

Abstract
We describe an ongoing pilot project in which newborn screening (NBS) for FMR1 mutations and subsequent cascade testing are performed by the MIND Institute at the University of California, Davis Medical Center (UCDMC). To date, out of 3042 newborns initially screened, 44 extended family members have been screened by cascade testing of extended family members once a newborn is identified. 14 newborns (7 males and 7 females) and 27 extended family members (5 males and 22 females) have been identified with FMR1 mutations. Three family histories are discussed in detail, each demonstrating some benefits and risks of NBS and cascade testing for FMR1 mutations in extended family members. While we acknowledge inherent risks, we propose that with genetic counseling, clinical follow-up of identified individuals and cascade testing, newborn screening (NBS) has significant benefits. Treatment for individuals in the extended family who would otherwise not have received treatment can be beneficial. In addition, knowledge of carrier status can lead to lifestyle changes and prophylactic interventions that are likely to reduce the risk of late onset neurological or psychiatric problems in carriers. Also with identification of carrier family members through NBS, reproductive choices become available to those who would not have known that they were at risk to have offspring with fragile X syndrome.
Keywords: Newborn Screening, FMR1 mutations, Genetic Counseling, Cascade Testing, Premutation
INTRODUCTION
Fragile X syndrome (FXS), the most common inherited cause of intellectual disability (ID), is due a CGG trinucleotide expansion (>200 repeats) in the promoter region of the FMR1 gene. The premutation (55-200 CGG repeats) in FMR1 can sometimes cause developmental problems in childhood or psychiatric, immunological and/or neurological problems in adulthood, including the fragile X-associated tremor ataxia syndrome (FXTAS) [Chonchaiya et al., 2009] in older carriers. Alleles harboring between 45 and 54 CGG repeats [Maddalena et al., 2001] are named intermediate/grey zone alleles and can show intergenerational instability. The University of California Davis MIND Institute is conducting a newborn screening pilot study (NBS) for FMR1 mutations in collaboration with Rush University Medical Center, Chicago (RUMC). Alleles harboring between 45 and 54 CGG repeats [Maddalena et al., 2001] are named intermediate/gray zone alleles and they can show intergenerational instability.
Screening for FMR1 mutations has long been controversial and currently no state includes FMR1 mutations in mandated newborn screening programs. A number of issues have prevented the inclusion of FMR1 testing in State mandated NBS programs. These have included technological limitations, cost of testing and a lack of effective treatments. However, the fragile X field is evolving rapidly in these areas [Hagerman et al., 2012]. Advances in diagnostic testing have led to the availability of blood spot screening which is relatively inexpensive and allows for the screening of a large sample size throughout the expanded CGG range and in both genders [Chen et al., 2011; Coffee et al., 2009; Tassone et al., 2008] have carried out newborn screening of a large population, but was limited to only males with a full mutation.
Although FXS is the most common inherited form of intellectual disabilities, individuals with FMR1 mutations have an increased risk of experiencing a myriad of physical, psychological, and social difficulties. FXS is associated with the FMR1 full mutation (greater than 200 CGG repeats) and it is characterized by specific clinical features such as prominent ears, macrocephaly, enlarged testicles, hyperextensiblity and behavioral features including gaze avoidance, hand-flapping, hyperactivity, impulsivity, anxiety and perseveration [Chonchaiya et al., 2009]. FXS is the most common single gene disorder associated with autism; 30% of those diagnosed with FXS have autism and an additional 30% have PDDNOS or an autism spectrum disorder (ASD) [Hall et al., 2008; Harris et al., 2008; Hatton et al., 2006; Kaufmann et al., 2004; Rogers et al., 2001]. Fifty to 70% of females with FXS have borderline to mild deficits in IQ and 30 to 50% have a normal IQ but experience learning disabilities particularly in mathematics, behavioral and/or emotional problems, such as increased anxiety, shyness, gaze avoidance, perseveration, and mood lability [Angkustsiri et al., 2008; de Vries et al., 1996].
Although those with the premutation (55-200 CGG repeats) usually have a normal IQ, approximately 10 to 20% of males with the premutation have ASD, and/or attention deficit hyperactivity disorder (ADHD), and heightened anxiety [Aziz et al., 2003; Bailey et al., 2008a; Bourgeois et al., 2011; Chonchaiya et al., 2012; Clifford et al., 2007; Cornish et al., 2005; Farzin et al., 2006; Hessl et al., 2011]. The risk for ASD in premutation carriers is increased in those who have seizures [Chonchaiya et al., 2012]. In adults it is well documented that the premutation is associated with Fragile X-associated Tremor/Ataxia syndrome (FXTAS) [Jacquemont et al., 2004b] and Primary Ovarian Insufficiency (FXPOI) [Sullivan et al., 2011]. However, understanding of premutation involvement has advanced beyond these disorders [Chonchaiya et al., 2010; Coffey S.M. et al., 2008; Hagerman and Hagerman 2004; Sullivan et al., 2011]. Hypertension [Hamlin et al., 2012], sleep apnea [Hamlin et al., 2011], psychiatric problems [Bourgeois et al., 2011; Roberts et al., 2009], and immune problems such as fibromyalgia [Coffey S.M. et al., 2008; Leehey et al., 2011], and hypothyroidism [Coffey S.M. et al., 2008; Rodriguez-Revenga et al., 2009] occur commonly in mid and late adulthood.
The aim of this study was to gather pilot data investigating the feasibility, benefits, and risks associated with newborn screening for FXS. The project also included collection of prevalence data, identification and monitoring of newborns with FMR1 mutations as well as identification and testing of at risk extended family members through cascade testing. Fourteen newborns screened positive for FMR1 mutations. In addition, 44 at risk relatives from 10 families were tested and 27 were found to carry the FMR1 premutation.
This paper discusses the experience of cascade testing and preliminary findings of the NBS pilot project at the University of California, Davis as well as the benefits and risks of screening newborns for FMR1 mutations. We report on three newborns identified as having the premutation. Subsequent cascade testing revealed premutation carriers within the families of the identified newborns. These three cases illustrate the impact of NBS, including the range of clinical involvement related to the premutation that was identified through cascade testing
MATERIALS AND METHODS
Using an Institutional Review Board (IRB) approved protocol, a blood sample was obtained on individual filter paper (extra spot card was provided to the nursery) from all newborns at the UC Davis Medical Center during the state mandated heel stick. Parents of newborns were approached daily by research assistants to request consent for participation in the fragile X newborn screening study. A prepared script used across both participating sites introduced parents to the project. IRB approved consent was used to obtain formal consent from those who agreed to join the study. Blood spots from newborns of parents who gave consent were collected and genotyped. Parents of newborns who screened positive for an FMR1 mutation (>55 CGG repeats) were contacted by the Genetic Associate at the UC Davis MIND Institute. Initial blood spot screen results were reported and briefly clarified by telephone and the parents were invited to bring the infant to the MIND Institute for blood testing. During the initial clinic visit genetic counseling was performed, a detailed family history taken, the family was introduced to the Developmental Pediatrician overseeing the project and a peripheral blood sample was obtained from the infant and parents to determine parent of origin. The parents were given the opportunity for the infant to participate in a longitudinal study that will be reported at a later date when completed. Members of the immediate and extended family of the positive newborn were offered genetic testing for FMR1 mutations according to the recommendations set by McConkie-Rosell et al. [2005]. Individuals who had an FMR1 mutation were offered genetic counseling, and a medical evaluation including a medical history and examination.
Molecular Measures
CGG repeat size on blood spots was performed by PCR analysis as described in Tassone [2008] with some modifications. CGG repeat size and methylation status for the confirmatory testing on both the identified newborn and extended family members was determined by isolating DNA from whole peripheral blood leukocytes using standard procedure from Qiagen (QIAxtractor) in Valencia, CA. Both Southern Blot and PCR analysis were performed as detailed in Tassone et al. [2008].
