

Abstract
Rod-cone dystrophy (RCD), also known as retinitis pigmentosa, is a progressive inherited retinal disorder characterized by photoreceptor cell death and genetic heterogeneity. Mutations in many genes have been implicated in the pathophysiology of RCD, but several others remain to be identified. Herein, we applied whole-exome sequencing to a consanguineous family with one subject affected with RCD and identified a homozygous nonsense mutation, c.226C>T (p.Arg76∗), in KIZ, which encodes centrosomal protein kizuna. Subsequent Sanger sequencing of 340 unrelated individuals with sporadic and autosomal-recessive RCD identified two other subjects carrying pathogenic variants in KIZ: one with the same homozygous nonsense mutation (c.226C>T [p.Arg76∗]) and another with compound-heterozygous mutations c.119_122delAACT (p.Lys40Ilefs∗14) and c.52G>T (p.Glu18∗). Transcriptomic analysis in mice detected mRNA levels of the mouse ortholog (Plk1s1) in rod photoreceptors, as well as its decreased expression when photoreceptors degenerated in rd1 mice. The presence of the human KIZ transcript was confirmed by quantitative RT-PCR in the retina, the retinal pigment epithelium, fibroblasts, and whole-blood cells (highest expression was in the retina). RNA in situ hybridization demonstrated the presence of Plk1s1 mRNA in the outer nuclear layer of the mouse retina. Immunohistology revealed KIZ localization at the basal body of the cilia in human fibroblasts, thus shedding light on another ciliary protein implicated in autosomal-recessive RCD.
Main Text
Rod-cone dystrophy (RCD), also known as retinitis pigmentosa (MIM 268000), is a heterogeneous group of inherited retinal disorders affecting rod photoreceptors in the majority of cases and causing secondary cone degeneration.1 Population-based studies have indicated that there are one million affected individuals worldwide.1 Subjects diagnosed with RCD initially complain of night blindness due to rod dysfunction, as well as subsequent progressive constriction of their visual field, abnormal color vision, and eventually loss of central vision due to cone photoreceptor involvement.1 RCD is inherited as a Mendelian trait in most cases; 30%–40% is autosomal dominant, 50%–60% is autosomal recessive, and 5%–15% is X-linked.1
Candidate-gene approaches—for example, those comparing human phenotypes to similar phenotypes observed in animal models—are widely used for identifying gene defects leading to inherited retinal diseases.2–5 However, with the emergence of massively parallel sequencing techniques, considerable efforts are now being made to report known and novel genes implicated in the pathophysiology of inherited retinal disease.6–9 In the present study, we applied whole-exome sequencing (WES) to five members (including one affected by RCD) of a consanguineous family (family A [128], Figure 1) to identify the underlying gene defect. We detected a homozygous nonsense mutation, c.226C>T (p.Arg76∗), in the third exon of KIZ, coding for centrosomal protein kizuna (KIZ).
Figure 1.
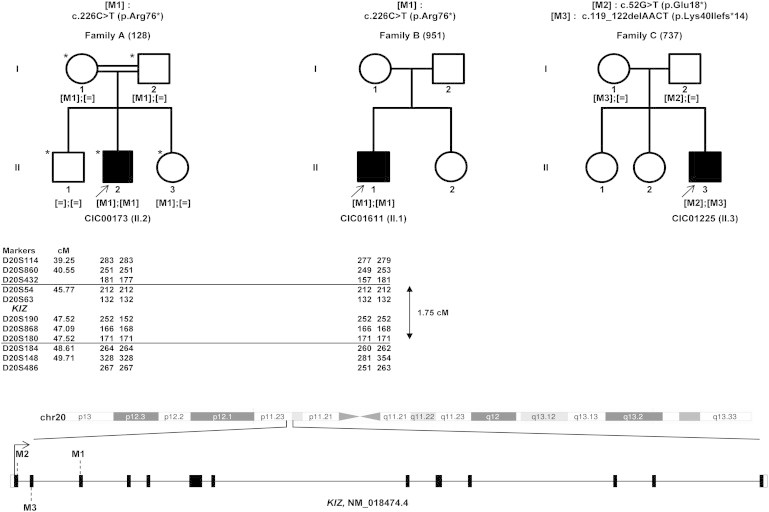
Identification of Homozygous and Compound-Heterozygous KIZ Variants in Three Unrelated Families Affected by Autosomal-Recessive RCD
Family A (left), family B (middle), and family C (right) were analyzed in the present study. Individuals highlighted with an asterisk were screened with WES. Affected individuals are marked with an arrow. A homozygous nonsense mutation, c.226C>T (p.Arg76∗), in KIZ was identified in the affected index subject from family A and cosegregated with the phenotype. Subsequent direct Sanger sequencing of KIZ in 340 individuals with autosomal-recessive RCD identified two other subjects with KIZ mutations: the affected index subject in family B had the homozygous c.226C>T (p.Arg76∗) mutation, and the affected index subject in family C had compound-heterozygous mutations c.52G>T (p.Glu18∗) and c.119_122delAACT (p.Lys40Ilefs∗14). Haplotype analysis performed for investigating whether the c.226C>T (p.Arg76∗) variant represents a founder mutation in families A and B is shown under each pedigree symbol. Microsatellite marker locations according to Marshfield genetic maps (Marshfield Laboratories) are shown in cM. Numbers indicate the allele size in nucleotides for each microsatellite. Both subjects were found to share a common haplotype of five polymorphic microsatellites (i.e., DS20S54, DS20S63, DS20S190, DS20S868, and DS20S180; delineated by two horizontal lines) flanking KIZ and spanning ≈1.75 cM (1.18 Mb). Filled and unfilled symbols indicate affected and unaffected status, respectively. Square boxes indicate males, and circles indicate females. In the schematic representation of the structure of KIZ (RefSeq NM_018474.4; harboring 13 exons), filled and unfilled boxes represent coding and noncoding exonic regions, respectively. M1, M2, and M3 depict the positions of the mutations identified in the current study.
