnar Z., Hayward B.E., Carr I.M., Springell K., Woods C.G., Ahmed M., Hattingh L. Mutation of the variant alpha-tubulin TUBA8 results in polymicrogyria with optic nerve hypoplasia. Am. J. Hum. Genet. 2009;85:737–744. doi: 10.1016/j.ajhg.2009.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Breuss M., Heng J.I., Poirier K., Tian G., Jaglin X.H., Qu Z., Braun A., Gstrein T., Ngo L., Haas M. Mutations in the β-tubulin gene TUBB5 cause microcephaly with structural brain abnormalities. Cell Rep. 2012;2:1554–1562. doi: 10.1016/j.celrep.2012.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Simons C., Wolf N.I., McNeil N., Caldovic L., Devaney J.M., Takanohashi A., Crawford J., Ru K., Grimmond S.M., Miller D. A de novo mutation in the β-tubulin gene TUBB4A results in the leukoencephalopathy hypomyelination with atrophy of the basal ganglia and cerebellum. Am. J. Hum. Genet. 2013;92:767–773. doi: 10.1016/j.ajhg.2013.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Poirier K., Lebrun N., Broix L., Tian G., Saillour Y., Boscheron C., Parrini E., Valence S., Pierre B.S., Oger M. Mutations in TUBG1, DYNC1H1, KIF5C and KIF2A cause malformations of cortical development and microcephaly. Nat. Genet. 2013;45:639–647. doi: 10.1038/ng.2613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mitchison T., Kirschner M. Dynamic instability of microtubule growth. Nature. 1984;312:237–242. doi: 10.1038/312237a0. [DOI] [PubMed] [Google Scholar]
- 12.Desai A., Mitchison T.J. Microtubule polymerization dynamics. Annu. Rev. Cell Dev. Biol. 1997;13:83–117. doi: 10.1146/annurev.cellbio.13.1.83. [DOI] [PubMed] [Google Scholar]
- 13.Guzik B.W., Goldstein L.S. Microtubule-dependent transport in neurons: steps towards an understanding of regulation, function and dysfunction. Curr. Opin. Cell Biol. 2004;16:443–450. doi: 10.1016/j.ceb.2004.06.002. [DOI] [PubMed] [Google Scholar]
- 14.Dehmelt L., Halpain S. Actin and microtubules in neurite initiation: are MAPs the missing link? J. Neurobiol. 2004;58:18–33. doi: 10.1002/neu.10284. [DOI] [PubMed] [Google Scholar]
- 15.Kumar R.A., Pilz D.T., Babatz T.D., Cushion T.D., Harvey K., Topf M., Yates L., Robb S., Uyanik G., Mancini G.M. TUBA1A mutations cause wide spectrum lissencephaly (smooth brain) and suggest that multiple neuronal migration pathways converge on alpha tubulins. Hum. Mol. Genet. 2010;19:2817–2827. doi: 10.1093/hmg/ddq182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Amrom D., Tanyalçin I., Verhelst H., Deconinck N., Brouhard G., Décarie J.C., Vanderhasselt T., Das S., Hamdan F., Lissens W. Polymicrogyria with dysmorphic basal ganglia? Think tubulin! Clin. Genet. 2013 doi: 10.1111/cge.12141. Published online March 15, 2013. [DOI] [PubMed] [Google Scholar]
- 17.Tischfield M.A., Engle E.C. Distinct alpha- and beta-tubulin isotypes are required for the positioning, differentiation and survival of neurons: new support for the ‘multi-tubulin’ hypothesis. Biosci. Rep. 2010;30:319–330. doi: 10.1042/BSR20100025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Leandro-García L.J., Leskelä S., Jara C., Gréen H., Avall-Lundqvist E., Wheeler H.E., Dolan M.E., Inglada-Perez L., Maliszewska A., de Cubas A.A. Regulatory polymorphisms in β-tubulin IIa are associated with paclitaxel-induced peripheral neuropathy. Clin. Cancer Res. 2012;18:4441–4448. doi: 10.1158/1078-0432.CCR-12-1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stottmann R.W., Donlin M., Hafner A., Bernard A., Sinclair D.A., Beier D.R. A mutation in Tubb2b, a human polymicrogyria gene, leads to lethality and abnormal cortical development in the mouse. Hum. Mol. Genet. 2013;22:4053–4063. doi: 10.1093/hmg/ddt255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Löwe J., Li H., Downing K.H., Nogales E. Refined structure of alpha beta-tubulin at 3.5 A resolution. J. Mol. Biol. 2001;313:1045–1057. doi: 10.1006/jmbi.2001.5077. [DOI] [PubMed] [Google Scholar]
- 21.Nogales E., Whittaker M., Milligan R.A., Downing K.H. High-resolution model of the microtubule. Cell. 1999;96:79–88. doi: 10.1016/s0092-8674(00)80961-7. [DOI] [PubMed] [Google Scholar]
- 22.Lewis S.A., Tian G., Cowan N.J. The alpha- and beta-tubulin folding pathways. Trends Cell Biol. 1997;7:479–484. doi: 10.1016/S0962-8924(97)01168-9. [DOI] [PubMed] [Google Scholar]
- 23.Poirier K., Keays D.A., Francis F., Saillour Y., Bahi N., Manouvrier S., Fallet-Bianco C., Pasquier L., Toutain A., Tuy F.P. Large spectrum of lissencephaly and pachygyria phenotypes resulting from de novo missense mutations in tubulin alpha 1A (TUBA1A) Hum. Mutat. 2007;28:1055–1064. doi: 10.1002/humu.20572. [DOI] [PubMed] [Google Scholar]
- 24.Tischfield M.A., Baris H.N., Wu C., Rudolph G., Van Maldergem L., He W., Chan W.M., Andrews C., Demer J.L., Robertson R.L. Human TUBB3 mutations perturb microtubule dynamics, kinesin interactions, and axon guidance. Cell. 2010;140:74–87. doi: 10.1016/j.cell.2009.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Leandro-García L.J., Leskelä S., Landa I., Montero-Conde C., López-Jiménez E., Letón R., Cascón A., Robledo M., Rodríguez-Antona C. Tumoral and tissue-specific expression of the major human beta-tubulin isotypes. Cytoskeleton (Hoboken) 2010;67:214–223. doi: 10.1002/cm.20436. [DOI] [PubMed] [Google Scholar]
- 26.Yoshida K., Watanabe M., Hatakeyama S. ZNRF1 interacts with tubulin and regulates cell morphogenesis. Biochem. Biophys. Res. Commun. 2009;389:506–511. doi: 10.1016/j.bbrc.2009.09.011. [DOI] [PubMed] [Google Scholar]
- 27.Wakatsuki S., Saitoh F., Araki T. ZNRF1 promotes Wallerian degeneration by degrading AKT to induce GSK3B-dependent CRMP2 phosphorylation. Nat. Cell Biol. 2011;13:1415–1423. doi: 10.1038/ncb2373. [DOI] [PubMed] [Google Scholar]
- 28.Mullins J.G., Chung S.K., Rees M.I. Fine architecture and mutation mapping of human brain inhibitory system ligand gated ion channels by high-throughput homology modeling. Adv Protein Chem Struct Biol. 2010;80:117–152. doi: 10.1016/B978-0-12-381264-3.00004-7. [DOI] [PubMed] [Google Scholar]
- 29.Prota A.E., Bargsten K., Zurwerra D., Field J.J., Díaz J.F., Altmann K.H., Steinmetz M.O. Molecular mechanism of action of microtubule-stabilizing anticancer agents. Science. 2013;339:587–590. doi: 10.1126/science.1230582. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.