Cognitive and Psychological Methods
Infants were assessed using the Vineland Adaptive Behavior Scales – Second Edition [Sparrow et al., 2005] and the Mullen Scales of Early Learning (MSEL) [Mullen 1995].
RESULTS
Clinical Report Family 1
The male proband screened positive for the presence of a fragile X premutation allele with 68 CGG repeats by blood spot. The Genetic Associate reported results to the proband’s mother over the phone and the initial visit was scheduled by the project coordinator. The proband was seen at the MIND Institute at 5 months of age accompanied by his parents and maternal grandparents. Genetic counseling, blood draw on proband and parents, and medical exam were performed at that time. DNA testing on peripheral blood confirmed the presence of a 68 CGG repeat allele. The initial cascade testing revealed that the proband’s mother had 23 and 67 CGG repeats and the maternal grandfather had 65 CGG repeats.
The prenatal period was significant because the proband’s mother experienced a ruptured aneurism at six months gestation and underwent surgery at that time. The mother continues to experience severe headaches reported to be a consequence of subsequent surgeries. The mother also had a history ptosis of her left eye and this can occasionally be seen in those with the full mutation [Hagerman 2002] and it is less common in those with the premutation.. Connective tissue problems have been associated with the both the full and the premutation including aneurysms of the aorta [Hagerman 2002; Riddle et al., 1998]. The proband was seen in clinic a second time one month later at six months of age. Developmental assessments were performed. The Vineland II Adaptive Behavior Composite (ABC) was 119. The Early Learning Composite (ELC) on the Mullen was 105.
Cascade testing of extended family members identified 12 other premutation carriers. In total, 22 family members were tested and a total of 14 premutation carriers were identified including the proband and his mother (Fig. 2). The proband’s maternal great grandmother had 30 and 63 CGG repeats and had a previous diagnosis of Alzheimer’s disease (AD). She underwent a medical evaluation in clinic. She reported good health until 81 years of age at which time she began to experience cognitive decline. During the medical exam the patient demonstrated difficulty with tandem walking and a subtle tremor with positioning. She also had unintelligible handwriting because of tremor. A brain MRI was performed and showed a significant degree of atrophy and white matter disease in the frontal and parietal regions (Fig. 3). She also had significant white matter disease in the insula bilaterally which is commonly seen in those with the Fragile X-associated tremor ataxia syndrome (FXTAS). She had increased vascular spaces and cortical white matter disease but no involvement of the middle cerebellar peduncles. The patient’s white matter disease and clinical history is consistent with FXTAS in females [Adams et al., 2007; Hagerman et al., 2004]. AD together with FXTAS occurs in approximately 50% of females who died with FXTAS on and is documented on neuropathological studies [Tassone et al., 2012].
Figure 2.
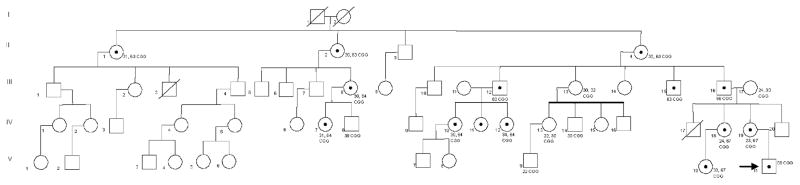
Pedigree for Family 1: The arrow indicates the proband (V-11). The proband’s mother (IV-19), maternal aunt (IV-18), maternal first cousin (V-10), maternal grandfather (III-16), and maternal great grandmother (II-4), carry the premutation. Nine other family members have been identified as premutation carriers (II-1, II-1, III-8, III-12, III-15, IV-7, IV-10, IV-11, IV-12). The CGG repeat sizes for each individual are noted.
Figure 3.
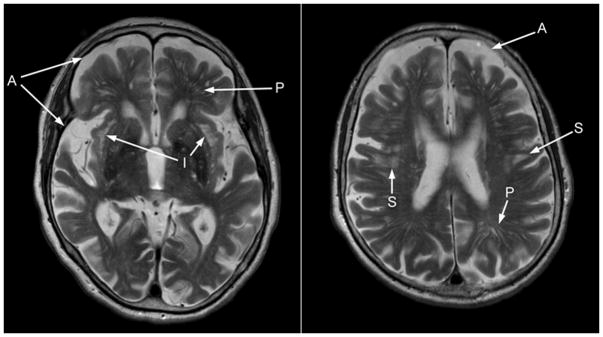
The subject presented with atrophy (A) and increased T2 signal intensities (S) in the deep white matter of the frontal and parietal regions. She also had a multitude of increased perivascular spaces (P), as well as a pronounced increase in T2 signal intensity in the sub-insular region (I).
The maternal great grandmother’s two sisters also carry the premutation and were seen in clinic. The oldest was 92 years of age (CGG 31, 62) and demonstrated no significant health problems. The youngest sister was 85 years of age (CGG 30, 62) and reported type II diabetes, congestive heart failure, coronary artery bypass surgery and a pacemaker because of arrythymias. We have noted an increase pacemakers in FXTAS presumably linked to autonomic dysfunction and FXTAS inclusions in pericardial ganglia and in cardiac tissue [Hunsaker et al., 2011]. She also reports bladder incontinence, which is associated with FXTAS [Jacquemont et al., 2003], although she did not have a clinically significant tremor or balance problem.
Two maternal great uncles tested positive for the premutation. The great uncles were 50 and 55 years of age, both carrying a premutation allele of 63 CGG repeats. Neither reported health problems. One obligate carrier, daughter of the proband’s maternal great uncle, reported experiencing panic attacks which are common in carriers [Bourgeois et al., 2011]. The proband’s maternal aunt is a carrier and reported sleep problems that was treated with alprazolam. Panic attacks and sleep problems have been reported as increased in carriers compared to controls [Bourgeois et al., 2011; Chonchaiya et al., 2010]. Otherwise the family history was negative.
Clinical Report Family 2
The female proband screened positive by blood spot as having a premutation allele with 59 CGG repeats. The results of the screening were reported by the Genetic Associate by phone and an initial visit was arranged by the project coordinator. At 6 months of age the proband and her parents were seen at the MIND Institute. Genetic counseling, blood draw on infant and parents to determine parent of origin, and medical assessment were performed. Peripheral blood confirmed that the proband had 59 CGG repeats, the mother had two normal alleles, and the father had a grey zone allele (52 CGG repeats). The proband underwent developmental assessments at 6 and 12 months of age. Scores on the Mullen and Vineland II were average at both time points. The Mullen ELC was 88 (6 months) and 93 (12 months), the Vineland II ABC was 86 and 94 respectively.
The proband’s father underwent neuropsychological testing, MRI, and medical exam. The medical evaluation revealed hypertension and treatment with antihypertensive meds was recommended in addition to exercise and antioxidants. Hypertension has been associated with the premutation [Hamlin et al., 2012]. The father reported a history of learning problems during primary school including ADHD symptoms. Scores on the WAIS-III were VIQ 92, PIQ 107, FSIQ 99. The split between the verbal and performance scores is significant.
Subsequent cascade testing revealed that the paternal grandmother carries a high premutation allele (109 CGG repeats). The difference between the repeat sizes of the father (52 CGG) and his mother (14, 109 CGG) is significant and it represents a remarkable contraction of CGG repeats from a high premutation to a gray zone allele thus demonstrating significant instability. Expansion of a maternal allele of 109 CGG repeats to a full mutation in the next generation is predicted to be 100% [Nolin et al., 2003; Yrigollen et al., 2011]. Contraction of CGG repeats from a premutation allele is documented but extremely rare (<1%) [Brown et al., 1996; Nolin et al., 2003; Rife et al., 2004]. This case demonstrates that newborn screening can uncover molecular results that challenge previously held tenets and would not have been seen in clinic without screening.