The study protocol was conducted in accordance with the Declaration of Helsinki, national guidelines, and the regional ethics committee. Prior to testing and after explanation of the study and its potential outcome, informed consent was obtained from RCD subjects and their family members. Each subject underwent an ophthalmic examination with clinical assessment as previously described.10 At a time when next-generation sequencing (NGS) approaches were not commonly used, index subject CIC00173 II.2 (family A, Figure 1) was excluded upon screening by microarray analysis and direct Sanger sequencing for known mutations in EYS and C2orf71 (a major and a minor gene, respectively, implicated in RCD).11,12 Because exon ORF15 in RPGR (MIM 312610) is not targeted by existing NGS panels,13 we also analyzed it by Sanger sequencing but found no pathogenic variant. Subsequently, we performed targeted NGS on the index subject’s DNA by using a panel of 120 genes previously found to carry mutations in retinal diseases (this panel was modified and improved since our previous study).13 This survey did not reveal any pathogenic variant, and we therefore proceeded to WES.
Exons of DNA samples were captured and investigated as reported before with in-solution enrichment methodology (SureSelect Human All Exon Kits version 3, Agilent) and NGS (Illumina HiSeq, Illumina). Image analysis and base calling were performed with real-time analysis software (Illumina).6,9,13 Bioinformatic analysis of sequencing data was based on a pipeline (Consensus Assessment of Sequence and Variation 1.8, Illumina) that performs alignment, variant calling (single-nucleotide variants [SNVs] and indels), and coverage analysis. Annotation of genetic variation was done by an in-house pipeline (IntegraGen). To rapidly identify the pathogenic variant, we applied WES to all five members of the consanguineous family A (Figure 1). For all subjects, the overall sequencing coverage of the captured regions was 94% and 85% for a 10× and 25× depth of coverage, respectively, resulting in a mean sequencing depth of 80× per base.
Filtering approaches were subsequently applied for identification of candidate mutation(s). Referenced variants that occurred homozygously or heterozygously with a minor allele frequency (MAF) ≥ 0.005 in dbSNP137, HapMap,14 1000 Genomes,15 and the NHLBI Exome Sequencing Project Exome Variant Server (EVS)16 were removed.6,7,9 This step reduced the number of variants from 4,572 to 0 indels and from 55,051 SNVs to two compound-heterozygous missense variants in OBSCN (MIM 608616), one homozygous missense variant in four different genes (CTNNA3 [MIM 607667], PRRX2 [MIM 604675], UNCX, and PTCD3 [MIM 614918]), and one nonsense exchange in KIZ. Sanger sequencing confirmed all variants except the UNCX mutation, which turned out to be a false positive. In order to identify a potential disease-causing effect of missense substitutions, we investigated their species conservation and their predicted impact on the protein structure.17,18 All missense variants, except the ones in OBSCN, were excluded after consideration of the previously mentioned characteristics. The compound-heterozygous mutations in OBSCN were absent in genetic public databases, conserved across species, and predicted to be probably damaging and deleterious by PolyPhen-2 and SIFT. However, given that consanguinity among parents was reported for family A (Figure 1), homozygous variants represent the most likely candidate, although this does not totally exclude underlying causative compound-heterozygous mutations (K.M. Bujakowska et al., 2011, ARVO, abstract).
The homozygous nonsense mutation (c.226C>T [p.Arg76∗]; RefSeq accession number NM_018474.4) located in exon 3 of KIZ cosegregated with the phenotype (family A in Figure 1 and Table 1). It represents a rare variant (rs202210819) that was detected heterozygously in 5 out of 5,920 individuals in the NHLBI EVS (MAF = 0.0004 in European Americans exclusively because it was not found among African Americans). After our discrete filtering approach, which identified candidate mutations in KIZ and OBSCN, we performed stratification on the basis of functional impact and gave a greater weight to the likelihood that the homozygous stop codon in KIZ was the most deleterious.
Table 1.
KIZ Mutations Causing RCD
| Index individual | Family | Exon | Nucleotide Exchangea | Allele State | Protein Effect |
|---|---|---|---|---|---|
| CIC00173 (II.2) | A (128) | 3 | c.226C>T | homozygous | p.Arg76∗ |
| CIC01611 (II.1) | B (951) | 3 | c.226C>T | homozygous | p.Arg76∗ |
| CIC01225 (II.1) | C (737) | 1 | c.52G>T | heterozygous | p.Glu18∗ |
| 2 | c.119_122delAACT | heterozygous | p.Lys40Ilefs∗14 |
RefSeq NM_018474.4.
To further evaluate which of the two genes might be the most likely to carry the pathogenic variants underlying autosomal-recessive RCD, we assessed genetic expression of both genes. Ubiquitous expression of KIZ, including in the eye, was found in UniGene. The KIZ mouse ortholog (Plk1s1, also known as Gm114) showed higher expression in rod photoreceptors of the retinal-cell-type comparative transcriptome atlas20 than in cone photoreceptors and horizontal, bipolar, amacrine, ganglion, and microglia cells (Figure 2A).20 The in-house rd1 mouse transcriptomic database revealed a significant decrease in the Plk1s1 mRNA level from day 12 to day 21 (p < 0.05) when photoreceptors degenerated, which was in keeping with rod photoreceptor expression (Figure 2B). In addition, Strunnikova et al.21 reported KIZ expression in the human retinal pigment epithelium (RPE).21 Similarly to KIZ, OBSCN was found to be ubiquitously expressed in UniGene. In contrast, mRNA expression was not reported in the mouse retinal-cell-type comparative transcriptome atlas.20 Furthermore, the in-house rd1 mouse transcriptomic database reported no changes in Obscn mRNA levels during photoreceptor degeneration (data available upon request). To support transcriptomic database results and further document the implication of KIZ in retinal physiology, we performed quantitative real-time PCR, which revealed that the KIZ transcript was most abundant in the retina, followed by the RPE, whole-blood cells, and fibroblasts (p ≤ 0.01, Figure 2C). Subsequent Sanger sequencing of the PCR products confirmed correct KIZ-fragment amplification. Because transcriptomic data were only reported in the mouse retina at postnatal day 7 and there was no available information on expression location,22 we performed RNA in situ hybridization in the adult mouse retina as previously described23,24 by using a riboprobe encompassing exons 8–14 of Plk1s1 mRNA (RefSeq NM_001033298.3). Plk1s1 was found to be expressed in the outer nuclear layer, corresponding to photoreceptor nuclei, in the mouse retina (Figure 2D and 2E). All together, these results support our hypothesis that the nonsense variant in KIZ is the most convincing underlying defect for RCD in subject II.2 of family A (Figure 1).