The paternal grandmother underwent a medical history and exam at 67 years of age. She reported early menarche at 10 years and a hysterectomy at 32 years for dysfunctional bleeding. She had a history of hypertension and an irregular heartbeat, which was treated by ablation. Strabismus was observed in the right eye. Finger to nose examination demonstrated a mild intention tremor of the left hand. She will be followed closely for any exacerbation of her neurological problems. Antioxidants have been recommended along with daily exercise.
Further cascade testing revealed that the 100 year old paternal great grandfather and one paternal uncle do not carry FMR1 mutations (Fig. 4). One additional paternal uncle and one paternal aunt are at risk to carry the premutation but have not yet been tested.
Figure 4.

Pedigree for Family 2: The arrow indicates the proband (IV-6). The proband’s older sister (IV-5), and paternal grandmother (II-2) are confirmed carriers of FMR1 premutations. The proband’s father (III-2) has a intermediate allele that represents a remarkable contraction during transmission. The CGG repeat sizes for each individual are noted.
Clinical Report Family 3
The female proband screened positive by blood spot for the FMR1 premutation with 55 CGG repeats. The Genetic Associate reported the result to the family by telephone and the initial visit was scheduled by the project coordinator. The first visit occurred when the proband was five months of age. At this time, the proband was seen with both parents who had blood drawn to determine parent of origin. A medical exam was performed on the newborn. Peripheral blood analysis confirmed that the proband had 55 CGG repeats and revealed that the mother had 19, 28 CGG repeats and the father had an intermediate allele (54 CGG repeats). The proband underwent subsequent developmental assessments at 6 and 12 months of age. Scores on the Mullen and Vineland II ranged from average to very high. The Mullen ELC was 98 (6 months) and 131 (12 months), the Vineland II ABC was 86 and 97 respectively.
Cascade testing revealed that the paternal grandmother has an intermediate allele (54 CGG repeats) and the paternal uncle and great uncle have normal alleles (Fig. 5).
Figure 5.
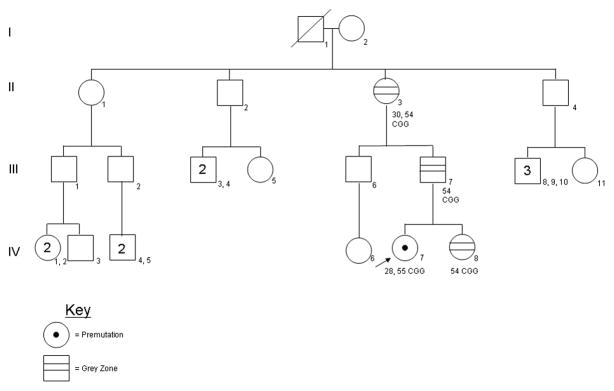
Pedigree for Family 3: The arrow indicates the proband (IV-7), who is a premutation carrier. The proband’s sister, father, and paternal grandmother all have intermediate alleles. The paternal uncle and one great uncle have tested negative for FMR1 mutations. The CGG repeat sizes for each individual are noted.
At the time of their initial visit, the parents of the proband conveyed their understanding that the likelihood of 54 CGG repeats expanding to the full mutation was impossible because the father passes only the premutation to his daughters. They continued to have concerns regarding the impact of this information upon a future pregnancy, however. The parents expressed concerns about gene stability, having another child with the premutation who would have to make her own reproductive decisions when of age, and the possibility of sex selection. The couple actively sought to address these issues by considering sperm sorting, conception without intervention and adoption. They explored these options with the Genetic Associate and other professionals.
During the time that the couple was considering their options, AGG anchor analysis became available [Yrigollen et al., 2011]. AGG analysis revealed the presence of 2 AGG interruptions in the paternal allele of 54 CGG repeats. The Genetic Associate explained to the couple that the presence of 2 anchors conferred FMR1 gene stability, although AGG analysis at the time was not considered to be diagnostic but instead a research finding [Yrigollen et al., 2012]. A few weeks later, the couple contacted the Genetic Associate to share the information that the wife was pregnant unexpectedly. Chorionic villus sampling was performed and revealed a female fetus with 54 CGG repeats.
DISCUSSION
Recent advances in targeted treatment and educational tools for those with fragile X emphasizes the need to reconsider NBS for fragile X mutations. The benefits and efficacy of new targeted treatment trials such as mGluR 5 antagonists, GABAA and B agonists and minocycline are beginning to be documented for the full mutation with FXS [Berry-Kravis et al., 2009; Hagerman et al., 2012; Jacquemont et al., 2011; Paribello et al., 2010]. The data in this area will grow in the next few years as new targeted treatments continue to be developed. However, most of the individuals identified in newborn screening for FMR1 mutations have the premutation. Since the premutation is less likely than the full mutation to cause intellectual disability and ASD but more likely than the full mutation to cause adult onset problems there is the risk that the family will worry about the late onset medical problems of their children. Typically we have seen that the families are very happy that their children with the premutation are doing well in the developmental testing and they are relieved that their child does not have the full mutation. The identification of other family members with the premutation allows for earlier treatment for some of the medical problems that are associated with the premutation including psychiatric problems, hypertension, cardiac arrythmias and tremor and ataxia.
The current NBS pilot project evaluates the benefits and risks associated with the identification of FMR1 mutations through newborn diagnosis and follow- up familial cascade testing. Without early diagnosis (as is the current experience) the benefits to both patient and family members are lost because diagnosis is delayed or, in the case of many premutation carriers, never made [Bailey 2004]. The landscape of weighing the risks against the benefits of fragile X NBS is a dynamic one that warrants continual reassessment [Bailey et al., 2006].
Benefits of Diagnosing those with the Full Mutation
Despite widespread clinical knowledge regarding the FMR1 gene, many families undergo a diagnostic odyssey before their child receives the diagnosis of FXS. Indeed a recent report from a national parent survey indicates that the age at diagnosis for the past 7 years continues to be approximately 35 to 37 months [Bailey et al., 2009]. This lack of early testing and diagnosis is often due to medical professionals’ dismissal of parental concern [Bailey et al., 2009]. NBS would eliminate the time between emerging clinical findings and parental concerns to genetic diagnosis. Early diagnosis through NBS simultaneously alleviates this anxiety and frustration currently experienced by too many parents and allows for immediate intervention to address and remediate the difficulties associated with fragile X in the newborn and early years of life. Early detection of a full mutation allows early and intensive educational and psychosocial interventions, which are well established and improve the outcome of infants and children [Bailey 2004]. In addition, psychologists and genetic counselors can also provide assistance to families following newborn diagnosis including future family planning. The recent utilization of AGG repeat anchors to further delineate for carriers the risk of expansion to a full in the next generation can benefit many families; when 2 AGG anchors are present in a premutation carrier the risk of expansion into the next generation drops remarkably compared to previous estimates [Yrigollen et al., 2012].
Benefits of Diagnosing Premutation Carriers
The prevalence of the premutation (55 to 200 CGG repeats) is 1 in 130–250 females and 1 in 250–810 males in the general population [Dombrowski et al., 2002; Fernandez-Carvajal et al., 2009; Song et al., 2003]. Until recently the premutation was largely ignored because it was not associated with clinical findings. Recent research suggests that, while most children with the premutation do well, approximately 10–20% of the males may experience learning problems, ADHD, anxiety and/ or an ASD diagnosis in childhood [Aziz et al., 2003; Bailey et al., 2008a; Chonchaiya et al., 2012; Clifford et al., 2007; Cornish et al., 2005; Farzin et al., 2006]. Seizures, experienced by approximately 10% of carriers [Bailey et al., 2008a] are associated with an increased risk for ASD in young males with the premutation [Chonchaiya et al., 2012]. Enhanced spikes compared to controls are also present in premutation neurons studied in culture [Cao et al., 2012].