Figure 2.
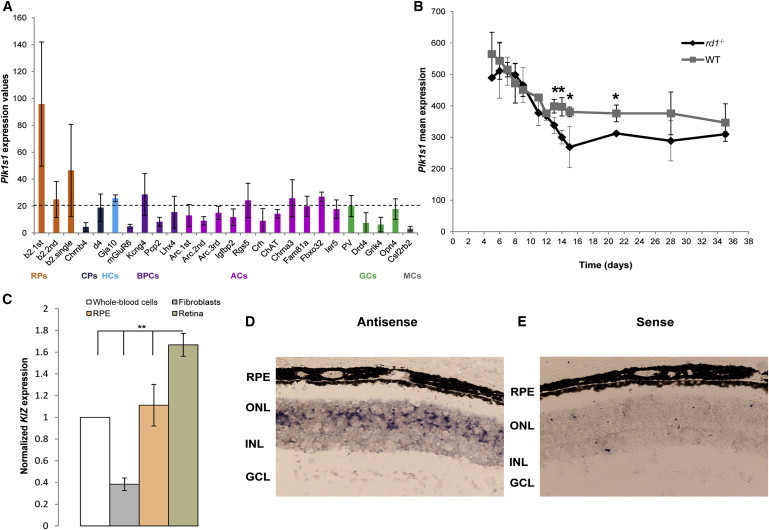
Plk1s1 Transcriptomic Analysis in the Mouse Retina
(A) Plk1s1 expression (1455558_at) in six different cell types from the mouse adult retina: rod photoreceptors (RPs), cone photoreceptors (CPs), horizontal cells (HCs), bipolar cells (BPCs), amacrine cells (ACs), ganglion cells (GCs), and microglia cells (MCs). The graph presents Plk1s1 normalized expression values. Only values higher than 20 can be considered significantly expressed. Retinal cell types were established from a library composed of 22 transgenic mouse lines.20 Each value on the x axis corresponds to a specific retinal cell type established from the following abbreviated mouse lines: RPs (b2), CPs (Chrnb4 and d4), HCs (Gja10), BPCs (mGluR6, Kcng4, Pcp2, and Lhx4), ACs (Arc, Igfbp2, Rgs5, Crh, ChAT, Chrna3, Fam81a, Fbxo32, and Ier5), GCs (Pv, Drd4, Grik4, and Opn4), and MCs (Csf2rb2). (According to Siegert et al.,20 RNA amplification was performed in different batches. For avoiding differences caused by variable amplification across batches, RNA samples of cell groups belonging to the same biological triplicate were amplified in different batches. As such, “1st,” “2nd,” “single,” and “3rd” correspond to the batch numbers.) Plk1s1, implicated in autosomal-recessive RCD, showed the highest expression in RPs.
(B) Plk1s1 expression (1455558_at) in rd1 and wild-type mice during RP degeneration. The rd1 mouse, carrying Pde6b mutations, is a naturally occurring RCD model leading to a complete loss of RPs by postnatal day 36 and a preserved inner retina. cDNAs of neural retinas from rd1 and wild-type mice on identical genetic backgrounds were hybridized to the mouse genome 430 2.0 array (Affymetrix). ∗p < 0.05.
(C) KIZ expression in four human tissues: retina, retinal pigment epithelium (RPE), fibroblasts, and whole-blood cells. Quantitative real-time PCR, normalized to the expression of 18S, revealed that KIZ had higher expression in the retina than in the RPE, whole-blood cells, and fibroblasts (n = 3, ∗∗p ≤ 0.01).
(D and E) An RNA in situ hybridization assay for Plk1s1 expression in the mouse retina. A riboprobe encompassing exons 8–14 of mouse Plk1s1 mRNA (RefSeq NM_001033298.3) was used. Antisense (D) and sense (E) probes are shown. Abbreviations are as follows: GCL, ganglion cell layer; INL, inner nuclear layer; ONL: outer nuclear layer; and RPE, retinal pigment epithelium.
Further screening of coding and flanking exonic regions of KIZ in 340 unrelated individuals with autosomal-recessive and sporadic RCD by direct Sanger sequencing (PCR protocol and primer sequences are available upon request) identified two other subjects with mutations in this gene. Interestingly, another subject (CIC01611 II.1 in family B [951]) had the same nonsense variant (homozygous c.226C>T [p.Arg76∗]) found in subject CIC00173 II.2 (Figure 1). Subject CIC00173 was of North African Sephardic Jewish ancestry, and CIC01611 was of Spanish ancestry, and neither was aware of any family connection. To investigate whether the stop variant represents a founder alteration, we performed haplotype analysis for each of the index subjects (CIC00173 II.2 from family A) and CIC01611 II.1 from family B) by selecting 11 microsatellite DNA markers flanking the KIZ locus.25 These markers were distributed over a physical distance of ≈7.38 Mb, corresponding to ≈10.5 cM (genetic distance according to Marshfield genetic maps). Both subjects were found to share a common haplotype of five polymorphic microsatellites (DS20S54, DS20S63, DS20S190, DS20S868, and DS20S180) flanking KIZ and spanning ≈1.75 cM (1.18 Mb) (Figure 1). This result suggests that c.226C>T (p.Arg76∗) is most likely a founder mutation causing autosomal-recessive RCD in the southern European population.