Therefore in children with the premutation the physician and family need to be vigilant for any developmental problems, seizures, ADHD or anxiety that may develop. NBS and cascade testing also reveals typically a variety of medical problems that may occur in multiple extended relatives as seen in our case reports.
Some premutation carriers are at increased risk for a variety of late-onset disorders. Both males and females have an increased risk for anxiety, ADHD, and mood disorders including depression in mid adulthood [Bourgeois et al., 2011; Hessl et al., 2005; Hessl et al., 2011; Roberts et al., 2009; Rodriguez-Revenga et al., 2009]. Males and females over age 50 are at risk for FXTAS, and additional medical problems, such as neuropathy, fibromyalgia, hypothyrodism, hypertension, arrhythmias and sleep apnea are common and may begin before the onset of FXTAS [Coffey S.M. et al., 2008; Goodrich-Hunsaker et al., 2011; Hamlin et al., 2011; Hamlin et al., 2012; Hunsaker et al., 2011]. There is also evidence of mitochondrial disease and enhanced oxidative stress in human cells with the premutation [Napoli et al., 2011; Ross-Inta et al., 2010] in addition to evidence of early premutation neuronal cell death compared to controls [Chen, 2010]. The cellular data suggests that antioxidants may be beneficial for the enhanced oxidative stress in those with the full mutation [de Diego-Otero et al., 2009; Romero-Zerbo et al., 2009] and in those with the premutation [Chen et al., 2010; Napoli et al., 2011; Ross-Inta et al., 2010], therefore antioxidants are routinely recommended for carriers. In addition, avoidance of environmental toxins, such as organophosphate insecticides, drugs of abuse, chemotherapy and smoking is recommended because they may worsen premutation medical problems [O’Dwyer et al., 2005; Paul et al., 2010]. Premutation carriers likely benefit from healthy lifestyles that avoid oxidative stress or environmental toxins. When a carrier is diagnosed, lifestyle changes should be recommended by the physician who is caring for the patient. These can include cessation of alcohol use and the implementation of a selective serotonin reuptake inhibitor (SSRI) and exercise to treat depression. Further, it is likely that avoidance of environmental exposures that cause enhanced toxicity to the neurons or other cells vulnerable to the premutation, such as the granulosa cells of the ovary, may decrease the risk of these late onset problems for individuals with the premutation [Hagerman et al., 2008; Jacquemont et al., 2004a; Paul et al., 2010; Sherman 2000; Sullivan et al., 2011]. The use of antioxidants in early childhood and throughout the life span may also be beneficial for those with the premutation and the full mutation because they have been helpful in animal and cellular [Cao et al., 2012; Chen et al., 2010; de Diego-Otero et al., 2009; Napoli et al., 2011; Romero-Zerbo et al., 2009; Ross-Inta et al., 2010]. However, controlled trials have not been carried out in patients regarding the benefits of antioxidants in preventing premutation disorders.
Females carry the additional risk for fragile X-associated Primary Ovarian Insufficiency (FXPOI), which is cessation of menstruation prior to age 40. The premutation is the most common inherited cause of POI [Sullivan et al., 2011]. These medical problems can also be addressed and even treated when the premutation is identified if treatment was not initiated before.
For the one third or more of male carriers who will develop FXTAS, cascade testing that identifies these individuals prior to the onset of symptoms can prevent misdiagnosis later in life [Jacquemont et al., 2004a]. This represents an improvement for many men with FXTAS who are misdiagnosed as having Parkinson’s, other ataxias, stroke, Alzheimer’s, multiple sclerosis and other movement disorders [Hall et al., 2005; Hedrich et al., 2005].
Benefits of Cascade Testing
The same benefits conferred by NBS to those with both the pre and full fragile X mutations can also accrue to family members of the identified infant, due to the X-linked inheritance pattern, through a program of cascade testing. A detailed family history must be obtained as it serves as a guide to identify family members at risk for having an FMR1 mutation. A newborn carrying the fragile X full mutation identifies the mother’s family as being at risk to have offspring with fragile X and autism. A newborn carrying the premutation necessitates one or both parents being tested to identify the carrier. Cascade testing thus represents an additional benefit of NBS in that it expands the circle of diagnosis of both the full and premutations and the benefits, discussed above, to be derived from such diagnosis. In the absence of NBS, the need for diagnostic testing goes undiscovered until symptoms begin, meaning that the opportunity for prevention is lost. In this way, the opportunity to offer cascade testing represents yet another benefit of NBS in the eyes of many. It is through cascade testing that these conditions are identified and diagnosed accurately and early and that treatments are delivered for the many problems associated with fragile X mutations.
Concerns Associated with FX Newborn Screening and Cascade Testing
Despite the benefits that expanded detection of individuals affected by FMR1 gene mutations can confer, detection is associated with very legitimate concerns. Newborn screening for any genetic condition can increase anxiety in parents who are approached for consent and for those who have an identified infant. It has been postulated that parental anxiety around genetic risks may interfere with infant bonding and change the parent-child relationship through increased parental vigilance, however, there is little evidence to support this [Bailey et al., 2008b; Bailey et al., 2008c]. Cascade testing may increase anxiety in extended family members because there is the prospect of diagnosing risks in asymptomatic children and adult relatives.
Questions remain about the extent of benefit a diagnosis confers on asymptomatic people. For many who test positive, the information may have no practical use. For example, the older sister, who is a premutation carrier, of the maternal great grandmother in Family 1 discussed above has lived into her 90s with no reported health problems of any significance. Based on what we know today about the FMR1 gene, she would represent the typical premutation carrier diagnosed by NBS or cascade testing. What benefit would a diagnosis at birth, or even in adulthood, have conferred to her? How might that information have affected her throughout the course of her life? She may have expected the onset of various symptoms for decades and it is hard to know what the psychological impact of such waiting might have been. Thus, what benefit would it have been for her to have known her risks?
A diagnosis can have real life consequences for people screened at birth or tested in childhood as they mature and learn this genetic information about themselves. Even though they may understand that testing was done out of concern for their health and future well-being, there is the possibility that some will nevertheless feel wronged by having been diagnosed because they would prefer not to know future health risk information about themselves, or have others know it about them either. There is also the possibility that an asymptomatic premutation carrier will feel the burden to disclose one’s premutation to a potential life partner, due to the premutation’s implications for reproductive decision-making or even one’s own future health.
Cascade testing also creates an additional challenge. Benefits to family members are achievable only if parents notify other family members, who in turn notify other family members. Test results are part of the confidential medical record but the well-being of other family members needs to be given some consideration in the counseling process. Thus, cascade testing may elicit the ethical issues that arise when parents of affected newborns do not want other family members to be notified [Yarborough et al., 1989].
In Defense of a Voluntary Approach to Expanded Detection
We think our review of the tangible benefits and concerns related to Fragile X NBS and cascade testing reaffirms the fact that, like so many other new medical technologies, DNA testing is an important medical advance that needs to be used with a measure of caution. Fragile X NBS has tremendous potential to increase the number of timely diagnoses. Each of these diagnoses will present opportunities to improve the health and quality of life of those who are diagnosed. Further, as our experience has shown, complementing NBS with a program of cascade testing is a largely untapped potential benefit of NBS. Cascade testing can detect many additional affected individuals whose risks would otherwise remain unknown. Our experience has also shown that such testing will identify people who will never suffer ill health effects related to their genetic diagnosis. Whether the knowledge that testing confers upon these individuals will be experienced as beneficial, benign, or harmful is unknown at this time and it requires further study.