CIC01225 II.3 in family C (737), an additional individual with sporadic RCD, carried compound-heterozygous mutations: nonsense mutation c.52G>T (p.Glu18∗) in exon 1 and deletion c.119_122delAACT (p.Lys40Ilefs∗14) in exon 3 (Table 1). Cosegregation analysis revealed that the father was heterozygous for the nonsense mutation and the mother was heterozygous for the frameshift deletion (Figure 1). The identified defects most likely result in nonsense-mediated mRNA decay or truncated KIZ with a loss of function as a putative disease mechanism. Additional KIZ polymorphisms identified from screening the RCD cohort and their respective frequencies are provided in Table S1.
Overall, KIZ mutations in the studied cohort would account for about 1% of autosomal-recessive RCD. This might be a slight overestimation given that a majority of the 340 affected individuals included in this work had already been investigated and excluded for carrying defects in known genes implicated in retinal diseases.
The three affected individuals with KIZ variants in this study were all diagnosed with RCD in their late teens on the basis of night blindness followed by changes in midperipheral visual fields and undetectable responses in a full-field electroretinogram by approximately 35 years of age. Index II.2 of family A (CIC00173, Figure 1) was a 50-year-old male subject of North African Jewish Sephardic descent and had unaffected first-cousin parents. The index subject was overweight and complained of moderate hearing difficulties. Best-corrected visual acuity (BCVA) was 20/800 in the right eye and 20/640 in the left eye. A kinetic visual-field test revealed decreased central retinal sensitivity in addition to bilateral peripheral-field constriction. Fundus changes were typical of RCD with additional macular thinning (Figures 3A–3C). Index II.1 of family B (CIC01611, Figure 1) was a 34-year-old subject of Spanish descent. His medical and familial history was noncontributory. BCVA was 20/20 in both eyes. A binocular kinetic visual field using the III4e stimulus showed an annular scotoma in the midperiphery and preservation of the peripheral isopter. Fundus changes were typical of RCD with macular preservation (Figures 3D–3F). Index II.3 of family C (CIC01225, Figure 1) was a 51-year-old male subject of a mixed Italian and French descent. His history was significant for a congenital ichthyosis that was well tolerated. There was no familial history of systemic or ocular disease. BCVA was 20/40 in the right eye and 20/32 in the left. A binocular kinetic visual field using the III4e stimulus was reduced to the central 10° with bitemporal islands of perception peripherally. Fundus changes were typical of RCD with relative macular preservation (Figures 3G–3I).
Figure 3.
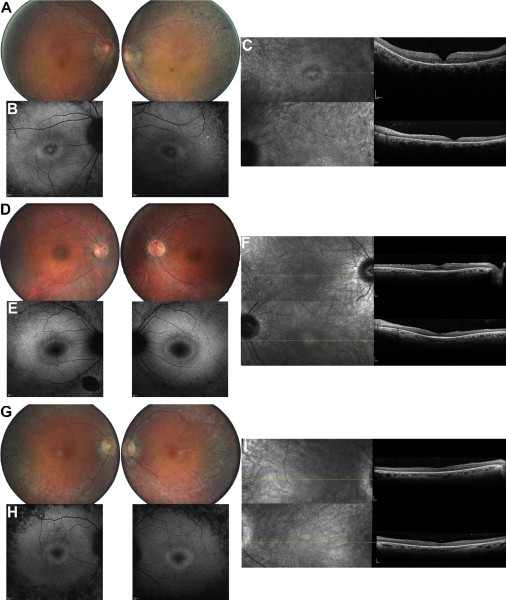
Retinal Imaging of Autosomal-Recessive-RCD Individuals Carrying KIZ Variants
(A–C) Fundus imaging of the right and left eye of CIC00173 from family A at 50 years of age. Fundus color photographs (FCPs; A), fundus autofluorescence (FAF; B), and macular horizontal scans of spectral domain optical coherence tomography (SD-OCT; C) show not only pigmentary changes in the peripheral retina but also atrophic changes in the central macula with a ring of hypoautofluorescence and foveal thinning with loss of outer-segment structures on SD-OCT.
(D–F) Fundus imaging of the right and left eye of CIC01611 from family B at 34 years of age. FCPs (D), FAF (E), and SD-OCT (F) show mild pigmentary changes in the peripheral retina in association with slight changes in FAF outside the vascular arcades and a perifoveal ring of increased autofluorescence (larger than observed for CIC01225), as well as normal foveal structure on OCT in keeping with normal central vision.
(G–I) Fundus imaging of the right and left eye of CIC01225 from family C at 51 years of age. FCPs (G), FAF (H), and SD-OCT (I) show pigmentary changes in the peripheral retina in association with a loss of FAF outside the vascular arcades and a perifoveal ring of increased autofluorescence and moderate thinning of the fovea on OCT.
KIZ is a 12 kb gene clustering on the short arm of chromosome 20 at 20p11.23 (Figure 1). It harbors 13 exons and encodes a 673 aa protein that belongs to the kizuna family (Figure 1). It plays a critical role during cell-cycle progression while it undergoes sequential phosphorylation.26,27 Oshimori et al.27 demonstrated that KIZ is a centrosomal substrate for PLK1, given that the latter mediates its phosphorylation at amino acid residue Thr379. During mitosis, KIZ localizes to mature centrioles and interacts with PLK1 in order to protect the centrosome from collapsing during spindle formation in a Thr379-phosphorylation-dependent manner.27 Stabilized centrosomes resist microtubule-mediated pulling and pushing forces and thus ensure the spindle bipolarity required for accurate separation of chromosomes during mitosis.28,29 At the plasma membrane, the mother centriole can serve as the basal body where ciliogenesis begins, thus giving rise to either motile or immotile cilia, which exist on most cells in the human body.30 Ciliogenesis can be either linked to the cell cycle or associated with cell differentiation.30 To investigate a potential association between KIZ and cilia in humans, we induced cilia formation in a serum-free human fibroblast cell culture as described elsewhere.31 Colocalization with a cilium marker (acetylated α-tubulin) (Figures 4A–4C) and a basal body marker (γ-tubulin) (Figures 4D–4F) demonstrated that KIZ (SAB2700541, Sigma Aldrich) localizes to the basal body of monocilia in human fibroblasts. This might suggest its location at the connecting cilium of photoreceptors. Interestingly, in the in-house rd1 mouse transcriptomic database, Rab28, which encodes a protein localized at the connecting cilium of photoreceptors, shows a profile similar to that of KIZ. In rod and cone photoreceptors, the connecting cilium bridges the inner segment to the outer segment, which harbors the photopigment.32 All components necessary for assembly, maintenance, and continuous turnover of the outer segments are synthesized in the cell body and are moved through the connecting cilium. KIZ might have a critical role in this trafficking process, and its dysfunction or absence would compromise molecular transport and therefore outer-segment homeostasis. Future studies are needed for documenting this potential role for KIZ in human and mouse retinas.