We have been able to traverse the challenges posed by fragile X NBS and cascade testing by making both of them voluntary. Even though NBS is typically thought of as a mandatory public health undertaking, it is important to keep in mind that it began as a voluntary health procedure [Baily and Murray 2008]. Voluntary NBS can play an important therapeutic role for conditions like fragile X that do not yet meet the current criteria for state mandated NBS, criteria that continue to generate controversy [Botkin et al., 2006]. Such voluntary testing is comparable in many respects to offering FMR1 screening to the pre-pregnant or pregnant populations, which pilot studies also suggest might be favorably received [Anido et al., 2007; Anido et al., 2005; Fanos et al., 2006; McConkie-Rosell et al., 2007; Metcalfe et al., 2008].
Voluntary testing is not the only safeguard required at this time to address the ethical complexities of fragile X NBS and cascade testing. It is important that it is combined with access to genetic counseling and medical follow-up, both of which have been central to our research project to date. Any program designed to increase detection of fragile X gene mutations should provide access to counseling and other resources that can mitigate concerns by providing follow-up assessments of the identified individuals. Follow-up can provide ongoing health monitoring, assuage anxiety and provide reassurance regarding the development of individuals. The voluntary aspect of our testing, coupled with the genetic counseling and appropriate medical follow-up at the heart of our program, has permitted us to serve and respect the individuals to whom testing has been offered.
CONCLUSION
We believe that fragile X NBS, accompanied by cascade testing, is the most effective means by which to avoid misdiagnosis and delay of treatment to affected patients. Yet, as is true of other disorders included in state newborn screening programs, fragile X NBS identifies significantly more carriers than individuals with FXS. Because many of these premutation carriers will likely develop some of the symptoms that occur in childhood, such as anxiety or in adulthood including FXPOI, psychiatric symptoms, hypertension, hypothyroidism or neurological problems we are in support of NBS and cascade testing of extended family members.
Our experience to date indicates both the benefits and concerns historically associated with fragile X NBS. As our clinical reports show, we have provided accurate and early diagnosis of premutation carrier infants and thus we are now able to provide important guidance and surveillance to their parents. We have also identified many additional premutation carriers through cascade testing who otherwise would be undiagnosed. The medical histories suggest that some of these premutation carriers could have benefitted from having this genetic knowledge but it is less clear how others, who have been healthy, could have benefited from a diagnosis. In short, our experience to date highlights both the benefits and drawbacks of screening for FMR1 mutations.
We caution that NBS for fragile X and cascade testing must be accompanied by genetic counseling that adheres to clinical guidelines previously established [McConkie-Rosell et al., 2007; McConkie-Rosell et al., 2005]. Furthermore, it is imperative that long-term clinical follow-up be offered to individuals who test positive for FMR1 mutations. These services temper the increase in anxiety that may be experienced by parents of identified infants and extended family members.
Figure 1.

Flow Chart: This chart indicates the progression of contact, enrollment, and participation in Newborn Screening study.
Acknowledgments
This work was supported by a National Institute of Health grants R01HD055510, 3P30-HD02274-35S1, HD036071 and 5R01HD040661-11. We would like to thank all the families that kindly participated in the research.
This work is dedicated to the memory of Matteo.
References
- Adams JS, Adams PE, Nguyen D, Brunberg JA, Tassone F, Zhang W, Koldewyn K, Rivera SM, Grigsby J, Zhang L, DeCarli C, Hagerman PJ, Hagerman RJ. Volumetric brain changes in females with fragile X-associated tremor/ataxia syndrome (FXTAS) Neurology. 2007;69(9):851–9. doi: 10.1212/01.wnl.0000269781.10417.7b. [DOI] [PubMed] [Google Scholar]
- Angkustsiri K, Wirojanan J, Deprey LJ, Gane LW, Hagerman RJ. Fragile X syndrome with anxiety disorder and exceptional verbal intelligence. Am J Med Genet A. 2008;146(3):376–9. doi: 10.1002/ajmg.a.32118. [DOI] [PubMed] [Google Scholar]
- Anido A, Carlson LM, Sherman SL. Attitudes toward fragile X mutation carrier testing from women identified in a general population survey. J Genet Couns. 2007;16(1):97–104. doi: 10.1007/s10897-006-9049-0. [DOI] [PubMed] [Google Scholar]
- Anido A, Carlson LM, Taft L, Sherman SL. Women’s attitudes toward testing for fragile X carrier status: a qualitative analysis. J Genet Couns. 2005;14(4):295–306. doi: 10.1007/s10897-005-1159-6. [DOI] [PubMed] [Google Scholar]
- Aziz M, Stathopulu E, Callias M, Taylor C, Turk J, Oostra B, Willemsen R, Patton M. Clinical features of boys with fragile X premutations and intermediate alleles. Am J Med Genet. 2003;121B(1):119–27. doi: 10.1002/ajmg.b.20030. [DOI] [PubMed] [Google Scholar]
- Bailey DB., Jr Newborn screening for fragile X syndrome. Ment Retard Dev Disabil Res Rev. 2004;10(1):3–10. doi: 10.1002/mrdd.20002. [DOI] [PubMed] [Google Scholar]
- Bailey DB, Jr, Beskow LM, Davis AM, Skinner D. Changing perspectives on the benefits of newborn screening. Ment Retard Dev Disabil Res Rev. 2006;12(4):270–79. doi: 10.1002/mrdd.20119. [DOI] [PubMed] [Google Scholar]
- Bailey DB, Jr, MR, Bishop E, Holiday D. No change in the age of diagnosis for fragile x syndrome: Findings from a national parent survey. Pediatrics. 2009;124(2):527–533. doi: 10.1542/peds.2008-2992. [DOI] [PubMed] [Google Scholar]
- Bailey DB, Jr, Raspa M, Olmsted M, Holiday DB. Co-Occurring Conditions Associated With FMR1 Gene Variations: Findings From a National Parent Survey. Am J Med Genet. 2008a;146A(16):2060–2069. doi: 10.1002/ajmg.a.32439. [DOI] [PubMed] [Google Scholar]
- Bailey DB, Jr, Sideris J, Roberts JE, Hatton D. Child and genetic variables associated with maternal adaptation to fragile X syndrome: a multidimensional analysis. Am J Med Genet A. 2008b;146(6):720–9. doi: 10.1002/ajmg.a.32240. [DOI] [PubMed] [Google Scholar]
- Bailey DB, Jr, Skinner D, Davis AM, Whitmarsh I, Powell C. Ethical, legal, and social concerns about expanded newborn screening: fragile X syndrome as a prototype for emerging issues. Pediatrics. 2008c;121(3):e693–704. doi: 10.1542/peds.2007-0820. [DOI] [PubMed] [Google Scholar]
- Baily MA, Murray TH. Ethics, evidence, and cost in newborn screening. Hastings Cent Rep. 2008;38(3):23–31. doi: 10.1353/hcr.0.0009. [DOI] [PubMed] [Google Scholar]