Figure 4.
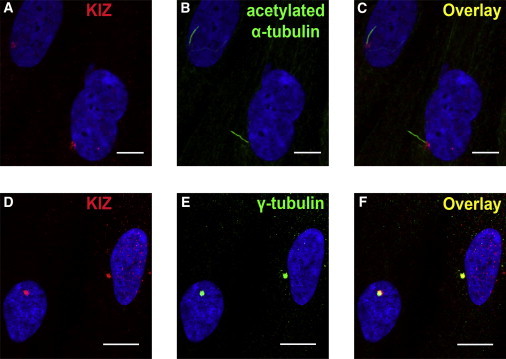
Subcellular Localization of KIZ in Monocilia from Human Fibroblasts
Primary fibroblasts derived from human skin biopsies were cultured via standard conditions. To induce formation of monocilia in human fibroblasts, we deprived cell culture from fetal bovine serum for 24 hr. Serum-starved fibroblasts were fixed 5 min with methanol at −20°C, and immunofluorescence staining was performed as previously described.31 Human fibroblasts were immunolabeled with a KIZ antibody (PLK1S1 rabbit-polyclonal antibody,1/250, Sigma Aldrich SAB2700541; A, C, D, and F), an acetyl α-tubulin mouse-monoclonal antibody (1/1,000, Sigma-Aldrich; B and C), and a γ-tubulin mouse-monoclonal antibody (1/1,000, Sigma-Aldrich; E and F), visualized with standard secondary antibodies, and counterstained with nuclear DAPI (blue, 1/1,000, Euromedex, Souffelweyersheim). The overlay view in (C) shows KIZ localization (A, red) at the bottom of the cilia (B, green) and colocalization (F) of KIZ (D, red) to the basal body of the cilium-associated centriole (E, green). Scale bars represent 20 μm.
An increasing number of Mendelian and complex diseases are associated with defects in ciliogenesis.29,33 These comprise a diverse group of pathologies, including isolated or syndromic retinal degeneration (e.g., Bardet-Biedl syndrome), cystic kidney disease, infertility, chronic respiratory problems, hypertension, and obesity.29,34 To date, mutations in at least 24 ciliary genes have been implicated in retinal degenerations, such as RCD, cone dystrophy, cone-rod dystrophy, Leber congenital amaurosis, and macular degeneration.35–39 For the majority of mutations, a clear genotype-phenotype correlation cannot be established, given that mutations in the same ciliary gene can cause both syndromic and nonsyndromic forms.37 However, retina-restricted disease was reported for mutations in autosomal-recessive RCD (in C2ORF71 [MIM 613425],35,40 FAM161A [MIM 613596],41,42 MAK [MIM 154235],43,44 RP1 [MIM 603937],45 and ARL2BP [MIM 615407]36), in X-linked RCD (in RPGR and RP2 [MIM 300757]46,47), and in autosomal-dominant RCD (in RP1 [MIM 603937]45 and TOPORS [MIM 609507]48,49). The most common inheritance patterns are autosomal recessive and X-linked.37
In conclusion, our study has identified KIZ mutations leading to autosomal-recessive RCD. We speculate that these defects are loss-of-function alleles, are sensitive to nonsense-mediated mRNA decay, or result in a truncated KIZ in photoreceptors and thus lead to a defect in photoreceptor-connecting cilia.32 The data presented here suggest a role for KIZ in photoreceptor homeostasis. However, further functional studies are needed for elucidating the exact role of KIZ in the retina and the underlying pathogenic mechanism(s).
Acknowledgments
We thank the affected subjects and family members who participated in this study. In addition, we would like to thank Caroline Moreau Fauvarque and Antoine Mialot for their fruitful technical support, Stéphane Fouquet and David Godefroy for their help in acquiring the images, and Cameron Parsa for critical reading. The project was supported by Foundation Fighting Blindness (FFB) grant CD-CL-0808-0466-CHNO (to I.A.), FFB center grant C-CMM-0907-0428-INSERM04, Foundation Voir et Entendre (C.Z.), Fondation Dalloz prix “pour la recherche en ophtalmologie” (to C.Z.), Ville de Paris and Region Ile de France, and Labex LIFESENSES (reference ANR-10-LABX-65), supported by French state funds managed by the Agence Nationale de la Recherche within the Investissements d’Avenir program (ANR-11-IDEX-0004-0).
Contributor Information
Christina Zeitz, Email: christina.zeitz@inserm.fr.