- Berry-Kravis EM, Hessl D, Coffey S, Hervey C, Schneider A, Yuhas J, Hutchison J, Snape M, Tranfaglia M, Nguyen DV, Hagerman R. A pilot open-label single-dose trial of fenobam in adults with fragile X syndrome. J Med Genet. 2009;46(4):266–271. doi: 10.1136/jmg.2008.063701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botkin JR, Clayton EW, Fost NC, Burke W, Murray TH, Baily MA, Wilfond B, Berg A, Ross LF. Newborn screening technology: proceed with caution. Pediatrics. 2006;117(5):1793–9. doi: 10.1542/peds.2005-2547. [DOI] [PubMed] [Google Scholar]
- Bourgeois JA, Seritan AL, Casillas EM, Hessl D, Schneider A, Yang Y, Kaur I, Cogswell JB, Nguyen DV, Hagerman RJ. Lifetime prevalence of mood and anxiety disorders in fragile x premutation carriers. J Clin Psychiatry. 2011;72(2):175–182. doi: 10.4088/JCP.09m05407blu. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown WT, Houck GE, Jr, Ding X, Zhong N, Nolin S, Glicksman A, Dobkin C, Jenkins EC. Reverse mutations in the fragile X syndrome. Am J Med Genet. 1996;64(2):287–92. doi: 10.1002/(SICI)1096-8628(19960809)64:2<287::AID-AJMG11>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- Cao Z, Hulsizer S, Tassone F, Tang HT, Hagerman RJ, Rogawski MA, Hagerman PJ, Pessah IN. Clustered burst firing in FMR1 premutation hippocampal neurons: amelioration with allopregnanolone. Hum Mol Genet. 2012;21(13):2923–35. doi: 10.1093/hmg/dds118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Hadd AG, Sah S, Houghton JF, Filipovic-Sadic S, Zhang W, Hagerman PJ, Tassone F, Latham GJ. High-resolution methylation polymerase chain reaction for fragile X analysis: evidence for novel FMR1 methylation patterns undetected in Southern blot analyses. Genet Med. 2011;13(6):528–38. doi: 10.1097/GIM.0b013e31820a780f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Tassone F, Berman RF, Hagerman PJ, Hagerman RJ, Willemsen R, Pessah IN. Murine hippocampal neurons expressing Fmr1 gene premutations show early developmental deficits and late degeneration. Hum Mol Genet. 2010;19(1):196–208. doi: 10.1093/hmg/ddp479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chonchaiya W, Au J, Schneider A, Hessl D, Harris SW, Laird M, Mu Y, Tassone F, Nguyen DV, Hagerman RJ. Increased prevalence of seizures in boys who were probands with the FMR1 premutation and comorbid autism spectrum disorder. Hum Genet. 2012;131(4):581–9. doi: 10.1007/s00439-011-1106-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chonchaiya W, Nguyen DV, Au J, Campos L, Berry-Kravis EM, Lohse K, Mu Y, Utari A, Hervey C, Wang L, Sorensen P, Cook K, Gane L, Tassone F, Hagerman RJ. Clinical involvement in daughters of men with fragile X-associated tremor ataxia syndrome. Clin Genet. 2010;78(1):38–46. doi: 10.1111/j.1399-0004.2010.01448.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chonchaiya W, Schneider A, Hagerman RJ. Fragile X: a family of disorders. Adv Pediatr. 2009;56:165–86. doi: 10.1016/j.yapd.2009.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clifford S, Dissanayake C, Bui QM, Huggins R, Taylor AK, Loesch DZ. Autism spectrum phenotype in males and females with fragile X full mutation and premutation. J Autism Dev Disord. 2007;37(4):738–47. doi: 10.1007/s10803-006-0205-z. [DOI] [PubMed] [Google Scholar]
- Coffee B, Keith K, Albizua I, Malone T, Mowrey J, Sherman SL, Warren ST. Incidence of fragile X syndrome by newborn screening for methylated FMR1 DNA. Am J Hum Genet. 2009;85(4):503–14. doi: 10.1016/j.ajhg.2009.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffey SM, Cook K, Tartaglia N, Tassone F, Nguyen DV, Pan R, Bronsky HE, Yuhas J, Borodyanskaya M, Grigsby J, Doerflinger M, Hagerman PJ, RJH Expanded clinical phenotype of women with the FMR1 premutation. Am J Med Genet A. 2008;146A(8):1009–16. doi: 10.1002/ajmg.a.32060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornish KM, Kogan C, Turk J, Manly T, James N, Mills A, Dalton A. The emerging fragile X premutation phenotype: Evidence from the domain of social cognition. Brain Cogn. 2005;57(1):53–60. doi: 10.1016/j.bandc.2004.08.020. [DOI] [PubMed] [Google Scholar]
- de Diego-Otero Y, Romero-Zerbo Y, el Bekay R, Decara J, Sanchez L, Rodriguezde Fonseca F, del Arco-Herrera I. Alpha-tocopherol protects against oxidative stress in the fragile X knockout mouse: an experimental therapeutic approach for the Fmr1 deficiency. Neuropsychopharmacology. 2009;34(4):1011–26. doi: 10.1038/npp.2008.152. [DOI] [PubMed] [Google Scholar]
- de Vries BB, Wiegers AM, Smits AP, Mohkamsing S, Duivenvoorden HJ, Fryns JP, Curfs LM, Halley DJ, Oostra BA, van den Ouweland AM, Niermeijer MF. Mental status of females with an FMR1 gene full mutation. Am J Hum Genet. 1996;58(5):1025–32. [PMC free article] [PubMed] [Google Scholar]
- Dombrowski C, Levesque ML, Morel ML, Rouillard P, Morgan K, Rousseau F. Premutation and intermediate-size FMR1 alleles in 10 572 males from the general population: loss of an AGG interruption is a late event in the generation of fragile X syndrome alleles. Hum Mol Genet. 2002;11(4):371–378. doi: 10.1093/hmg/11.4.371. [DOI] [PubMed] [Google Scholar]
- Fanos JH, Spangner KA, Musci TJ. Attitudes toward prenatal screening and testing for Fragile X. Genet Med. 2006;8(2):129–33. doi: 10.1097/01.gim.0000200158.66554.7f. [DOI] [PubMed] [Google Scholar]
- Farzin F, Perry H, Hessl D, Loesch D, Cohen J, Bacalman S, Gane L, Tassone F, Hagerman P, Hagerman R. Autism spectrum disorders and attention-deficit/hyperactivity disorder in boys with the fragile X premutation. J Dev Behav Pediatr. 2006;27(2 Suppl):S137–44. doi: 10.1097/00004703-200604002-00012. [DOI] [PubMed] [Google Scholar]
- Fernandez-Carvajal I, Walichiewicz P, Xiaosen X, Pan R, Hagerman PJ, Tassone F. Screening for expanded alleles of the FMR1 gene in blood spots from newborn males in a Spanish population. J Mol Diagn. 2009;11(4):324–9. doi: 10.2353/jmoldx.2009.080173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodrich-Hunsaker NJ, Wong LM, McLennan Y, Tassone F, Harvey D, Rivera SM, Simon TJ. Adult Female Fragile X Premutation Carriers Exhibit Age- and CGG Repeat Length-Related Impairments on an Attentionally Based Enumeration Task. Front Hum Neurosci. 2011;5:63. doi: 10.3389/fnhum.2011.00063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagerman PJ. FMR1 Gene Expression and Prospects for Gene Therapy. In: Hagerman RJ, Hagerman PJ, editors. Fragile X Syndrome: Diagnosis, Treatment and Research. 3. Baltimore: The Johns Hopkins University Press; 2002. pp. 465–494. [Google Scholar]