Isabelle Audo, Email: isabelle.audo@inserm.fr.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
GeneCards, http://www.genecards.org/
Human Gene Mutation Database (HGMD), http://www.hgmd.org/
Leiden Open Variation Database (LOVD), http://www.lovd.nl/3.0/home
Mammalian Genotyping Service, Marshfield genetic maps, http://research.marshfieldclinic.org/genetics/GeneticResearch/compMaps.asp
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
PolyPhen-2, http://genetics.bwh.harvard.edu/pph2/
Roska, http://fmi.ch/roska.data/
UCSC Genome Browser, http://genome.ucsc.edu/
UniGene, http://www.ncbi.nlm.nih.gov/UniGene/ESTProfileViewer.cgi?uglist=Hs.187635
UniProt, http://www.uniprot.org/
References
- 1.Hartong D.T., Berson E.L., Dryja T.P. Retinitis pigmentosa. Lancet. 2006;368:1795–1809. doi: 10.1016/S0140-6736(06)69740-7. [DOI] [PubMed] [Google Scholar]
- 2.Audo I., Kohl S., Leroy B.P., Munier F.L., Guillonneau X., Mohand-Saïd S., Bujakowska K., Nandrot E.F., Lorenz B., Preising M. TRPM1 is mutated in patients with autosomal-recessive complete congenital stationary night blindness. Am. J. Hum. Genet. 2009;85:720–729. doi: 10.1016/j.ajhg.2009.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zeitz C., van Genderen M., Neidhardt J., Luhmann U.F., Hoeben F., Forster U., Wycisk K., Mátyás G., Hoyng C.B., Riemslag F. Mutations in GRM6 cause autosomal recessive congenital stationary night blindness with a distinctive scotopic 15-Hz flicker electroretinogram. Invest. Ophthalmol. Vis. Sci. 2005;46:4328–4335. doi: 10.1167/iovs.05-0526. [DOI] [PubMed] [Google Scholar]
- 4.Wycisk K.A., Zeitz C., Feil S., Wittmer M., Forster U., Neidhardt J., Wissinger B., Zrenner E., Wilke R., Kohl S., Berger W. Mutation in the auxiliary calcium-channel subunit CACNA2D4 causes autosomal recessive cone dystrophy. Am. J. Hum. Genet. 2006;79:973–977. doi: 10.1086/508944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zeitz C., Kloeckener-Gruissem B., Forster U., Kohl S., Magyar I., Wissinger B., Mátyás G., Borruat F.X., Schorderet D.F., Zrenner E. Mutations in CABP4, the gene encoding the Ca2+-binding protein 4, cause autosomal recessive night blindness. Am. J. Hum. Genet. 2006;79:657–667. doi: 10.1086/508067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Audo I., Bujakowska K., Orhan E., El Shamieh S., Sennlaub F., Guillonneau X., Antonio A., Michiels C., Lancelot M.E., Letexier M. The familial dementia gene revisited: a missense mutation revealed by whole-exome sequencing identifies ITM2B as a candidate gene underlying a novel autosomal dominant retinal dystrophy in a large family. Hum. Mol. Genet. 2014;23:491–501. doi: 10.1093/hmg/ddt439. [DOI] [PubMed] [Google Scholar]
- 7.Audo I., Bujakowska K., Orhan E., Poloschek C.M., Defoort-Dhellemmes S., Drumare I., Kohl S., Luu T.D., Lecompte O., Zrenner E. Whole-exome sequencing identifies mutations in GPR179 leading to autosomal-recessive complete congenital stationary night blindness. Am. J. Hum. Genet. 2012;90:321–330. doi: 10.1016/j.ajhg.2011.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Siemiatkowska A.M., van den Born L.I., van Hagen P.M., Stoffels M., Neveling K., Henkes A., Kipping-Geertsema M., Hoefsloot L.H., Hoyng C.B., Simon A. Mutations in the mevalonate kinase (MVK) gene cause nonsyndromic retinitis pigmentosa. Ophthalmology. 2013;120:2697–2705. doi: 10.1016/j.ophtha.2013.07.052. [DOI] [PubMed] [Google Scholar]
- 9.Zeitz C., Jacobson S.G., Hamel C.P., Bujakowska K., Neuillé M., Orhan E., Zanlonghi X., Lancelot M.E., Michiels C., Schwartz S.B., Congenital Stationary Night Blindness Consortium Whole-exome sequencing identifies LRIT3 mutations as a cause of autosomal-recessive complete congenital stationary night blindness. Am. J. Hum. Genet. 2013;92:67–75. doi: 10.1016/j.ajhg.2012.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Audo I., Manes G., Mohand-Saïd S., Friedrich A., Lancelot M.E., Antonio A., Moskova-Doumanova V., Poch O., Zanlonghi X., Hamel C.P. Spectrum of rhodopsin mutations in French autosomal dominant rod-cone dystrophy patients. Invest. Ophthalmol. Vis. Sci. 2010;51:3687–3700. doi: 10.1167/iovs.09-4766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Audo I., Sahel J.A., Mohand-Saïd S., Lancelot M.E., Antonio A., Moskova-Doumanova V., Nandrot E.F., Doumanov J., Barragan I., Antinolo G. EYS is a major gene for rod-cone dystrophies in France. Hum. Mutat. 2010;31:E1406–E1435. doi: 10.1002/humu.21249. [DOI] [PubMed] [Google Scholar]
- 12.Audo I., Lancelot M.E., Mohand-Saïd S., Antonio A., Germain A., Sahel J.A., Bhattacharya S.S., Zeitz C. Novel C2orf71 mutations account for ∼1% of cases in a large French arRP cohort. Hum. Mutat. 2011;32:E2091–E2103. doi: 10.1002/humu.21460. [DOI] [PubMed] [Google Scholar]