- Hagerman PJ, Hagerman RJ. The fragile-X premutation: a maturing perspective. Am J Hum Genet. 2004;74(5):805–816. doi: 10.1086/386296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagerman R, Lauterborn J, Au J, Berry-Kravis E. Fragile X syndrome and targeted treatment trials. Results Probl Cell Differ. 2012;54:297–335. doi: 10.1007/978-3-642-21649-7_17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagerman RJ, Hall DA, Coffey S, Leehey M, Bourgeois J, Gould J, Zhang L, Seritan A, Berry-Kravis E, Olichney J, Miller JW, Fong AL, Carpenter R, Bodine C, Gane LW, Rainin E, Hagerman H, Hagerman PJ. Treatment of fragile X-associated tremor ataxia syndrome (FXTAS) and related neurological problems. Clin Interv Aging. 2008;3(2):251–62. doi: 10.2147/cia.s1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagerman RJ, Leavitt BR, Farzin F, Jacquemont S, Greco CM, Brunberg JA, Tassone F, Hessl D, Harris SW, Zhang L, Jardini T, Gane LW, Ferranti J, Ruiz L, Leehey MA, Grigsby J, Hagerman PJ. Fragile-X-associated tremor/ataxia syndrome (FXTAS) in females with the FMR1 premutation. Am J Hum Genet. 2004;74(5):1051–1056. doi: 10.1086/420700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall DA, Berry-Kravis E, Jacquemont S, Rice CD, Cogswell J, Zhang L, Hagerman RJ, Hagerman PJ, Leehey MA. Initial diagnoses given to persons with the fragile X associated tremor/ataxia syndrome (FXTAS) Neurology. 2005;65(2):299–301. doi: 10.1212/01.wnl.0000168900.86323.9c. [DOI] [PubMed] [Google Scholar]
- Hall SS, Lightbody AA, Reiss AL. Compulsive, self-injurious, and autistic behavior in children and adolescents with fragile X syndrome. Am J Ment Retard. 2008;113(1):44–53. doi: 10.1352/0895-8017(2008)113[44:CSAABI]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- Hamlin A, Liu Y, Nguyen DV, Tassone F, Zhang L, Hagerman RJ. Sleep apnea in fragile X premutation carriers with and without FXTAS. Am J Med Genet B Neuropsychiatr Genet. 2011;156B(8):923–928. doi: 10.1002/ajmg.b.31237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamlin AA, Sukharev D, Campos L, Mu Y, Tassone F, Hessl D, Nguyen DV, Loesch D, Hagerman RJ. Hypertension in FMR1 premutation males with and without fragile X-associated tremor/ataxia syndrome (FXTAS) Am J Med Genet A. 2012;158A(6):1304–9. doi: 10.1002/ajmg.a.35323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris SW, Hessl D, Goodlin-Jones B, Ferranti J, Bacalman S, Barbato I, Tassone F, Hagerman PJ, Herman H, Hagerman RJ. Autism profiles of males with fragile X syndrome. American Journal of Mental Retardation. 2008;113(6):427–38. doi: 10.1352/2008.113:427-438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatton DD, Sideris J, Skinner M, Mankowski J, Bailey DB, Jr, Roberts JE, Mirrett P. Autistic behavior in children with fragile X syndrome: Prevalence, stability, and the impact of FMRP. Am J Med Genet A. 2006;140(17):1804–1813. doi: 10.1002/ajmg.a.31286. [DOI] [PubMed] [Google Scholar]
- Hedrich K, Pramstaller PP, Stubke K, Hiller A, Kabakci K, Purmann S, Kasten M, Scaglione C, Schwinger E, Volkmann J, Kostic V, Vieregge P, Martinelli P, Abbruzzese G, Klein C, Zuhlke C. Premutations in the FMR1 gene as a modifying factor in Parkin-associated Parkinson’s disease? Mov Disord. 2005;20(8):1060–2. doi: 10.1002/mds.20512. [DOI] [PubMed] [Google Scholar]
- Hessl D, Tassone F, Loesch DZ, Berry-Kravis E, Leehey MA, Gane LW, Barbato I, Rice C, Gould E, Hall DA, Grigsby J, Wegelin JA, Harris S, Lewin F, Weinberg D, Hagerman PJ, Hagerman RJ. Abnormal elevation of FMR1 mRNA is associated with psychological symptoms in individuals with the fragile X premutation. Am J Med Genet B Neuropsychiatr Genet. 2005;139(1):115–121. doi: 10.1002/ajmg.b.30241. [DOI] [PubMed] [Google Scholar]
- Hessl D, Wang JM, Schneider A, Koldewyn K, Le L, Iwahashi C, Cheung K, Tassone F, Hagerman PJ, Rivera SM. Decreased Fragile X Mental Retardation Protein Expression Underlies Amygdala Dysfunction in Carriers of the Fragile X Premutation. Biol Psychiatry. 2011;70(9):859–865. doi: 10.1016/j.biopsych.2011.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunsaker MR, Greco CM, Spath MA, Smits AP, Navarro CS, Tassone F, Kros JM, Severijnen LA, Berry-Kravis EM, Berman RF, Hagerman PJ, Willemsen R, Hagerman RJ, Hukema RK. Widespread non-central nervous system organ pathology in fragile X premutation carriers with fragile X-associated tremor/ataxia syndrome and CGG knock-in mice. Acta Neuropathol. 2011;122(4):467–479. doi: 10.1007/s00401-011-0860-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacquemont S, Curie A, des Portes V, Torrioli MG, Berry-Kravis E, Hagerman RJ, Ramos FJ, Cornish K, He Y, Paulding C, Neri G, Chen F, Hadjikhani N, Martinet D, Meyer J, Beckmann JS, Delange K, Brun A, Bussy G, Gasparini F, Hilse T, Floesser A, Branson J, Bilbe G, Johns D, Gomez-Mancilla B. Epigenetic modification of the FMR1 gene in fragile X syndrome is associated with differential response to the mGluR5 antagonist AFQ056. Sci Transl Med. 2011;3(64):64ra1. doi: 10.1126/scitranslmed.3001708. [DOI] [PubMed] [Google Scholar]
- Jacquemont S, Farzin F, Hall D, Leehey M, Tassone F, Gane L, Zhang L, Grigsby J, Jardini T, Lewin F, Berry-Kravis E, Hagerman PJ, Hagerman RJ. Aging in individuals with the FMR1 mutation. Am J Ment Retard. 2004a;109(2):154–64. doi: 10.1352/0895-8017(2004)109<154:AIIWTF>2.0.CO;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacquemont S, Hagerman RJ, Leehey M, Grigsby J, Zhang L, Brunberg JA, Greco C, Des Portes V, Jardini T, Levine R, Berry-Kravis E, Brown WT, Schaeffer S, Kissel J, Tassone F, Hagerman PJ. Fragile X premutation tremor/ataxia syndrome: molecular, clinical, and neuroimaging correlates. Am J Hum Genet. 2003;72(4):869–878. doi: 10.1086/374321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacquemont S, Hagerman RJ, Leehey MA, Hall DA, Levine RA, Brunberg JA, Zhang L, Jardini T, Gane LW, Harris SW, Herman K, Grigsby J, Greco CM, Berry-Kravis E, Tassone F, Hagerman PJ. Penetrance of the fragile X-associated tremor/ataxia syndrome in a premutation carrier population. JAMA. 2004b;291(4):460–469. doi: 10.1001/jama.291.4.460. [DOI] [PubMed] [Google Scholar]
- Kaufmann WE, Cortell R, Kau AS, Bukelis I, Tierney E, Gray RM, Cox C, Capone GT, Stanard P. Autism spectrum disorder in fragile X syndrome: communication, social interaction, and specific behaviors. Am J Med Genet. 2004;129A(3):225–234. doi: 10.1002/ajmg.a.30229. [DOI] [PubMed] [Google Scholar]
- Leehey MA, Legg W, Tassone F, Hagerman R. Fibromyalgia in fragile X mental retardation 1 gene premutation carriers. Rheumatology (Oxford) 2011;50(12):2233–2236. doi: 10.1093/rheumatology/ker273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddalena A, Richards CS, McGinniss MJ, Brothman A, Desnick RJ, Grier RE, Hirsch B, Jacky P, McDowell GA, Popovich B, Watson M, Wolff DJ. Technical standards and guidelines for fragile X: the first of a series of disease-specific supplements to the Standards and Guidelines for Clinical Genetics Laboratories of the American College of Medical Genetics. Quality Assurance Subcommittee of the Laboratory Practice Committee. Genet Med. 2001;3(3):200–5. doi: 10.1097/00125817-200105000-00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McConkie-Rosell A, Abrams L, Finucane B, Cronister A, Gane LW, Coffey SM, Sherman S, Nelson LM, Berry-Kravis E, Hessl D, Chiu S, Street N, Vatave A, Hagerman RJ. Recommendations from multi-disciplinary focus groups on cascade testing and genetic counseling for fragile X-associated disorders. J Genet Couns. 2007;16(5):593–606. doi: 10.1007/s10897-007-9099-y. [DOI] [PubMed] [Google Scholar]