- 13.Audo I., Bujakowska K.M., Léveillard T., Mohand-Saïd S., Lancelot M.E., Germain A., Antonio A., Michiels C., Saraiva J.P., Letexier M. Development and application of a next-generation-sequencing (NGS) approach to detect known and novel gene defects underlying retinal diseases. Orphanet J. Rare Dis. 2012;7:8. doi: 10.1186/1750-1172-7-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Altshuler D.M., Gibbs R.A., Peltonen L., Altshuler D.M., Gibbs R.A., Peltonen L., Dermitzakis E., Schaffner S.F., Yu F., Peltonen L., International HapMap 3 Consortium Integrating common and rare genetic variation in diverse human populations. Nature. 2010;467:52–58. doi: 10.1038/nature09298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Abecasis G.R., Altshuler D., Auton A., Brooks L.D., Durbin R.M., Gibbs R.A., Hurles M.E., McVean G.A., 1000 Genomes Project Consortium A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tennessen J.A., Bigham A.W., O’Connor T.D., Fu W., Kenny E.E., Gravel S., McGee S., Do R., Liu X., Jun G., Broad GO. Seattle GO. NHLBI Exome Sequencing Project Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science. 2012;337:64–69. doi: 10.1126/science.1219240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Adzhubei I.A., Schmidt S., Peshkin L., Ramensky V.E., Gerasimova A., Bork P., Kondrashov A.S., Sunyaev S.R. A method and server for predicting damaging missense mutations. Nat. Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kumar P., Henikoff S., Ng P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009;4:1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 20.Siegert S., Cabuy E., Scherf B.G., Kohler H., Panda S., Le Y.Z., Fehling H.J., Gaidatzis D., Stadler M.B., Roska B. Transcriptional code and disease map for adult retinal cell types. Nat. Neurosci. 2012;15:487–495. doi: 10.1038/nn.3032. S1–S2. [DOI] [PubMed] [Google Scholar]
- 21.Strunnikova N.V., Maminishkis A., Barb J.J., Wang F., Zhi C., Sergeev Y., Chen W., Edwards A.O., Stambolian D., Abecasis G. Transcriptome analysis and molecular signature of human retinal pigment epithelium. Hum. Mol. Genet. 2010;19:2468–2486. doi: 10.1093/hmg/ddq129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Magdaleno S., Jensen P., Brumwell C.L., Seal A., Lehman K., Asbury A., Cheung T., Cornelius T., Batten D.M., Eden C. BGEM: an in situ hybridization database of gene expression in the embryonic and adult mouse nervous system. PLoS Biol. 2006;4:e86. doi: 10.1371/journal.pbio.0040086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Di Meglio T., Nguyen-Ba-Charvet K.T., Tessier-Lavigne M., Sotelo C., Chédotal A. Molecular mechanisms controlling midline crossing by precerebellar neurons. J. Neurosci. 2008;28:6285–6294. doi: 10.1523/JNEUROSCI.0078-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Orhan E., Prézeau L., El Shamieh S., Bujakowska K.M., Michiels C., Zagar Y., Vol C., Bhattacharya S.S., Sahel J.A., Sennlaub F. Further insights into GPR179: expression, localization, and associated pathogenic mechanisms leading to complete congenital stationary night blindness. Invest. Ophthalmol. Vis. Sci. 2013;54:8041–8050. doi: 10.1167/iovs.13-12610. [DOI] [PubMed] [Google Scholar]
- 25.Goldstein D.B., Ruiz Linares A., Cavalli-Sforza L.L., Feldman M.W. An evaluation of genetic distances for use with microsatellite loci. Genetics. 1995;139:463–471. doi: 10.1093/genetics/139.1.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dephoure N., Zhou C., Villén J., Beausoleil S.A., Bakalarski C.E., Elledge S.J., Gygi S.P. A quantitative atlas of mitotic phosphorylation. Proc. Natl. Acad. Sci. USA. 2008;105:10762–10767. doi: 10.1073/pnas.0805139105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Oshimori N., Ohsugi M., Yamamoto T. The Plk1 target Kizuna stabilizes mitotic centrosomes to ensure spindle bipolarity. Nat. Cell Biol. 2006;8:1095–1101. doi: 10.1038/ncb1474. [DOI] [PubMed] [Google Scholar]
- 28.Bettencourt-Dias M., Glover D.M. Centrosome biogenesis and function: centrosomics brings new understanding. Nat. Rev. Mol. Cell Biol. 2007;8:451–463. doi: 10.1038/nrm2180. [DOI] [PubMed] [Google Scholar]
- 29.Conroy P.C., Saladino C., Dantas T.J., Lalor P., Dockery P., Morrison C.G. C-NAP1 and rootletin restrain DNA damage-induced centriole splitting and facilitate ciliogenesis. Cell Cycle. 2012;11:3769–3778. doi: 10.4161/cc.21986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dawe H.R., Farr H., Gull K. Centriole/basal body morphogenesis and migration during ciliogenesis in animal cells. J. Cell Sci. 2007;120:7–15. doi: 10.1242/jcs.03305. [DOI] [PubMed] [Google Scholar]
- 31.Schmid F., Glaus E., Barthelmes D., Fliegauf M., Gaspar H., Nürnberg G., Nürnberg P., Omran H., Berger W., Neidhardt J. U1 snRNA-mediated gene therapeutic correction of splice defects caused by an exceptionally mild BBS mutation. Hum. Mutat. 2011;32:815–824. doi: 10.1002/humu.21509. [DOI] [PubMed] [Google Scholar]
- 32.Fliegauf M., Benzing T., Omran H. When cilia go bad: cilia defects and ciliopathies. Nat. Rev. Mol. Cell Biol. 2007;8:880–893. doi: 10.1038/nrm2278. [DOI] [PubMed] [Google Scholar]
- 33.Afzelius B.A. Microtubules in the spermatids of stick insects. J. Ultrastruct. Mol. Struct. Res. 1988;98:94–102. doi: 10.1016/s0889-1605(88)80937-6. [DOI] [PubMed] [Google Scholar]
- 34.Ansley S.J., Badano J.L., Blacque O.E., Hill J., Hoskins B.E., Leitch C.C., Kim J.C., Ross A.J., Eichers E.R., Teslovich T.M. Basal body dysfunction is a likely cause of pleiotropic Bardet-Biedl syndrome. Nature. 2003;425:628–633. doi: 10.1038/nature02030. [DOI] [PubMed] [Google Scholar]
- 35.Collin R.W., Safieh C., Littink K.W., Shalev S.A., Garzozi H.J., Rizel L., Abbasi A.H., Cremers F.P., den Hollander A.I., Klevering B.J., Ben-Yosef T. Mutations in C2ORF71 cause autosomal-recessive retinitis pigmentosa. Am. J. Hum. Genet. 2010;86:783–788. doi: 10.1016/j.ajhg.2010.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Davidson A.E., Schwarz N., Zelinger L., Stern-Schneider G., Shoemark A., Spitzbarth B., Gross M., Laxer U., Sosna J., Sergouniotis P.I. Mutations in ARL2BP, encoding ADP-ribosylation-factor-like 2 binding protein, cause autosomal-recessive retinitis pigmentosa. Am. J. Hum. Genet. 2013;93:321–329. doi: 10.1016/j.ajhg.2013.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Estrada-Cuzcano A., Roepman R., Cremers F.P., den Hollander A.I., Mans D.A. Non-syndromic retinal ciliopathies: translating gene discovery into therapy. Hum. Mol. Genet. 2012;21(R1):R111–R124. doi: 10.1093/hmg/dds298. [DOI] [PubMed] [Google Scholar]
- 38.Nishiguchi K.M., Tearle R.G., Liu Y.P., Oh E.C., Miyake N., Benaglio P., Harper S., Koskiniemi-Kuendig H., Venturini G., Sharon D. Whole genome sequencing in patients with retinitis pigmentosa reveals pathogenic DNA structural changes and NEK2 as a new disease gene. Proc. Natl. Acad. Sci. USA. 2013;110:16139–16144. doi: 10.1073/pnas.1308243110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Roosing S., Rohrschneider K., Beryozkin A., Sharon D., Weisschuh N., Staller J., Kohl S., Zelinger L., Peters T.A., Neveling K., European Retinal Disease Consortium Mutations in RAB28, encoding a farnesylated small GTPase, are associated with autosomal-recessive cone-rod dystrophy. Am. J. Hum. Genet. 2013;93:110–117. doi: 10.1016/j.ajhg.2013.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nishimura D.Y., Baye L.M., Perveen R., Searby C.C., Avila-Fernandez A., Pereiro I., Ayuso C., Valverde D., Bishop P.N., Manson F.D. Discovery and functional analysis of a retinitis pigmentosa gene, C2ORF71. Am. J. Hum. Genet. 2010;86:686–695. doi: 10.1016/j.ajhg.2010.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bandah-Rozenfeld D., Mizrahi-Meissonnier L., Farhy C., Obolensky A., Chowers I., Pe’er J., Merin S., Ben-Yosef T., Ashery-Padan R., Banin E., Sharon D. Homozygosity mapping reveals null mutations in FAM161A as a cause of autosomal-recessive retinitis pigmentosa. Am. J. Hum. Genet. 2010;87:382–391. doi: 10.1016/j.ajhg.2010.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Langmann T., Di Gioia S.A., Rau I., Stöhr H., Maksimovic N.S., Corbo J.C., Renner A.B., Zrenner E., Kumaramanickavel G., Karlstetter M. Nonsense mutations in FAM161A cause RP28-associated recessive retinitis pigmentosa. Am. J. Hum. Genet. 2010;87:376–381. doi: 10.1016/j.ajhg.2010.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ozgül R.K., Siemiatkowska A.M., Yücel D., Myers C.A., Collin R.W., Zonneveld M.N., Beryozkin A., Banin E., Hoyng C.B., van den Born L.I., European Retinal Disease Consortium Exome sequencing and cis-regulatory mapping identify mutations in MAK, a gene encoding a regulator of ciliary length, as a cause of retinitis pigmentosa. Am. J. Hum. Genet. 2011;89:253–264. doi: 10.1016/j.ajhg.2011.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tucker B.A., Scheetz T.E., Mullins R.F., DeLuca A.P., Hoffmann J.M., Johnston R.M., Jacobson S.G., Sheffield V.C., Stone E.M. Exome sequencing and analysis of induced pluripotent stem cells identify the cilia-related gene male germ cell-associated kinase (MAK) as a cause of retinitis pigmentosa. Proc. Natl. Acad. Sci. USA. 2011;108:E569–E576. doi: 10.1073/pnas.1108918108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pierce E.A., Quinn T., Meehan T., McGee T.L., Berson E.L., Dryja T.P. Mutations in a gene encoding a new oxygen-regulated photoreceptor protein cause dominant retinitis pigmentosa. Nat. Genet. 1999;22:248–254. doi: 10.1038/10305. [DOI] [PubMed] [Google Scholar]
- 46.Schwahn U., Lenzner S., Dong J., Feil S., Hinzmann B., van Duijnhoven G., Kirschner R., Hemberger M., Bergen A.A., Rosenberg T. Positional cloning of the gene for X-linked retinitis pigmentosa 2. Nat. Genet. 1998;19:327–332. doi: 10.1038/1214. [DOI] [PubMed] [Google Scholar]
- 47.Evans R.J., Schwarz N., Nagel-Wolfrum K., Wolfrum U., Hardcastle A.J., Cheetham M.E. The retinitis pigmentosa protein RP2 links pericentriolar vesicle transport between the Golgi and the primary cilium. Hum. Mol. Genet. 2010;19:1358–1367. doi: 10.1093/hmg/ddq012. [DOI] [PubMed] [Google Scholar]
- 48.Chakarova C.F., Khanna H., Shah A.Z., Patil S.B., Sedmak T., Murga-Zamalloa C.A., Papaioannou M.G., Nagel-Wolfrum K., Lopez I., Munro P. TOPORS, implicated in retinal degeneration, is a cilia-centrosomal protein. Hum. Mol. Genet. 2011;20:975–987. doi: 10.1093/hmg/ddq543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chakarova C.F., Papaioannou M.G., Khanna H., Lopez I., Waseem N., Shah A., Theis T., Friedman J., Maubaret C., Bujakowska K. Mutations in TOPORS cause autosomal dominant retinitis pigmentosa with perivascular retinal pigment epithelium atrophy. Am. J. Hum. Genet. 2007;81:1098–1103. doi: 10.1086/521953. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.