- McConkie-Rosell A, Finucane BM, Cronister AC, Abrams L, Bennett RL, Pettersen BJ. Genetic counseling for fragile X syndrome: Updated recommendations of the National Society of Genetic Counselors. J Genet Couns. 2005;14(4):249–270. doi: 10.1007/s10897-005-4802-x. [DOI] [PubMed] [Google Scholar]
- Metcalfe S, Jacques A, Archibald A, Burgess T, Collins V, Henry A, McNamee K, Sheffield L, Slater H, Wake S, Cohen J. A model for offering carrier screening for fragile X syndrome to nonpregnant women: results from a pilot study. Genet Med. 2008;10(7):525–35. doi: 10.1097/gim.0b013e31817c036e. [DOI] [PubMed] [Google Scholar]
- Mullen EM. Mullen Scales of Early Learning. Circle Pines: American Guidance Service; 1995. [Google Scholar]
- Napoli E, Ross-Inta C, Wong S, Omanska-Klusek A, Barrow C, Iwahashi C, Garcia-Arocena D, Sakaguchi D, Berry-Kravis E, Hagerman R, Hagerman PJ, Giulivi C. Altered zinc transport disrupts mitochondrial protein processing/import in fragile X-associated tremor/ataxia syndrome. Hum Mol Genet. 2011;20(15):3079–92. doi: 10.1093/hmg/ddr211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolin SL, Brown WT, Glicksman A, Houck GE, Gargano AD, Sullivan A, Biancalana V, Brondum-Nielsen K, Hjalgrim H, Holinski-Feder E, Kooy F, Longshore J, Macpherson J, Mandel JL, Matthijs G, Rousseau F, Steinbach P, Vaisanen ML, von Koskull H, Sherman S. Expansion of the fragile X CGG repeat in females with premutation or intermediate alleles. Am J Hum Genet. 2003;72:454–464. doi: 10.1086/367713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Dwyer JP, Clabby C, Crown J, Barton DE, Hutchinson M. Fragile X-associated tremor/ataxia syndrome presenting in a woman after chemotherapy. Neurology. 2005;65(2):331–2. doi: 10.1212/01.wnl.0000168865.36352.53. [DOI] [PubMed] [Google Scholar]
- Paribello C, Tao L, Folino A, Berry-Kravis E, Tranfaglia M, Ethell IM, Ethell DW. Open-label add-on treatment trial of minocycline in fragile X syndrome. BMC Neurol. 2010;10:91. doi: 10.1186/1471-2377-10-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul R, Pessah IN, Gane L, Ono M, Hagerman PJ, Brunberg JA, Tassone F, Bourgeois JA, Adams PE, Nguyen DV, Hagerman R. Early onset of neurological symptoms in fragile X premutation carriers exposed to neurotoxins. Neurotoxicology. 2010;31(4):399–402. doi: 10.1016/j.neuro.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riddle JE, Cheema A, Sobesky WE, Gardner SC, Taylor AK, Pennington BF, Hagerman RJ. Phenotypic involvement in females with the FMR1 gene mutation. Am J Ment Retard. 1998;102(6):590–601. doi: 10.1352/0895-8017(1998)102<0590:piifwt>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- Rife M, Badenas C, Quinto L, Puigoriol E, Tazon B, Rodriguez-Revenga L, Jimenez L, Sanchez A, Mila M. Analysis of CGG variation through 642 meioses in Fragile X families. Mol Hum Reprod. 2004;10(10):773–6. doi: 10.1093/molehr/gah102. [DOI] [PubMed] [Google Scholar]
- Roberts JE, Bailey DB, Jr, Mankowski J, Ford A, Sideris J, Weisenfeld LA, Heath TM, Golden RN. Mood and anxiety disorders in females with the FMR1 premutation. Am J Med Genet B Neuropsychiatr Genet. 2009;150B(1):130–9. doi: 10.1002/ajmg.b.30786. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Revenga L, Madrigal I, Pagonabarraga J, Xuncla M, Badenas C, Kulisevsky J, Gomez B, Mila M. Penetrance of FMR1 premutation associated pathologies in fragile X syndrome families. Eur J Hum Genet. 2009;17(10):1359–62. doi: 10.1038/ejhg.2009.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers SJ, Wehner EA, Hagerman RJ. The behavioral phenotype in fragile X: Symptoms of autism in very young children with fragile X syndrome, idiopathic autism, and other developmental disorders. J Dev Behav Pediatr. 2001;22(6):409–417. doi: 10.1097/00004703-200112000-00008. [DOI] [PubMed] [Google Scholar]
- Romero-Zerbo Y, Decara J, el Bekay R, Sanchez-Salido L, Del Arco-Herrera I, de Fonseca FR, de Diego-Otero Y. Protective effects of melatonin against oxidative stress in Fmr1 knockout mice: a therapeutic research model for the fragile X syndrome. J Pineal Res. 2009;46(2):224–34. doi: 10.1111/j.1600-079X.2008.00653.x. [DOI] [PubMed] [Google Scholar]
- Ross-Inta C, Omanska-Klusek A, Wong S, Barrow C, Garcia-Arocena D, Iwahashi C, Berry-Kravis E, Hagerman RJ, Hagerman PJ, Giulivi C. Evidence of mitochondrial dysfunction in fragile X-associated tremor/ataxia syndrome. Biochem J. 2010;429(3):545–52. doi: 10.1042/BJ20091960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman SL. Premature ovarian failure among fragile X premutation carriers: Parent-of-origin effect? Am J Med Genet. 2000;67(1):11–13. doi: 10.1086/302985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song FJ, Barton P, Sleightholme V, Yao GL, Fry-Smith A. Screening for fragile X syndrome: a literature review and modelling study. Health Technol Assess. 2003;7(16):1–106. doi: 10.3310/hta7160. [DOI] [PubMed] [Google Scholar]
- Sparrow SS, Cicchetti DV, Balla DA. Vineland Adaptive Behavior Scales. 2. Circle Pines: AGS Publishing; 2005. [Google Scholar]
- Sullivan SD, Welt C, Sherman S. FMR1 and the continuum of primary ovarian insufficiency. Semin Reprod Med. 2011;29(4):299–307. doi: 10.1055/s-0031-1280915. [DOI] [PubMed] [Google Scholar]
- Tassone F, Greco CM, Hunsaker MR, Seritan AL, Berman RF, Gane LW, Jacquemont S, Basuta K, Jin LW, Hagerman PJ, Hagerman RJ. Neuropathological, clinical and molecular pathology in female fragile X premutation carriers with and without FXTAS. Genes Brain Behav. 2012;11(5):577–585. doi: 10.1111/j.1601-183X.2012.00779.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tassone F, Pan R, Amiri K, Taylor AK, Hagerman PJ. A rapid polymerase chain reaction-based screening method for identification of all expanded alleles of the fragile X (FMR1) gene in newborn and high-risk populations. J Mol Diagn. 2008;10(1):43–9. doi: 10.2353/jmoldx.2008.070073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarborough M, Scott JA, Dixon LK. The role of beneficence in clinical genetics: non-directive counseling reconsidered. Theor Med. 1989;10(2):139–49. doi: 10.1007/BF00539879. [DOI] [PubMed] [Google Scholar]
- Yrigollen CM, Durbin-Johnson B, Gane L, Nelson DL, Hagerman R, Hagerman PJ, Tassone F. AGG interruptions within the maternal FMR1 gene reduce the risk of offspring with fragile X syndrome. Genet Med. 2012 doi: 10.1038/gim.2012.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yrigollen CM, Tassone F, Durbin-Johnson B. The role of AGG interruptions in the transcription of FMR1 premutation alleles. PLoS One. 2011;6(7):e21728. doi: 10.1371/journal.pone.0021728. [DOI] [PMC free article] [PubMed] [Google Scholar]